

Abstract
To investigate the contribution of the folding cores to the thermodynamic stability of RNases H, we used rational design to create two chimeras composed of parts of a thermophilic and a mesophilic RNase H. Each chimera combines the folding core from one parent protein and the remaining parts of the other. Both chimeras form active, well-folded RNases H. Stability curves, based on CD-monitored chemical denaturations, show that the chimera with the thermophilic core is more stable, has a higher midpoint of thermal denaturation, and a lower change in heat capacity (ΔCp) upon unfolding than the chimera with the mesophilic core. A possible explanation for the low ΔCp of both the parent thermophilic RNase H and the chimera with the thermophilic core is the residual structure in the denatured state. On the basis of the studied parameters, the chimera with the thermophilic core resembles a true thermophilic protein. Our results suggest that the folding core plays an essential role in conferring thermodynamic parameters to RNases H.
Keywords: Thermodynamic stability of proteins, chimera, stability curves, heat capacity, folding core, ribonuclease H
How proteins from thermophilic organisms (i.e., thermophilic proteins) achieve high thermostability is a question central to our understanding of proteins. Structures of thermophilic proteins have not revealed any obvious stabilization strategies; they are usually very similar to those of homologous proteins found in mesophilic organisms. An example of two structurally similar but thermodynamically distinct proteins is a pair of two ribonucleases H (RNase H), one from Escherichia coli (a mesophile), and one from Thermus thermophilus (a thermophile). These RNases H are small, single-domain proteins, with 52% sequence identity (Fig. 1 ▶).
Fig. 1.

Sequence alignment of T. thermophilus and E. coli RNases H* (cysteine-free variants of RNase H). Vertical lines separate the core region from the remaining part (outside) of the protein. The bold letters indicate the sequence of TCEO chimera (T. thermophilus Core E. coli Outside), whereas the non-bold letters correspond to the sequence of ECTO chimera (E. coli Core T. thermophilus Outside). A cartoon of secondary structural elements (rectangles labeled αA–αE correspond to helices; arrows labeled β1–β5 correspond to strands) is shown below the sequences.
Although T. thermophilus and E. coli RNases H have indistinguishable architectures (Fig. 2 ▶) (r. m. s. d. = 1.4Å), they are thermodynamically very different (J. Hollien and S. Marqusee 1999b). Stability curves and native-state hydrogen exchange experiments have been used to map the folding energy landscapes of the two RNases H (Hollien and Marqusee 1999a,b). The thermophilic RNase H is more stable than the mesophilic protein at all temperatures; however, its stability has a shallower temperature dependence, that is, a lower change in heat capacity upon unfolding (ΔCp). The result is that the thermophilic RNase H undergoes thermal denaturation 20°C above the melting point of the E. coli RNase H. This is particularly surprising given the similarity in sequence and structure.
Fig. 2.

Crystal structures of (left) E. coli RNase H* (Goedken et al. 2000) and (right) T.thermophilus RNase H (Ishikawa et al. 1993). Folding cores are shown in dark grey.
In both the E. coli and T. thermophilus RNases H, native-state hydrogen exchange reveals that two central helices (helix A and D) are significantly more stable than the rest of the protein (Chamberlain et al. 1996; Hollien and Marqusee 1999a). Studies on the kinetics of folding have shown that for both proteins, these two helices fold first, forming an early folding intermediate (Raschke and Marqusee 1997; Hollien and Marqusee, unpubl.). Models of these intermediates as isolated fragments were found to fold independently (Chamberlain 1999; K.F. Fisher, unpubl.). All of these observations lead to a model in which the two central helices (helix A and D) compose the crucial folding core of RNase H (Fig. 2 ▶).
Given the core's apparent importance for the stability and folding of RNase H, we questioned whether the core might also be essential in conferring some of the thermophilic characteristics to T. thermophilus RNase H. To test this hypothesis, we designed a system of two chimeric RNase H molecules, one containing the core of the mesophilic RNase H, surrounded by the remaining residues from thermophilic RNase H; and the other with the thermophilic core combined with the remaining residues from the mesophilic RNase H. By incorporating a thermophilic core into a mesophilic protein, and vice versa, we were able to dissect the contributions of the core and periphery to the thermodynamic stabilities of these RNases H. Our studies show that the folding core is more important in conferring the thermodynamic stability profile to T. thermophilus RNase H than the remaining parts of the protein.
Results
The designed chimeras are folded and active
We designed two chimeric RNase H molecules, each of which is a combination of fragments from a mesophilic (E. coli) and a thermophilic (T. thermophilus) RNase H. Each chimera contains the core region from one protein, and the remaining residues, corresponding to the outer region, from the other. To define the sequence boundaries of this core, we used the program RAFT, which searches for fragments as potential autonomous folding units using the structure of the full-length protein on the basis of an evaluation of contact density. In previous studies, RAFT identified a region representing the folding core of RNase H (Fischer and Marqusee 2000).
