

Abstract
Crystal structures have shown that the HIV-1 protease flaps, domains that control access to the active site, are closed when the active site is occupied by a ligand. Although flap structures ranging from closed to semi-open are observed in the free protease, crystal structures reveal that even the semi-open flaps block access to the active site, indicating that the flaps are mobile in solution. The goals of this paper are to characterize the secondary structure and fast (sub-ns) dynamics of the flaps of the free protease in solution, to relate these results to X-ray structures and to compare them with predictions of dynamics calculations. To this end we have obtained nearly complete backbone and many sidechain signal assignments of a fully active free-protease construct that is stabilized against autoproteolysis by three point mutations. The secondary structure of this protein was characterized using the chemical shift index, measurements of 3hJNC` couplings across hydrogen bonds, and NOESY connectivities. Analysis of these measurements indicates that the protease secondary structure becomes irregular near the flap tips, residues 49–53. Model-free analysis of 15N relaxation parameters, T1, T2 (T1ρ) and 15N-{1H} NOE, shows that residues in the flap tips are flexible on the sub-ns time scale, in contrast with previous observations on the inhibitor-bound protease. These results are compared with theoretical predictions of flap dynamics and the possible biological significance of the sub-ns time scale dynamics of the flap tips is discussed.
Keywords: AIDS, NMR, secondary structure, relaxation, hydrogen bonds
The HIV protease has served as an effective target for antiretroviral drug therapy because this enzyme is indispensable for viral maturation and propagation. Currently, several potent protease inhibitors are in clinical use (Shafer et al. 1999). However, the emergence of active, drug-resistant mutants of the protease has limited the long-term effectiveness of these inhibitors. For this reason, current interest lies in obtaining inhibitors directed at sites not targeted by current inhibitors. Combination therapies that target multiple protease sites are expected to hinder the selection of drug-resistant protein molecules.
All protease inhibitors currently used as anti-HIV drugs target the substrate-binding site of the enzyme, the large vacant region of the free protease ribbon structure shown in Figure 1 ▶. However, at least two other sites in the protein appear to be attractive drug targets. The first is the dimer interface, a four-stranded anti-parallel β-sheet at the base of the molecule (Fig. 1 ▶) made up of N- and C-terminal residues (1–4 and 96–99) of each monomer (Zutshi et al. 1997; Zutshi and Chmielewski 2000).The second is the flaps, residues 32–63 (Rose et al. 1998), which contain β-hairpin structures (residues 43–58) that form part of the substrate binding site (Fig. 1 ▶). X-ray studies of the ligand-bound protease show that the flap β-hairpins are well-ordered and interact with inhibitors and substrate analogs (Wlodawer and Erickson 1993). However, crystal structures of the free protease reveal more heterogeneous flap structures. These structures range from closed flap conformations (Rick et al. 1998; Pillai et al. 2001), such as those observed when an inhibitor is present, to semi-open conformations (Lapatto et al. 1989; Wlodawer et al. 1989; Spinelli et al. 1991) in which the β-hairpins structures have moved apart by several angstroms compared to the closed conformation. However, it is interesting that even the semi-open flaps do not allow substrates access to the active site (Rick et al. 1998). These conformations have been termed "open" (Rick et al. 1998), but we prefer the term "semi-open" because of this restricted access.
Fig. 1.

A colored ribbon representation of the backbone structure of the free HIV-1 protease homodimer. In the left monomer, the flap domain, residues 32`–63` is represented by the green ribbon, whereas the ribbon representing the remaining residues is cyan. In the right monomer, the flap is represented by a tri-colored ribbon: red, residues in the tip of the flap β-hairpin, 49–52; yellow, residues 43–48 and 53–58 in the two-stranded sheet portion of the flap β-hairpin; green, residues 37–42, referred to as the flap elbow. Several residues in the flap domain, are identified by number in the right monomer. The drawing was generated by INSIGHT using the X-ray coordinates PDB accession code 3PHV (Lapatto et al. 1989).
The variation in flap conformations observed in X-ray studies of the free protease indicates that the flaps in the free protease are flexible in solution. Calculations have predicted a variety of flap motions on various time scales (Harte et al. 1990; York et al. 1993; Collins et al. 1995; Liu et al. 1996; Rick et al. 1998; Scott and Schiffer 2000). The earlier calculations indicated that the flaps moved as relatively rigid levers that opened out and away from the protein, whereas more recent calculations predict a partial melting of the flap β-hairpins. For example, a reaction path calculation (Rick et al. 1998) predicts a conversion from semi-open to closed flap structures takes place through a melting of the β-hairpins at the flap tips, whereas a recent 10 ns solvated molecular dynamics simulation (Scott and Schiffer 2000), starting from the coordinates of an unliganded crystal structure (Spinelli et al. 1991), predicts that the protease flaps curl inward and make the active site accessible within a few ns.