One chimera, ECTO (E. coli Core T. thermophilus Outside), consists of the core region (43–122) of E. coli, and the outer residues (−5–42 and 123–166, based on E. coli RNase H numbering) from T. thermophilus RNase H (Fig. 1 ▶). The other chimera, TCEO (T. thermophilus Core E. coli Outside), is the converse of ECTO, it contains the core from T. thermophilus RNase H combined with the remaining pars of E. coli RNase H. Analysis of contacting residues in the potential interface of the chimeras suggested that very few unfavorable interactions would be introduced in the chimeric proteins, assuming that the structures of the chimeras would not differ from the parent proteins (this was confirmed for the TCEO chimera, see below). The analysis revealed that there are only three core-periphery pairs for which the pair of interacting residues showed significantly different chemistries between the two parent proteins. Contacting residue pairs V54–F35, D66–A6, and R117–R4 in E. coli RNase H are analogous to E54–L35, I66–E6, and K117–Q4 in T. thermophilus RNase H. In the chimeras, these pairs are split, so that one residue originating from E. coli contacts the corresponding interaction partner from T. thermophilus RNase H. Of these interfacial contacts, only one introduces a potentially unfavorable electrostatic interaction to the TCEO chimera. D66–E6 introduces a potential charge–charge repulsion, not found in either of the parent proteins. No such interactions were predicted to be introduced into ECTO chimera. The lack of additional potentially unfavorable interactions encouraged us to proceed with the creation and characterization of the chimeras.
Both chimeras were over-expressed in E. coli and purified as soluble proteins (see Materials and Methods). Low-resolution studies [circular dichroism (CD) and activity assays] suggest that both fold into active RNases H. RNase H activity was monitored by a UV-absorbance-based activity assay, using a DNA–RNA hybrid as substrate (data not shown). Figure 3 ▶ shows the far-UV CD spectra of all four proteins (two chimeras plus two parent proteins). These data are difficult to interpret due to the fact that, in spite of having the same three-dimensional structure, the CD spectra of the parent proteins are different. The spectrum of ECTO overlays that of T. thermophilus RNase H. The CD spectrum of TCEO differs from both of the parent proteins; however, X-ray crystallography confirmed that it adopts the RNase H fold (see below). Equilibrium sedimentation experiments confirmed that TCEO and ECTO are monomeric proteins (data not shown). In sum, ECTO and TCEO both appear to be well folded and functional RNases H.
Fig. 3.

CD spectra of E. coli RNase H* (□) T. thermophilus RNase H* (○), ECTO (▾) and TCEO(▴) chimera.
Crystal structure of TCEO shows the RNase H fold
Diffraction-quality TCEO crystals were obtained in the presence of 50 mM Tris (ph 8.0) and 18% PEG 600, using the hanging-drop method. The protein crystallized in the space group P21, and diffracted to 1.8Å (Table 1), with four TCEO molecules in each asymmetric unit. The structure was solved by molecular replacement, using E. coli RNase H* as the starting model (Table 1). Residues 3–152 were modeled into the density map of two molecules in the asymmetric unit; the remaining two molecules, however, showed no clear density in the basic-helix-loop region (residues 80–97), which is distal to the interface generated by combining the fragments. The final refined structure has a crystallographic R-factor (Rcryst) of 24.7, and a free R-factor (Rfree) of 28.5%.
Table 1.
Crystallographic and refinement data
| Space group | P21 |
| Cell dimensions (Å) | a = 56.2, b = 87.0, c = 60.7 |
| α = 90°, β = 91.90°, γ = 90° | |
| Number of molecules per ASU | 4 |
| Resolution range (Å) | 20–1.76 |
| Observed reflections | 62607 (6102)a |
| Unique reflections | 5255 (189) |
| Completeness (%) | 97.7 (95.7) |
| I/σ | 18.8 (2.52) |
| Rsymb | 6.1 (27.7) |
| Rcyst (%)c | 24.7 |
| Rfree (%) | 28.5 |
| Number of atoms | 4709 |
| Number of water molecules | 281 |
| R.m.s. deviation of bonds (Å) | 0.005 |
| R.m.s. deviation of angles (°) | 1.25 |
a Values within parentheses are for the highest resolution shell (1.83–1.76 Å).
b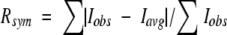 in which summation is over all reflections.
in which summation is over all reflections.
c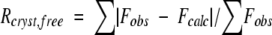 in which R factors are calculated using working and free reflection sets, respectively. The free reflections comprise a random 10% of the data set.
in which R factors are calculated using working and free reflection sets, respectively. The free reflections comprise a random 10% of the data set.
TCEO adopts the same fold as E. coli RNase H (Fig. 4 ▶). The backbone root mean square displacement between the two proteins is only 0.9 Å. Systematic visual comparison between TCEO and E. coli RNase H reveals that there are no obvious packing defects along the interface between the core and the outside region.
Fig. 4.

Ribbon diagram of the crystal structure of TCEO (shown in black) overlaid with the structure of E. coli RNase H* (shown in light gray) (Goedken et al. 2000).