Although the dynamics simulations have provided a variety of interesting and thought-provoking views of the movement of the flaps, it is desirable to compare the theoretical predictions with experimental measurements. High-resolution NMR is an experimental method that is well-suited to study the internal dynamics of proteins in solution on a wide range of time scales (Kay 1998; Ishima and Torchia 2000). However, such studies of the wild-type HIV-protease have been restricted to the inhibitor-bound protein because of rapid autoproteolysis of the free wild-type protease. Recently, we measured transverse spin relaxation rates of a fully active free-protease construct (Ishima et al. 1999) containing three mutations, Q7K, L33I, and L63I, that impede autoproteolysis (Rose et al. 1993; Mildner et al. 1994). Analysis of the transverse relaxation rates showed that residues in the protease N- and C-terminal dimer interface and in the flap hairpins were flexible on the chemical (conformational) exchange time scale, ms-μs. In the previous study (Ishima et al. 1999), we noted that preliminary relaxation data also indicated the presence of flap motions on the sub-ns time scale. Here, we present a detailed study of sub-ns backbone motions in the free protease and derive model-free (Lipari and Szabo 1982 a,b) order parameters and effective correlation times for most backbone amide sites. In addition we use chemical shifts, NOESY, and hydrogen bond measurements to characterize the secondary structure of the flap β-hairpins. These results on flap structural fluctuations complement those obtained previously using NMR (Ishima et al. 1999), fluorescence (Furfine et al. 1992; Rodriguez et al. 1993), and structural information provided by X-ray studies (Lapatto et al. 1989; Wlodawer et al. 1989; Spinelli et al. 1991; Wlodawer and Erickson 1993; Pillai et al. 2001). Theoretical predictions of the motions of the flaps of the free protease (Harte et al. 1990; Collins et al. 1995; Rick et al. 1998; Scott and Schiffer 2000) are compared with the experimental results.
Results and Discussion
Secondary structure of the flaps of the free protease
Nearly complete backbone signal assignments (>97%) were obtained using standard triple resonance experiments (see Supplemental Material); the amide proton and 15N assignments are shown on the 1H-15N HSQC spectrum of the free protease (Fig. 2 ▶). The signal assignments were used in conjunction with the chemical shift index (CSI; Wishart and Sykes 1994) to identify the elements of regular secondary structure in the protein. The CSI shows that, in solution, the backbone of the free protease, like that of the inhibitor-bound protease, is composed primarily of β-strands. As seen in Figure 3 ▶, the β-strand regions identified in solution are very similar to those identified in crystal structures of the free protease. It is interesting that this observation applies to flap residues 43–58. These residues are in the regions of the protease flaps that cover the active site. The CSI indicates that these residues form a pair of β-hairpin structures in solution that become disordered at the flaps tips (residues 49–53). This conclusion was verified by a 15N-separated 1H–1H NOESY spectrum (Fig. 4 ▶), which revealed cross-strand NH–NH NOE's K43–Q58, K45–V56, I47–I54, and a very weak G49–G52 NOE. These NOE's are characteristic of a two-stranded anti-parallel β-sheet structure and, together with the CSI, demonstrate the presence of β-hairpin-type conformations in solution that are well-ordered except at the tips of the flaps.
Fig. 2.

A 1H– 15N HSQC 500 MHz spectrum of the free HIV-1 protease recorded at pH 5.8 and 25°C. Backbone amide crosspeaks are labeled with the one letter amino acid code and number in the amino acid sequence. Sidechain 15N–1H2 signal pairs of Asn and Gln residues are connected by horizontal lines.
Fig. 3.

Comparison of the secondary structure of the free HIV-protease in solution determined by NMR (A) with secondary structures (B–E) in four independent crystal structures of the free protein. Secondary structure in solution was identified using the consensus chemical shift index (CSI). The secondary structures in (B–E) were derived using PROCHECK (Laskowski et al. 1993) and the following atomic coordinates: (B) Bhat and Erickson, pers. comm.; (C) 3HVP (Wlodawer et al. 1989); (D) 3PHV (Lapatto et al. 1989); and (E) 1HHP (Spinelli et al. 1991). The β-strands found in solution agree with those found in the crystal structures, at least to the extent that β-strand regions in the crystal structures agree among themselves. The Cα CSI identified a short helix spanning residues 87–90, although the consensus CSI did not.
Fig. 4.

(A) Strips taken from the amide proton region of the 500 MHz 3D 15N-edited 1H–1H NOESY spectrum of the free protease, recorded with a mixing time of 70 ms. The strips reveal the amide proton NOE's observed for residues 43–58, which are mapped on to the idealized secondary structure of the β-hairpin depicted in (B). In (A), the intense signal in each strip is the (diagonal) amide NH signal of the amino acid numbered at the bottom of the spectrum. The numbered cross-peak in each strip is a long range dNN connectivity between the diagonal amide proton and the amide proton of the residue identified by the number. In (B), the double arrows indicate amide protons that are linked by a pair of observed dNN connectivities. Single arrows identify long range dαN connectivites, linking α- and amide protons, that were observed in the upfield portion of the NOESY spectrum (not shown). A pair of very weak G49–G52 dNN connectivities, just above the noise level, are observed when the NOESY data are plotted at a lower threshold. Of the total of ten inter-strand hydrogen bonds expected in an ideal β-hairpin flap structure, only one, Q58NH–OC`K43, was identified by the hydrogen bond experiment discussed in the text.
To characterize the β-hairpin flap structures further, we measured 3hJNC` connectivities that arise from 15N–13C scalar couplings across NH–CO` hydrogen bonds (Cordier and Grzesiek 1999). Typically, 3hJNC` is <1 Hz in proteins (Cornilescu et al. 1999) making it a challenge to observe 3hJNC` connectivities in proteins with molecular weights >10 kD. Nonetheless, using the approach of Wang et al. (Wang et al. 1999) we would expect to detect 3hJNC` connectivities for most of the strong NH–CO` hydrogen bonds in a highly ordered β-hairpin structure. Each protease monomer contains two β-hairpin structures in addition to the flap β-hairpin. In crystal structures, the β-hairpin formed by strands P9–I15 and Q18–L24 is stabilized by seven NH–CO` hydrogen bonds, whereas the one formed by strands R57–I66 and H69–V77 is stabilized by ten hydrogen bonds. We observed 3hJNC` connectivities for 13 of the 17 expected hydrogen bonds in these two β-hairpins. In contrast with this result, we observed only one 3hJNC` connectivity in the flap β-hairpins, corresponding to the Q58NH–OC`K43 hydrogen bond, which is distant from the flap tips. In crystal structures of the free protease, 6–8 NH-CO` hydrogen bonds are observed in each flap β-hairpin. Because the magnitude of the 3hJNC` couplings decreases exponentially with increasing hydrogen bond length (Cornilescu et al. 1999), these observations immediately indicate that the hydrogen bonds in the flap β-hairpins are, on average, weaker than in the other two β-hairpins of the protease.
We emphasize that the lack of a measurable 3hJNC` connectivity does not imply that a hydrogen bond is absent. This is particularly the case in slower-tumbling, larger proteins in which small T2 values reduce the sensitivity of the hydrogen bond experiment. The problem of signal sensitivity is exacerbated for residues in the free protease flaps, because residues 49–54 have small amide 1H and 15N T2 values; as a consequence, internal motions are on the ms-μs time scale (Ishima et al. 1999). Rapid amide spin relaxation causes significant loss of magnetization during the long periods required for evolution by 3hJNC` couplings (Cordier and Grzesiek 1999; Wang et al. 1999) and is the reason that the magnitudes of signal intensities in the hydrogen bond reference spectrum are nearly ten-fold smaller for flap residues 49–54 than for residues in the two other β-hairpins. Hence, signal attenuation attributable to rapid spin relaxation could explain the absence of the expected 3hJNC` connectivities for these residues. We note, however, that reference signal intensities of residues 43, 45, and 56 are approximately equal to those of residues in the two other β-hairpins. Thus, the lack of 3hJNC` connectivities for these flap residues indicates that, on average, they form weaker hydrogen bonds than residues in the two other β-hairpins.
In the case of the protease bound to DMP323, six 3hJNC` connectivities are observed in the flap β-hairpins in hydrogen bond spectra. In comparing this result with that obtained for the free protease, it should be kept in mind that in the protease–DMP323 complex, only flap residues 50 and 51 have their T2's diminished by chemical exchange (Nicholson et al. 1995; Ishima et al. 1999). A comparison of hydrogen bonds observed in crystal structures with those identified in solution spectra of free and DMP323-bound protease is provided in Supplemental Material. Taken together, the NMR data indicate that flap residues 43–57 form β-hairpins in the free protease, whose structures are somewhat less ordered, particularly at the flap tips, and with weaker hydrogen bonds than is the case when the protease is bound to a potent inhibitor. This indicates that the flaps of the free protease may be flexible on a time scale faster than overall tumbling of the protein. We investigated this possibility using amide 15N relaxation measurements.
Qualitative analysis of relaxation data
Whereas measurements of conformational exchange have shown that the flaps of the free protease are flexible on the ms-μs time scale (Ishima et al. 1999), the 15N T1, T2, and NOE data presented here indicate that the flaps are also flexible on a time scale faster than overall tumbling, ∼10 ns, referred to as the sub-ns time scale. Profiles of these parameters measured at 25°C, plotted as a function of position in the amino acid sequence (Fig. 5 ▶), show rather uniform values of each of these parameters for most amide sites in the protein. However, several flap residues have 15N NOE's significantly below the average value, indicating internal motion on the sub-ns time scale. In contrast with the NOE results, the T1's of the flap residues are close to the average T1 values seen for most other residues. Together, these observations indicate that the T1's resulting from flap motion on the sub-ns time scale are approximately the same as the T1's resulting from overall tumbling on the ∼10 ns time scale. In contrast with their effect on T1 values, internal motions on the sub-ns time scale can cause an increase only in T2. However, Figure 5 ▶ shows that some flap residues have T2's that are larger than average, whereas other flap residues have smaller than the average T2's. This observation shows that motions on the ms-μs time scale, as well as on the sub-ns time scale, affect the T2's of the flap residues. This circumstance complicates analysis of the relaxation data.
Fig. 5.

A comparison of backbone amide 15N T1, T2, and NOE values, measured at 25°C and plotted as a function of amino acid residue number of the free HIV-1 protease. The relaxation parameters and uncertainties shown are the averages of two independent data sets. The errors in the T1, T2, and NOE parameters, when averaged over all residues, are 3.2, 2.7, and 8 %, respectively. The T2 values were recorded using a CPMG sequence, with a 1 ms separation between 180° pulses, corresponding to a 500 Hz effective RF field. As described in Materials and Methods, the NOE's are corrected for incomplete recovery of 1H and 15N magnetization.
To simplify the analysis of the T2 measurements, the relaxation parameters were also measured at 20°C (see Materials and Methods for a discussion of the temperatures chosen to carry out the various experiments), using a 2 kHz spin lock field in place of the CPMG sequence, to measure T2. The spin lock field is ∼4-fold larger than the effective field used in the CPMG experiment; our previous studies of ms-μs dynamics of the free protease showed that chemical exchange contributions to T2 are greatly reduced, if not completely eliminated, at the higher RF field strength. Indeed, this expectation is borne out by the profiles of relaxation parameters, measured at 20°C and plotted in Figure 6 ▶. The T1, T2, and NOE profiles are similar to those found at 25°C, except for residues 49–53 in the tips of the flaps. As anticipated, in the case of the spin-lock experiment, the T2 values of these residues are significantly above the average T2 observed for the other residues in the protease. This result indicates clearly that the chemical exchange contributions to the T2's of residues 49–53 are significantly reduced by using the spin-lock. In Figure 6 ▶, the T1's, T2's, and NOE's all indicate sub-ns time scale motions of the flap residues 49–53.