TCEO is more stable than ECTO
The thermodynamic stabilities of both chimeras were measured by monitoring the thermal- and chemical-induced denaturation by CD, using both urea and guanidinium chloride (GdmCl) as denaturants. All denaturation curves were fit assuming a two-state transition with a linear extrapolation model (Santoro and Bolen 1988).
For ECTO, both GdmCl and urea denaturation profiles resulted in a ΔGunf of 5.6 ± 0.3 kcal mole−1 and m values of 3.9 ± 0.4 and 1.8 ± 0.2 kcal mole−1 M−1, respectively, at 25°C (Fig. 5 ▶; Table 2). Thermal denaturation was reversible, with a Tm of 61°C (Fig. 5 ▶; Table 2).
Fig. 5.



Thermodynamic stability of TCEO (▴) and ECTO (▾) compared with E. coli (□) and T. thermophilus RNases H* (○): (a) Thermal melts in 1 M guanidium chloride (E. coli RNase H thermal denaturation is not reversible in the absence of denaturants); (b) Guanidinium chloride melts, (c) Urea melts. The error bars correspond to errors of fits to a two-state model.
Table 2.
Thermodynamic parameters for ECTO and TCEO chimera, compared with E. coli and T. thermophilus RNases H*
| E. coli RNase H* | T. thermophilus RNase H* | ECTO | TCEO | |
| Tm (°C) | 66 ± 1 | 86 ± 1 | 61 ± 1 | 76 ± 1 |
| ΔGH20 at 25°C (kcal mole−1) (guanidine melt) | 7.6 ± 0.2 | 12.2 ± 0.3 | 5.6 ± 0.3 | 7.5 ± 0.4 |
| m value (kcal mole−1 M−1) (guanidine melt) | 4.9 ± 0.2 | 4.4 ± 0.1 | 3.9 ± 0.4 | 3.8 ± 0.3 |
| ΔGH20 at 25°C (kcal mole−1) (urea melt) | 9.7 ± 0.3 | 12.8 ± 0.7 | 5.6 ± 0.5 | 11.9 ± 0.8 |
| m value (kcal mole−1 M−1) (urea melt) | 2.1 ± 0.1 | 1.9 ± 0.2 | 1.8 ± 0.2 | 2.0 ± 0.2 |
| ΔCp (kcal mole−1 K−1) | 2.7 ± 0.2 | 1.8 ± 0.1 | 2.4 ± 0.3 | 1.6 ± 0.2 |
TCEO is thermodynamically more stable than ECTO. GdmCl denaturation yields a ΔGunf of 7.5 ± 0.5 kcal mole−1 with an m value of 3.8 ± 0.3 kcal mole−1 M−1, whereas urea denaturation yields an even higher ΔGunf of 11.9 ± 0.8 kcal mole−1, with an m value of 2.0 ± 0.2 kcal mole−1 M−1 (Fig. 5 ▶; Table 2). The Tm of TCEO is 76°C (Fig.5 ▶; Table 2). Because TCEO has a higher Tm and a higher thermodynamic stability (as determined by urea denaturation) than E. coli RNase H, TCEO resembles a thermophilic protein.
Stability curves indicate that TCEO has a lower ΔCp than ECTO
Denaturation studies were carried out as a function of temperature to further characterize the thermodynamic stability of the two chimeras. CD-monitored GdmCl-induced denaturations were followed at eight different temperatures for ECTO and seven different temperatures for the TCEO chimera; each profile was fit to a two-state model to estimate the stability (ΔGunf) at a given temperature. Stability curves for ECTO and TCEO were generated by plotting stabilities as a function of temperature (Pace and Laurents 1989; Fig. 6 ▶). Because both chimeras undergo reversible thermal denaturation in the absence of denaturants, data from thermal denaturations were also used (squares in Fig. 6 ▶). All of the data points were then fit to the Gibbs-Helmholtz equation (see Materials and Methods).
Fig. 6.

Stability curves TCEO (• and ▪) and ECTO (○ and □) compared with the stability curves of T. thermophilus (broken line) and E. coli RNases H (dotted line) (Hollien and Marqusee 1999b). Each circle corresponds to a ΔG obtained from a guanidine denaturation experiment. Squares correspond to ΔGs obtained from temperature denaturation experiments. The dash-dot plot represents the constrained fit of TCEO stability data to the Gibbs-Helmholtz equation, in which ΔCp is fixed at 2.2 kcal mole−1 K−1. Fitting the stability curves to Gibbs-Helmholtz equation (without any constraints) results in ΔCp of 1.6 ± 0.2 kcal mole–1 K−1, Tm of 76 ± 1°C, and ΔH of 68 ± 2 kcal for TCEO; and ΔCp of 2.4 ± 0.3 kcal mole−1 K−1, Tm of 61 ± 1°C, and ΔH of 89 ± 2 kcal for TCEO.