Fig. 6.

A comparison of backbone amide 15N T1, T2(T1ρ), and NOE values, measured at 20°C and plotted as a function of amino acid residue number of the free HIV-1 protease. The T2(T1ρ) values were recorded using a 2 kHz spin lock. As described in Materials and Methods, the T1ρ values are corrected for off-resonance effects and the NOE's are corrected for incomplete recovery of 1H and 15N magnetization.
Model-free analyses of the 15N relaxation data
These qualitative statements regarding the internal dynamics of the free protease were confirmed by generalized order parameters S2 and effective internal correlation times τe derived from model-free analyses of the 15N relaxation data. As described in Materials and Methods, X-ray coordinates of the free protease were used to calculate the orientations of the NH bonds in the crystalline molecular frame. These coordinates and the 15N T1/T2 ratios (of residues having NOE's > 0.65) were then used to derive the mean correlation time (τ), the anisotropy (D∥/D⊥) and orientation (θ,ϕ) of the rotational diffusion tensor in the molecular frame. Table 1 lists the values of D∥/D⊥ and obtained from the data measured at 20° and 25°C. As expected, a smaller value of τ is found at the higher temperature whereas the values of D∥/D⊥ are nearly the same at the two temperatures and close to the value obtained for the protease bound to DMP323 (Tjandra et al. 1996). This latter result is not surprising, because the differences in the X-ray structures of free and inhibitor-bound protease are limited to flexible parts of the protein. Residues in these regions are relatively few in number and typically have small 15N NOE's that exclude them from the determination of the diffusion tensor.
Table 1.
Comparison of diffusion tensor components derived from T1/T1ρ and T1/T2 ratios a
| Temperatureb | (θ,ϕ)c | τd | D∥/D⊥ | (χ2)e |
| 20° | (18.2, 225.0) | 12.8 | 1.32 | 1.89 |
| 25° | (13.4, 207.6) | 12.1 | 1.27 | 3.23 |
a T1/T1ρ and T1/T2 ratios measured at 20°C and 25°C respectively, and the X-ray coordinates of the free HIV-1 protease (Lapatto et al. 1989), PDB accession code 3PHV.
b In centrigrade.
c Polar angles, in degrees, defining the orientation of the unique axis of the diffusion tensor in the X-ray (Lapatto et al. 1989) coordinate frame, PDB 3PHV.
d In ns, defined as 1/(4D⊥ + 2 D∥).
e Defined as E/(N − m) where E is given in Equation (2), N is the number of T1/T2 ratios fitted, and m is the number of parameters (m = 4) determined by the fit. Attempts to fit the T1/T2 ratios assuming isotropic overall tumbling resulted in significantly larger values of χ2. Application of the F-test (Tjandra et al. 1996) showed that the probability that the reduction in χ2 (obtained using the anisotropic model) could have occurred by chance was less than 10−4.
Using the coordinates of the semi-open X-ray structure depicted in Figure 1 ▶ and the corresponding global parameters listed in Table 1, model-free analyses were performed on the relaxation data obtained at 20° and 25°C. Profiles of S2 and τe derived in this manner are plotted in Figures 7 and 8 ▶ ▶ as a function of position in the amino acid sequence. Except for T80, only residues in tips of the flap β-hairpins (residues 49–53) and in the flap elbows (residues 37–41) have S2 < ∼0.75. In contrast, the remaining residues have considerably larger order parameters, having average values of 0.86 and 0.87 at 20° and 25°C, respectively. This observation indicates that at both temperatures the protease backbone has highly restricted flexibility on the sub-ns time scale, except for residues in the flaps and T80. The sub-ns flexibility of the flap elbows (residues 37–41) and of T80 is not surprising, as it was observed in the protease bound to potent inhibitors (Nicholson et al. 1995; Freedberg et al. 1998). However, flexibility on the sub-ns time scale was not observed in the tips of the β-hairpins when the protease was bound to the inhibitors (Nicholson et al. 1995; Freedberg et al. 1998). In contrast, in both Figures 7 and 8 ▶ ▶, all residues in the flap tips have S2 < 0.8. The more significant reduction in S2 is seen in the data recorded with the spin-lock (at 20°C) because, as noted earlier, the spin-lock reduces chemical exchange contributions to T2. As seen in Figure 8 ▶, residues 49–53 all have S2 < 0.75 and the average value of S2 for these five residues is 0.62, nearly 40% less than the average S2 found for residues not in the flaps. The effective correlation times for the motions of these residues are <∼1 ns.
Fig. 7.

Model-free parameters S2 and τe of backbone amides, derived from a model-free analysis of the 25°C relaxation data presented in Figure 5 ▶. After using T1/T2 values and the X-ray coordinates of the protease structure depicted in Figure 1 ▶, PDB accession code 3PHV, to derive the components of the rotational diffusion tensor describing global tumbling of the protease (Tjandra et al. 1996), a model-free analysis was carried out using D∥/D⊥ = 1.27 and τ = 12.1 ns (Table 1). Satisfactory fits to the data (χ2 < 7) were obtained using: the simple model-free (circle), simple model-free plus exchange (triangle), and local diffusion plus extended model-free (square) as discussed in the text. Within the experimental errors shown, the same values of S2 were obtained for residues 49–53 using either coordinates of the closed flap structure (Bhat and Erickson, pers. comm.) or averages of angles obtained from the coordinates of the four free-protease structures discussed in Figure 3 ▶. In lower panel, a symbol appearing at 1 ps without error bars signifies a τe value of <∼100 ps that has a large uncertainty.