The differences in the curvature of the stability curves (Fig. 6 ▶) suggest that the change in heat capacity upon unfolding (ΔCp) is lower for TCEO compared with ECTO. TCEO has a ΔCp of 1.6 ± 0.2 kcal mole−1 K−1, whereas ECTO has a ΔCp of 2.4 ± 0.3 kcal mole−1 K−1, compared with 1.8 kcal mole−1 K−1 for T. thermophilus RNase H, and 2.7 kcal mole−1 K−1 for E. coli RNase H (Hollien and Marqusee 1999b). The lower ΔCp of TCEO mirrors that of T. thermophilus RNase H (Table 2). Hence, the lower ΔCp, which is an important contributor to the thermophilic profile of T. thermophilus RNase H, tracks with its core and contributes to the thermophile-like profile of the TCEO chimera.
Discussion
Studying chimeric proteins is a powerful way to investigate the functional and structural relevance of protein domains and fragments. In this study, we investigated the role of the folding core in the thermodynamic profile of RNase H, that is, how the folding core contributes to the thermodynamic stability of RNases H. In particular, we wanted to know what role the core played in the thermophilic nature of T. thermophilus RNase H. To this end, we generated two chimeras by swapping the cores between the T. thermophilus and E. coli RNases H.
The use of chimeras to evaluate contributions of protein segments to the stability and folding of the whole protein poses its own challenges and complications. Packing defects, resulting from creation of new interfaces between protein fragments, often reduce the overall thermodynamic stability (Kenig et al. 2001). This masks any potential positive contribution of the studied fragment to the stability of the parent protein. Creation of new interfaces within chimeras can even change the folding mechanism (Numata et al. 1999). Disruption of the folding mechanism and introduction of packing defects can be minimized by careful selection of boundaries of fragments used to construct a chimeric protein.
Sequence alignments, protease digestions, and analyses of high-resolution structures are often used to determine domain boundaries. In this study, the rational design of the chimeric proteins was based on the RAFT algorithm, which predicts fragments of a protein likely to fold independently (Fischer and Marqusee 2000). We chose the RNase H fragment with the highest RAFT score for both T. thermophilus and E. coli RNase H. This fragment contains the two central helices, which play an essential role both in the thermodynamic stability (Chamberlain et al. 1996; Hollien and Marqusee 1999b) and the kinetic folding pathway of the two RNases H (Raschke and Marqusee 1997; J. Hollien and S. Marqusee, unpubl.) forming the thermodynamic and the kinetic folding core of RNase H.
Characterization of both proteins (ECTO, E. coli Core T. thermophilus Outside and TCEO, T. thermophilus Core E. coli Outside) shows that, despite the introduction of numerous new, potentially unfavorable interactions along the interface, both chimeras adopt the normal RNase H fold. This shows that in addition to design of small protein fragments (its original purpose), the RAFT score is also a useful criterion for choosing boundaries for chimeras. Successful generation of two well-folded chimeras, which combine cores and outside regions of two different proteins, implies that the folding core of RNase H is indeed an independent folding module. Not only can the core fold by itself, but also, it can be incorporated successfully into a different RNase H homolog.
The two functional chimeric proteins provide a system to assess relative contributions of core and outside regions to the stability and folding of RNase H. If all residues in the sequence were equally important for thermodynamic stability, we would expect the ECTO chimera to be more thermostable than TCEO. The core region is more conserved between E. coli and T. thermophilus RNases H, than the remaining part of the protein (58% vs. 48% identity for core and outside regions, respectively), and consequently ECTO shares more of its sequence with T. thermophilus than does TCEO (80% vs. 70%). Despite this, the TCEO chimera has a higher Tm than the ECTO chimera, and is also more thermodynamically stable at room temperature, as determined by both urea and guanidinium chloride denaturant titrations.
Interestingly, unlike ECTO, which has the same stability in urea and GdmCl (5.6 kcal mole−1), TCEO is much more stable in urea compared with GdmCl (11.9 vs. 7.5 kcal mole−1) at room temperature (Table 2). Although discrepancies between ΔGunf values, determined with urea and guanidinium chloride, have been noted for some proteins, it is usually the value obtained from GdmCl denaturation that is higher than the value obtained in urea (Makhatadze 1999). Perhaps TCEO has a lower stability in GdmCl because salts destabilize its native state. However, urea denaturation of TCEO in the presence of 1 M KCl yields an even higher stability than the value obtained from urea denaturation in the absence of salt (data not shown), suggesting that it is not the salt that preferentially destabilizes the folded state of TCEO.
The discrepancy between stability measurements in urea and GdmCl can be explained by either preferential binding of urea to the native TCEO, or by preferential binding of GdmCl to the denatured state of the TCEO chimera. E. coli RNase H also has a higher stability in urea than in GdmCl, although the difference is not as large (9.7 vs. 7.6 kcal mole−1 for urea and GdmCl, respectively). Perhaps, the periphery of E. coli RNase H (which is also present in TCEO) has more or tighter denaturant-binding sites than most proteins. The peripheries of E. coli and T. thermophilus RNases H do not differ significantly in the number and distribution of charged residues (E. coli RNase H has 10 negatively charged and 9 positively charged residues in the periphery, whereas T. thermophilus RNase H has 9 negatively and 11 positively charged residues in the periphery), and, hence, it is difficult to asses whether guanidine is acting to destabilize TCEO or whether urea stabilizes it.