Fig. 8.

Model-free parameters S2 and τe of backbone amides, derived from a model-free analysis of the 20°C relaxation data presented in Figure 6 ▶. After using T1/T1ρ values and the X-ray coordinates of the protease having a semi-open flap structure, PDB accession code 3PHV, to derive the components of the rotational diffusion tensor describing global tumbling of the protease (Tjandra et al. 1996), a model-free analysis was carried out using D∥/D⊥ = 1.32 and τ = 12.8 ns (Table 1). Satisfactory fits to the data (χ2 < 7) were obtained using: the simple model-free (circle), simple model-free plus exchange (triangle), and local diffusion plus extended model-free (square) as discussed in the text. Within the experimental errors shown, the same values of S2 were obtained for residues 49–53 using either coordinates of the closed flap structure (T. N. Bhat and John Erickson, pers. comm.) or averages of angles obtained from the coordinates of the four free-protease structures discussed in Figure 3 ▶. In lower panel, a symbol appearing at 1 ps without error bars signifies a τe value of <∼100 ps that has a large uncertainty.
Interpreting the NMR results in terms of structural fluctuations of the protease flaps
Taken together, the CSI, NOESY, hydrogen bond, and relaxation data indicate that residues 43–58 of the free protease form an ensemble of β-hairpin structures in solution. We suggest that the semi-open β-hairpin structures observed in crystal structures of the free protease are typical members of the ensemble of solution structures. In addition, we propose that the sub-ns structural fluctuations of the flap tips (residues 49–53) revealed by the relaxation data reflect a rapid dynamic equilibrium among members of the ensemble of semi-open structures. It is possible that the closed conformation observed in crystal structures is a minor component of the ensemble of solution structures; this conformation may be the source of the very weak G49-G52 NOE discussed earlier. A calculation (Rick et al. 1998) of the free energy along the reaction path connecting the semi-open and closed flap conformations also predicts a low population for the latter conformation. The calculated free energy of the closed conformation was several kcal/mole larger than that of the semi-open conformations, the latter being stabilized by their favorable configurational entropy.
Comparison of NMR results with molecular dynamics predictions of flap dynamics
In the previous paragraph, the discussion focused on flap fluctuations connecting semi-open and closed flap conformations. Of course, there has been much interest in characterizing the conformational fluctuations that result in flap opening. It has been reported (York et al. 1993; Collins et al. 1995) that the flaps do not open during the course of a several hundred ps molecular dynamics trajectory. To observe flap opening on this time scale, it was necessary to employ an activated molecular dynamics approach (Collins et al. 1995). A more recent solvated molecular dynamics simulation of the free protease extending for 10 ns (Scott and Schiffer 2000) predicts that the flaps open within a few ns. Examination of the 10 ns trajectory shows that five residues at the tips of the flaps, G48–G52, curl rapidly inward toward the body of the protein (Scott and Schiffer 2000) and thus bury exposed hydrophobic residues while making the active site accessible to ligands.
It is not possible to make a direct comparison of the predictions of the 10 ns dynamics trajectory (Scott and Schiffer 2000) and the experimental results in Figures 7 and 8 ▶ ▶, because root-mean square deviations (RMSD) of the α-carbon positions rather than order parameters were calculated from the trajectory (Scott and Schiffer 2000). However, the comparison of the calculated RMSD and the experimentally determined order parameters is interesting and informative. The large RMSD predicted for residues G49–G52 agrees with the small order parameters observed for these residues (Figs. 7, 8 ▶ ▶). Furthermore, the trajectory predicts that the hydrophobic cluster formed when the flaps curl involves a conformational change of P79–T80–P81. This prediction agrees with the small order parameter observed for the highly conserved T80 residue (unfortunately, data is not available for the flanking Pro residues, which lack amide protons).
Although these predictions (Scott and Schiffer 2000) agree with the small order parameters observed for the aforementioned residues, significant RMSD are predicted for numerous residues that have large order parameters. For example, residues 14–17, 34–36, and 45–47 are predicted to undergo significant conformational changes (Scott and Schiffer 2000). However, the order parameters of all of these residues exceed 0.8 (Figs. 7, 8 ▶ ▶). In addition, both chemical shift and NOESY data indicate that flap residues 45–47 are in a β-sheet conformation (Figs. 3, 4 ▶ ▶). Finally, the calculation predicts that the flaps open in 1–2 ns, and remain open during the 10 ns trajectory. This prediction indicates that the calculated open conformations are more stable than the starting semi-open flap structure. However, to our knowledge, structures resembling the predicted open conformations have not been observed experimentally.
These considerations indicate that the 10 ns simulation (Scott and Schiffer 2000) overestimates the number of free-protease residues that are flexible on the time scale <10 ns. Figures 7 and 8 ▶ ▶ show that large amplitude angular fluctuations of flap residues on this time scale are restricted to residues 27–41 and 49–53 at the tips of the flaps. In our previous study of flap dynamics of the free protease (Ishima and Torchia 2000), we observed that residues 48–55 are flexible on the ms-μs time scale. We suggested that motions on this time scale reflect an equilibrium between semi-open flap conformations (observed in crystal structures) and open conformations that permit access to the active site. Stopped-flow fluorescence studies of the kinetics of ligand binding to the protease have been interpreted in terms of a two-step process in the case of inhibitors whose binding affinity mimics that of substrates. The first step involves rapid formation of a protease-ligand collision complex, which is followed by a slow (ms time scale) conformational change associated with ligand binding within the active site (Furfine et al. 1992; Rodriguez et al. 1993). We suggest that the ms-μs time scale motions observed by NMR (Ishima and Torchia 2000) involve open conformations that play a role in the slow conformational change inferred from the fluorescence data. As noted earlier, we think that the more localized, fast fluctuations of the flap tips reported here reflect a dynamic equilibrium among semi-open conformations (and possibly the closed conformation) observed in crystal structures (Rick et al. 1998).
Biological significance of fast flap motions in the free protease
As we have mentioned, none of the five residues in the flap tips that are flexible on the sub-ns time scale in the free protease are flexible on this time scale when the protease is bound to a variety of inhibitors (Nicholson et al. 1995; Freedberg et al. 1998). Three of the five flexible residues are highly conserved glycines G49, G51, and G52. It has been suggested (Scott and Schiffer 2000) that these residues are highly conserved, because when the protease binds to a ligand, the flap tips assume a conformation which places these residues in regions of the Ramachandran map that are outside the regions most favored by non-glycine residues. One possible function of the flexibility of the glycine-rich flap tips in the free protease is that product release is facilitated by the configurational entropy increase in the flap tips that occurs when the active site is vacated.
Although further experiments and calculations are needed to obtain a consensus regarding the details of flap dynamics of the free protease, information currently available indicates that flap conformations ranging from fully closed to open forms are present in solution. Because the closed conformation is well-defined crystallographically, it presents a well-defined target for novel drugs that need not bind at the active site, unlike all currently available drugs. Alternatively, a drug that stabilizes a more open conformation would also interfere with substrate binding. The fact that all of the flexible residues in the flap tips are highly conserved indicates that it would be difficult for the virus to evolve active protease variants containing mutations at these sites. In any event, if inhibitors directed specifically against the flaps interact with the protease differently than the current crop of inhibitors, which bind at the active site, it is unlikely that a single mutation would confer resistance against both types of drugs.
Materials and methods
Protein expression
The HIV-1 protease construct used for signal assignments, NOESY, and relaxation experiments contained mutations Q7K, L33I, and L63I to retard autoproteolysis (Rose et al. 1993; Mildner et al. 1994). The protease was expressed in E. coli using minimal media containing either 15NH4Cl or 15NH4Cl and D-(13C6)glucose. A second protease construct, used for the hydrogen bond experiments, contained two additional mutations, C67A and C95A, which permitted us to obtain spectra without adding dithiothreitol to the sample. This construct was expressed in E. coli using minimal media containing 2H2O, 15NH4Cl, and D-(13C6)glucose. These two constructs are called TMPR and PMPR to indicate that they contain three and five mutations, respectively, relative to the wild-type protein. Both constructs were purified from inclusion bodies as described previously (Ishima et al. 1999); the protein was folded by rapid dilution into 20 mM sodium phosphate buffer (pH 5.8–6.0; also containing 2.5 mM dithiothreitol in the case of the TMPR). The enzymatic activity of both constructs was the same as that of wild-type protease (Mildner et al. 1994).
NMR solution conditions
NMR spectra were recorded on 250 μL solutions in Shigemi microcells (Allison Park, PA) at dimer concentrations ranging from 0.25 to 0.64 mM in H2O/2H2O (95%/5%), 10–20 mM phosphate buffer (pH 5.8–6.0). The solutions of the TMPR also contained 2.5–5 mM deuterated dithiothreitol to inhibit oxidation of Cys residues. Spectra were recorded at temperatures ranging from 20°–35°C. To maximize sensitivity, it is desirable to record spectra at the highest protein concentration and temperature (the latter to increase transverse relaxation times) compatible with protein stability. Unfortunately, even the protease constructs used here undergo slow autoproteolysis; the autoproteolysis rate increases with concentration and temperature. Therefore, the conditions used to record the NMR spectra reflect an unavoidable compromise in which sensitivity was balanced against autoproteolysis. The sensitivity of the hydrogen bond experiments decreases sharply as the amide transverse relaxation time times decrease; to attain acceptable sensitivity, it was necessary record spectra at a protein (dimer) concentration of 0.6 mM and a temperature of 35°C. Although observable autoproteolysis had occurred by the end of the experiment, the presence of small signals from hydrolyzed protein fragments did not interfere significantly with the measurements, because the signals are well-dispersed in the three-dimensional HNCO spectra used to acquire the data.
Relaxation data were acquired at 25°C and dimer concentrations of 0.5 mM to maximize signal-to-noise and at 20°C and a protein concentration of 0.3 mM to minimize autoproteolysis. Two NMR data sets were recorded at 25°C, and relaxation parameters were measured in the following order: (1) T1, T2, NOE and (2) T2, T1, NOE. Because the NOE was recorded last in both sets of experiments, autoproteolysis has the greatest impact on the NOE data and may have caused some of the NOE values measured at 25°C to exceed the theoretical maximum value of 0.82 (Fig. 5 ▶).
Signal assignment and NOESY experiments
A variety of double and triple resonance experiments recorded on Bruker DMX500 and DMX600 spectrometers, using triple resonance probes equipped with shielded gradient coils, were used to obtain signal assignments and NOESY data. The chemical shifts of the free protease and the pulse sequences and parameters used to record the various spectra are tabulated in Supplemental Material. For completeness, an updated tabulation of the chemical shifts of the protease bound to DMP323, containing several corrections of the original assignments (Yamazaki et al. 1996), is also provided. Data were processed using the NMRPipe (Delaglio et al. 1995) and PIPP (Garrett et al. 1991) programs.