Despite the lower stability in guanidium (compared with urea), the stability curve shows that TCEO is more stable than ECTO at all temperatures (Fig. 6 ▶). This, along with high Tm shows that the core region plays a significant role in conferring thermophilic character to the profile of this chimeric RNase H. Furthermore, the stability curve (temperature dependence of ΔGunf) is shallower for TCEO than ECTO. Therefore, like the parent thermophile, TCEO has a low ΔCp, whereas ECTO has a higher ΔCp, similar to E. coli RNase H. It appears that the ΔCp of each chimera correlates with that of the parent protein from which the core originates. This correlation, as well as the difference in ΔCp between ECTO and TCEO (2.4 ± 0.3 vs. 1.6 ± 0.2, respectively), is surprising. The m values and ΔCps are correlated for a large set of mesophilic proteins (Myers et al. 1995), and the m values from both urea and guanidium chloride denaturations do not differ significantly between TCEO and ECTO (Table 2). If we use this correlation to predict the ΔCp based on the measured m values, we predict a ΔCp close to the observed one for ECTO, but overestimate the ΔCp for TCEO. For ECTO we expect the ΔCp of 2.2.or 2.1 kcal mole−1K−1 (based on GdmCl and urea m values, respectively), which is within the calculated error of the value obtained from fitting the stability curves (2.4 ± 0.3 kcal mole−1K−1). For TCEO the correlation predicts a ΔCp of 2.2 or 2.3 kcal mole−1K−1 (based on GdmCl and urea m values, respectively), which is significantly different from 1.6 ± 0.2 kcal mole−1K−1 determined from the stability curves (Table 2). The dash-dot plot in Figure 6 ▶ shows that constraining the ΔCp to the value predicted by the m value (2.2 kcal mole−1 K−1) yields a poor fit, which does not account for the measured stability data.
Our observations lead us to question the way we interpret the change in heat capacity upon protein unfolding. The Δ4Cp and the m values are both interpreted in terms of the change in the accessible surface area (ASA) upon unfolding (Livingstone et al. 1991). We determined the stability curves by measuring the ΔGunf at different temperatures. If the change in ASA upon unfolding were temperature dependent (i.e., the denatured state ensemble changed significantly different under different conditions), we might not expect the ΔCp derived from a stability curve to correlate with the m values measured at room temperature. However, this does not appear to be the case, because for both TCEO and ECTO, the m values do not vary systematically with temperature (data not shown). In addition, at any given temperature, the m values are not significantly different between the two chimeras.
Perhaps the unusually low ΔCp is a result of a change in the native state. In the crystal structure of TCEO, residues 80–97 were disordered in two of the four molecules in the asymmetric unit. This local disorder could lower the ASA of the native state; the smaller change in ASA would account for the smaller ΔCp in TCEO. However, this fails to account for the fact that the m values are not significantly different in TCEO and ECTO. It also fails to explain the unusually low ΔCp for T. thermophilus RNase H (Hollien and Marqusee 1999b), in which there is no such disorder. Thus, a change in the structure of the native state is unlikely to account for the unusual ΔCp.
Another possible explanation for the lower ΔCp of the T. thermophilus RNase H and TCEO chimera is a difference in the structure of the denatured state. Perhaps for T. thermophilus RNase H and TCEO, the denatured state under native conditions contains residual structure and is more compact. The m values are derived from the transition data in higher denaturant, and therefore, might not be expected to follow the same trend. As the common feature between these two proteins is the core, this suggests that the core itself may contain residual structure. We are currently investigating this possibility using both differential scanning calorimetry and hydrogen-deuterium exchange in the denatured state.
In conclusion, the structural and thermodynamic properties of the two chimeras show that the previously identified folding cores of RNase H are independent folding units that can be combined successfully with regions from homologous proteins. Whereas both chimeras are folded and active, only the chimera with the thermophilic core region has thermodynamic parameters that resemble those of the thermophilic RNase H. These include higher Tm and lower ΔCp, and higher stability, as determined from urea denaturations. This is consistent with the core region playing a dominant role in the thermostability profile of RNases H.
Materials and methods
Design and construction of synthetic genes for ECTO and TCEO chimeric RNases H
To define the boundaries for the folding core and the chimeras, we used the RAFT score (Fischer and Marqusee 2000), which identifies autonomous folding units of proteins, on the basis of the density of contacts in the three-dimensional structure of the native protein. The core was defined as the protein fragment with the highest RAFT score. For E. coli RNase H, the highest scoring fragment contained residues 43–122 (Fischer and Marqusee 2000). A homologous fragment (residues 43–122, based on E. coli RNase H amino acid numbering, in which the fifth residue of T. thermophilus RNase H corresponds to the first residue of E. coli RNase H), also scores highest among all possible contiguous T. thermophilus RNase H fragments. Prior to construction of ECTO and TCEO chimeras, we analyzed the crystal structures of the two parent proteins to investigate whether we would introduce any obvious destabilizing interactions by combining the parts of two different proteins. The program Contacts (Bailey 1994) was used to identify all core residues that are between 0.5 Å and 5 Å from a non-core residue. Only non-backbone, non-Cα, non-Cβ atoms were considered in this analysis.
Plasmids encoding cysteine-free variants of E. coli (pSM101) (Dabora and Marqusee 1994) and T. thermophilus (pJH109) (Hollien and Marqusee 1999b) RNase H were used to construct the two chimeric RNases H. The core regions (residues 43–122, based on E. coli sequence) were amplified by PCR from both parent plasmids. The plasmid PJH109 and the E. coli core amplicon were digested with StuI and MluI restriction enzymes, in order to remove the core-coding sequence from pJH109 and generate the insertion site for the E. coli RNase H core sequence. The digested vector and insert were gel purified, ligated together, and sequenced. The resulting plasmid (pSR102) encodes the ECTO chimera (E. coli Core T. thermophilus Outside). Similarly, the plasmid PSM101 and the T. thermophilus core amplicon were digested with BstEII and MluI. The digested vector and insert were gel purified, ligated, and the products of the ligations were sequenced. The resulting plasmid (pSR202) encodes the TCEO chimera (T. thermophilus Core E. coli Outside).
Expression and purification of ECTO and TCEO proteins
Plasmids encoding the TCEO and ECTO chimeras were transformed into E. coli BL21 pLys S (Novagen) cells. TCEO transformants were grown at 30°C (no colonies appeared if grown at 37°C), whereas ECTO transformants were grown at 37°C. Liquid cultures, started from individual colonies grown in Luria broth, with 200 μg/mL ampicillin were induced with 1 mM IPTG at an A600 of 0.5. Cells expressing ECTO were induced for 3 h at 37°C before harvesting, whereas cells expressing TCEO were induced for 4.5 h at 30°C.
Cells were harvested by centrifugation and lysed by sonication in 50 mM Tris (pH 8.0), 20 mM NaCl, 0.1 mM EDTA (buffer A). Both TCEO and ECTO were found in the soluble fraction, which was loaded onto a FPLC heparin column pre-equilibrated with buffer A. The bound ECTO protein was eluted by a linear gradient between 20 and 600 mM NaCl (protein eluted at 400 mM NaCl), in 50 mM Tris (pH 8.0) and 0.1 mM EDTA. The pH of the pooled, protein-containing fractions was adjusted to pH 5.5 using concentrated acetic acid, and the solution was diluted to a final concentration of 200 mM NaCl. The protein sample was then applied to a Source S15 FPLC column pre-equilibrated with 20 mM NaOAc (pH 5.5), 200 mM NaCl, and 0.1 mM EDTA, and was eluted with a linear gradient between 200 and 400 mM NaCl (protein eluted at 300 mM NaCl). TCEO was purified over a heparin FPLC column at pH 8.0, following the same protocol as for ECTO (see above). ECTO eluted at 400 mM NaCl. Pooled heparin fractions were concentrated (by ammonium sulfate precipitation or Amicon concentrators), to the final volume of 5–10 mL, and applied to a gravity-flow Sephadex G-75 gel filtration column. The molecular weight of pure ECTO and TCEO RNases H (as determined by SDS-PAGE) was confirmed by mass spectrometry. Pure protein was dialyzed into 50 mM ammonium bicarbonate (pH 7.0), lyophilized, and stored in powder form.
Circular dichroism studies
Circular dichroism (CD) spectra of ECTO and TCEO chimera were collected on an Aviv 62DS spectrometer, in a 1-cm pathlength cuvette at 25°C. The spectra were taken in 5 mM NaOAC (pH5.5). Data points were recorded from 300 to 200 nm, at 0.5-nm intervals. Each data point was averaged for 3 sec.
Thermal and chemical (urea and guanidium chloride) denaturations were monitored by CD at 222 nm. All experiments were performed in 1-cm pathlength cuvettes, using 50 μg/mL of protein in 20 mM NaOAc and 50 mM KCL (pH 5.5). For thermal denaturation, data were gathered every 3°C, with a 3-min equilibration time, and each data point was averaged for 1 min. To test the reversibility of thermal denaturation, a CD spectrum was taken at room temperature after thermal denaturation, and compared with the spectrum taken prior to denaturation. Reversibility was defined as preservation of more than 95% of CD signal between 220 and 225 nm. For guanidinium chloride and urea-induced denaturation, individual samples with various concentrations of denaturant were prepared and equilibrated for 24 h at room temperature. The CD signal of each sample was averaged for 1 min. Denaturant concentrations were verified using a refractometer (Pace et al. 1989).