Hydrogen bond experiments
Hydrogen bond measurements were made using modified HNCO experiments (Wang et al. 1999) and recorded on a Bruker DMX750 spectrometer operating at a 1H frequency of 749.5 MHz using a triple resonance probe equipped with shielded gradient coils. Both the hydrogen bond and reference spectra were recorded with the same total time, 2T, for the 15N to 13C` INEPT and reverse INEPT periods to equalize relaxation losses. However, the 13C` π pulses were shifted by 16.6 ms relative to the 15N 180° pulses in both INEPT steps (Cordier and Grzesiek 1999) in the reference spectra. Eight scans per complex time-domain point were used to record the hydrogen bond spectra and two scans were used for the reference spectra. Loss of magnetization, caused by spin relaxation, during de/rephasing delays was reduced by recording the spectra at 35°C and by using a perdeuterated protein sample (Wang et al. 1999).
Relaxation experiments
All relaxation spectra at 25°C were recorded on a Bruker DMX500 spectrometer operating at a 1H frequency of 500.13 MHz using triple resonance probes equipped with shielded gradient coils. Backbone amide T1, T2, and NOE values were measured twice at 25°C using 90° pulses of 40 μs and ∼7 μs for 15N and 1H, respectively. T2's were measured using a CPMG sequence (Kay et al. 1992) with a 1 ms period between the centers of the 180° CPMG pulses, corresponding to a 500 Hz transverse RF field. It is known that off-resonance effects cause systematic errors in T2 values measured using CPMG sequences (Davis et al. 1994; Ross et al. 1997). Using our acquisition parameters, we calculate a maximum error of ∼3% in our 15N T2's caused by these resonance-offset effects. The data were not corrected for these errors because they are typically less than the random errors in our T2 data.
All relaxation spectra at 20°C were recorded on a Bruker DMX500 spectrometer operating at a 1H frequency of 500.13 MHz using triple resonance probes equipped with shielded gradient coils. Backbone amide T1, T2(T1ρ), and NOE values (the last measured twice) were measured in interleaved manner (Tjandra et al. 1996) using 90° pulses of 43 μs and 7.5 μs for 15N and 1H, respectively. To reduce the contribution of chemical exchange to transverse 15N relaxation, T1ρ was measured using a 2.0 kHz RF field in the 15N rotating frame (a spin-lock) in place of the CPMG pulse train. T1 values were also measured and used to correct the measured T1ρ values for resonance-offset effects (Davis et al. 1994). In the absence of such a correction, the T1ρ values of amide signals having the largest resonance offsets would be overestimated by ∼10%. 15N–{1H} NOE's were also measured at 20°C. An overlap of the amide signals of F53 and L97 that occurred at 25°C was relieved at 20°C
15N T1's were measured using relaxation delays of 32, 56, 88, 320, 640, and 960 ms at 25°C and 8, 16, 128, 256, 384, 512, and 640 ms at 20°C. T2 and T1ρ were measured with relaxation delays of 8, 16 24, 32, 56, 88, and 120 ms, and of 6, 12, 18, 24, 36, 48, and 60 ms, respectively. 15N–{1H} NOE experiments were performed using a water flip-back sequence (Grzesiek and Bax 1993). NOE values were measured by taking the ratio of peak intensities from experiments performed with and without 1H presaturation. The proton resonance frequency was shifted by 2 MHz (at 25°C) or 0.4 MHz, with 1H power attenuated by 10 dB (at 20°C) during the ∼3 s "presaturation" period for the unsaturated measurements. Corrected NOE's were derived from the measured NOE's, NOEm according to
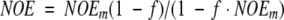 |
1 |
in which,
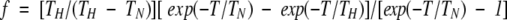 |
and T is the recycle delay and TN and TH are the T1's of 15N and 1H, respectively. This equation generalizes a previous formula (Grzesiek and Bax 1993) by accounting for the incomplete recovery of both 15N and 1H magnetization. For reasons of sensitivity and limited sample stability owing to autoproteolysis, the recycle delay T (2.8–3 s) is not much larger than either TN (0.7–1.2 s) or TH (∼1.4 s).
All data were processed using the NMRPipe software package (Delaglio et al. 1995) and peak heights measured in the processed spectra were fitted with a two-parameter exponential function to extract relaxation rates. Errors in T1 and T2 (or T1ρ) were determined by Monte-Carlo simulations (Kamath and Shriver 1989). The average pair-wise RMSD values of the T1, T2, and NOE for the two data sets recorded at 25°C were 6.4%, 5.3%, and 16%, respectively. These average pair-wise errors are approximately equal to the average error for each data set multiplied by root 2, indicating that errors in the measured data arise primarily from random (Gaussian) noise. The two data sets were merged into a single data set in the following way. The T1, T2, and NOE values were averaged for each residue and the average error in each relaxation parameter was obtained by dividing the pair-wise error of each parameter by two, yielding average T1, T2, and NOE errors of 3.2%, 2.7%, and 8%, respectively. This single merged data set was used for model-free analyses. The average errors in the T1, T1ρ, and NOE in the data set recorded at 20°C were 6.0, 5.8, and 5.2%, respectively. Two NOE data sets were recorded at 20°C and were averaged as described for the data obtained at 25°C.