To generate stability curves for TCEO and ECTO, GdmCl-induced denaturation experiments (see above), were performed at different temperatures (Pace and Laurents 1989), ranging from 4 to 45°C. GdmCl was chosen as the denaturant for comparison with previous studies on the parent proteins (Hollien and Marqusee 1999b). Samples were equilibrated at appropriate temperatures (in a heat block or in an ice-water bath) between 4 and 24 h prior to CD measurements (longer at lower temperatures). Each sample was further equilibrated for 3 min more, after it was placed in the CD sample holder with a Peltier temperature regulator.
Denaturation free energies (ΔGunf) were determined from GdmCl-induced denaturation experiments at different temperatures, assuming a two-state model and a linear dependence of ΔGunf on the concentration of GdmCl (Santoro and Bolen 1988). Thermal melts were fit to a two-state model, to determine the Tm of each protein, and the ΔGunf in the transition range of the thermal denaturation profile. The free energies of unfolding, obtained from both GdmCl and thermal denaturation experiments, were plotted as a function of temperature. Each point on the stability curve is the average of at least two experiments. The stability curve data were fit to the Gibbs-Helmholtz equation:
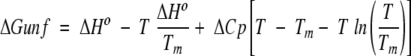 |
1 |
in which ΔHo is the enthalpy at Tm, and ΔCp is the change in heat capacity upon unfolding (Becktel and Schellman 1987). We assumed that ΔCp is constant in the experimental temperature range. All curve fitting was done using Sigma Plot (Jandel Scientific).
Activity assay
A UV-based spectrophotometric RNase H assay was used to test the activity of the two chimeras (partially on the basis of Black and Cowan 1994). The assay measures the loss of hypochromic effect resulting from the cleavage of the RNA moiety in DNA–RNA hybrids. Reactions were initiated by addition of 5 nM of ECTO or TCEO to a solution containing 25 μg/mL of an RNA/DNA hybrid (poly-rA/poly-dT) in the presence of 10 mM MgCl2 and 50 mM Tris (pH8.0) at 25°C. The loss of hypochromic effect was measured by monitoring the increase of absorbance at 260 nM. Activity was determined from the slope of the initial linear phase of the kinetic profile.
Equilibrium sedimentation
Ultracentrifugation experiments were performed in a Beckman Optima XL-A ultracentrifuge. Measurements were performed at three different concentrations of each chimera (ranging from 50 to 400 μg/mL), in 20 mM NaOAC and 50 mM KCl (pH5.5) at 25°C (the same conditions as the CD experiments).
Crystal structure of TCEO
The hanging-drop method was used to set crystal trays with TCEO and ECTO chimeras. Lyophilized protein was dissolved in water and filtered through 0.2-μm HT Tuffryn filters (Pall Gelman Laboratory), resulting in the final protein concentration of 6 mg/mL. Precipitant (PEG and Ammonium Sulfate) concentration, pH, and salt concentration were varied around the conditions in which E. coli RNase H* crystallized (Goedken et al. 2000). Octahedral crystals of TCEO were grown in 50 mM Tris (pH8.0) and 18% PEG600, flash-frozen directly from the drop into liquid nitrogen, and screened for diffraction on a Rigaku RU-200 generator equipped with a IIc detector. The full X-ray diffraction data set (280 frames) was collected at Stanford Synchrotron Radiation Laboratory Beamline 9.2. The data were integrated and scaled using Denzo and Scale Pack (Otwinowski and Minor 1997). The number of molecules per asymmetric unit (ASU) was calculated assuming the Matthews coefficient of 2.5 Å3/Da (Matthews 1968).
The crystal structure was solved by molecular replacement using AMoRe (Navaza 1993) and CNS (Brunger et al. 1998), with the cysteine-free variant of E. coli RNase H structure as the starting model (Protein Data Bank [PDB] code 1F121). All residues that differed between E. coli RNase H* and TCEO were replaced with serines in the starting model. Models for each one of the four TCEO molecules in the ASU were built in O (Jones et al. 1991). The model was further refined using RefMac (Murshudov et al. 1997) and CNS (Brunger et al. 1998). Cycles of model building and refinement were repeated until the free R-factor converged. The coordinates have been deposited in the PDB (PDB code 1JL2).
Acknowledgments
This work was supported by NIH grant GMS0945. We thank Eric Goedken and James Holton for assistance with X-ray crystallography; Sarah McWhirter for synchrotron data collection; Giulietta Spudich for critical reading of this manuscript; Kael Fisher for RAFT scores for RNase H fragments, help with equilibrium centrifugations, discussions about chimera design, and reading of this manuscript; and David King for mass spectromentry.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.38602.
References
- Bailey, S. 1994. The Ccp4 suite – programs for protein crystallography. Acta Crystallogr. Sect. D-Biol. Crystallogr. 50 760–763. [DOI] [PubMed] [Google Scholar]
- Becktel, W.J. and Schellman, J.A. 1987. Protein stability curves. Biopolymers 26 1859–1877. [DOI] [PubMed] [Google Scholar]
- Black, C.B. and Cowan, J.A. 1994. Magnesium aactivation of ribonuclease H – evidence for one catalytic metal ion. Inorganic Chem. 33 5805–5808. [Google Scholar]
- Brunger, A.T., Adams, P.D., Clore, G.M., Delano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography and NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. Sect. D Biol.l Crystallogr. 54 905–921. [DOI] [PubMed] [Google Scholar]
- Chamberlain, A.K., Fisher, K.F., Reardon, D., Handel, T.M., and Marqusee, S. 1999. Folding of an isolated ribonuclease H core fragment. Protein Science 8 2251–2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain, A.K., Handel, T.M., and Marqusee, S. 1996. Detection of rare partially folded molecules in equilibrium with the native conformation of RNaseH. Nat. Struct. Biol. 3 782–787. [DOI] [PubMed] [Google Scholar]
- Dabora, J.M. and Marqusee, S. 1994. Equilibrium unfolding of Escherichia coli ribonuclease H: Characterization of a partially folded state. Protein Sci. 3 1401–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer, K.F. and Marqusee, S. 2000. A rapid test for identification of autonomous folding units in proteins. J.Mol. Biol. 302 701–712. [DOI] [PubMed] [Google Scholar]
- Goedken, E.R., Keck, J.L., Berger, J.M., and Marqusee, S. 2000. Divalent metal cofactor binding in the kinetic folding trajectory of Escherichia coli ribonuclease HI. Protein Sci. 9 1914–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollien, J. and Marqusee, S. 1999a. Structural distribution of stability in a thermophilic enzyme. Proc. Natl. Acad. Sci. 96 13674–13678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 1999b. A thermodynamic comparison of mesophilic and thermophilic ribonucleases H. Biochemistry 38 3831–3836. [DOI] [PubMed] [Google Scholar]
- Ishikawa, K., Okumura, M., Katayanagi, K., Kimura, S., Kanaya, S., Nakamura, H., and Morikawa, K. 1993. Crystal structure of ribonuclease H from Thermus thermophilus HB8 refined at 2.8 A resolution. J. Mol. Biol. 230 529–542. [DOI] [PubMed] [Google Scholar]
- Jones, T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. Sect. a-Found. Crystallogr. 47 110–119. [DOI] [PubMed] [Google Scholar]
- Kenig, M., Jerala, R., Kroon-Zitko, L., Turk, V., and Zerovnik, E. 2001. Major differences in stability and dimerization properties of two chimeric mutants of human stefins. Proteins 42 512–522. [PubMed] [Google Scholar]
- Livingstone, J.R., Spolar, R.S., and Record, Jr., M.T. 1991. Contribution to the thermodynamics of protein folding from the reduction in water-accessible nonpolar surface area. Biochemistry 30 4237–4244. [DOI] [PubMed] [Google Scholar]
- Makhatadze, G.I. 1999. Thermodynamics of protein interactions with urea and guanidinium hydrochloride. J. Phys. Chem. B 103 4781–4785. [Google Scholar]
- Matthews, B.W. 1968. Solvent content of protein crystals. J. Mol. Biol, 33 491–497. [DOI] [PubMed] [Google Scholar]
- Murshudov, G.N., Vagin, A.A., and Dodson, E.J. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. Sect. D Biol. Crystallogr. 53 240–255. [DOI] [PubMed] [Google Scholar]
- Myers, J.K., Pace, C.N., and Scholtz, J.M. 1995. Denaturant m values and heat capacity changes: Relation to changes in accessible surface areas of protein unfolding. Protein Sci. 4 2138–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numata, K., Hayashiiwasaki, Y., Yutani, K., and Oshima, T. 1999. Studies on interdomain interaction of 3-isopropylmalate dehydrogenase from an extreme thermophile, Thermus thermophilus, by constructing chimeric enzymes. Extremophiles 3 259–262. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276 307–326. [DOI] [PubMed] [Google Scholar]
- Pace, C.N. and Laurents, D.V. 1989. A new method for determining the heat capacity change for protein folding. Biochemistry 28 2520–2525. [DOI] [PubMed] [Google Scholar]
- Pace, C.N., Shirley, B.A., and Thomson, J.A. 1989. Protein structure : A practical approach. In: Protein structure : A practical approach. (ed. T. Creighton), pp. 330–331. Oxford IRL Press, Washington, DC.
- Raschke, T.M. and Marqusee, S. 1997. The kinetic folding intermediate of ribonuclease H resembles the acid molten globule and partially unfolded molecules detected under native conditions. Nature Struct. Biol. 4 298–304. [DOI] [PubMed] [Google Scholar]
- Santoro, M.M. and Bolen, D.W. 1988. Unfolding free energy changes determined by the linear extrapolation method. 1. Unfolding of phenylmethanesulfonyl alpha-chymotrypsin using different denaturants. Biochemistry 27 8063–8068. [DOI] [PubMed] [Google Scholar]