Determination of components of the rotational diffusion tensor
T1/T2 ratios were used to determine the components of the overall rotational diffusion (the average correlation time τ, the anisotropy, D∥/D⊥, and the orientation, (θ, ϕ), of the unique diffusion axis in the molecular frame) as described previously (Tjandra et al. 1995, 1996). Initially, a coarse four-dimensional grid search was performed to obtain approximate values of the four diffusion tensor parameters by minimizing the quantity E given by
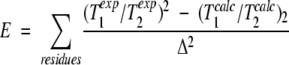 |
2 |
where (T1exp/T2exp) is the experimental T1/T2 ratio, Δ is the error in the T1/T2 ratio, and (T1calc/T2calc) is the T1/T2 ratio calculated with the assumption that effects of internal motion are negligible. Next, Powell optimization (Press et al. 1988) was used to determine the final set of parameters (θ, ϕ, τ, and D∥/D⊥) which minimized E. The values of the diffusion tensor components derived using the T1/T2 ratios measured at 20 and 25°C are listed in Table 1. Only residues having NOE values >0.65 were used in the determination of the diffusion tensor to exclude effects of fast internal motion on the relaxation parameters. A second round of minimization, which excluded, in addition to these residues, all residues for which the experimental and calculated ratios differed by three standard deviations after the first minimization, yielded values of τ and D∥/D⊥ within 1% of the values listed in Table 1.
Model-free analysis of relaxation data
After the components of the diffusion tensor were determined, the simple (two parameters, with Rex = 0) model-free approach (Lipari and Szabo 1982 a,b), appropriate to an axially symmetric rotor, was used to fit the measured T1, T2, and NOE values of individual residues. S2 and τe were allowed to vary until χ2 reached a minimum, in which
 |
3 |
Fits were considered acceptable when χ2 < 7. We think it is reasonable to accept fits with relatively large χ2 values because relaxation data have small systematic errors (e.g., owing to resonance offset effects discussed above and because of uncertainties and variations in 15N chemical shift anisotropy (Fushman et al. 1998; Kroenke et al. 1999) and in NH bond lengths) that are not included in our random error estimate. For residues with χ2 > 7 the relaxation data were fit using either (1) the simple model-free plus exchange, which uses three parameters, S2, τe, and Rex, or (2) using the extended model-free spectral density functions (Clore et al. 1990) employing three parameters, Sf2, Ss2, and τe(= τs), in conjunction a single overall correlation time given by the local diffusion approach (Bruschweiler et al. 1995). The local diffusion approach accounts for the effect of anisotropic overall motion by means of an orientation dependent (local) overall correlation time τi given by
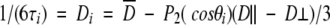 |
4 |
in which 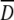 = (4D⊥ + 2 D∥/6 and θi is the angle made by the NH bond of the ith residue and the unique axis of the diffusion tensor.
= (4D⊥ + 2 D∥/6 and θi is the angle made by the NH bond of the ith residue and the unique axis of the diffusion tensor.
Acknowledgments
We thank Dan Garrett, Frank Delaglio, and Nico Tjandra for processing and analysis software and T. N. Bhat and John Erickson for the coordinates of their free protease crystal structure. This work was supported by the AIDS Targeted Antiviral Program of the Office of the Director of National Institutes of Health.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
HIV, human immunodeficiency virus
ms, millesecond
μs, microsecond
ns, nanosecond
ps, picosecond
NOE, nuclear Overhauser effect
HSQC, heteronuclear single quantum coherence
NOESY, NOE spectroscopy
CSI, Chemical Shift Index
CPMG, Carr-Purcell-Meiboom-Gill
TMPR and PMPR, triple- and penta-mutant HIV-1 protease constructs, respectively
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.33202.
References
- Bruschweiler, R., Liao, X.B., and Wright, P.E. 1995. Long-range motional restrictions in a multidomain zinc-finger protein from anisotropic tumbling. Science 268: 886–889. [DOI] [PubMed] [Google Scholar]
- Clore, G.M., Szabo, A., Bax, A., Kay, L.E., Driscoll, P.C., and Gronenborn, A. 1990. Deviation from the simple two-parameter model-free approach to the interpretation of nitrogen-15 nuclear magnetic relaxation of proteins. J. Am. Chem. Soc. 112: 4989–4991. [Google Scholar]
- Collins, J.R., Burt, S.K., and Erickson, J.W. 1995. Flap opening in HIV-1 protease simulated by `activated' molecular dynamics. Nat. Struct. Biol. 2: 334–338. [DOI] [PubMed] [Google Scholar]
- Cordier, F. and Grzesiek, S. 1999. Direct observation of hydrogen bonds in proteins by interresidue 3hJNC` scalar couplings. J. Am. Chem. Soc. 121: 1601–1602. [DOI] [PubMed] [Google Scholar]
- Cornilescu, G., Ramirez, B.E., Frank, M.K., Clore, G.M., Gronenborn, A.M., and Bax, A. 1999. Correlation between 3hJNC` and hydrogen bond length in proteins. J. Am. Chem. Soc. 121: 6275–6279. [Google Scholar]
- Davis, D.G., Perlman, M.E., and London, R.E. 1994. Direct measurements of the dissociation-rate constant for inhibitor-enzyme complexes via the T1r and T2(CPMG) methods. J. Magn. Reson. B 104: 266–275. [DOI] [PubMed] [Google Scholar]
- Delaglio, F., Grzesiek, S., Vuister, G.W., Zhu, G., Pfeifer, J., and Bax, A. 1995. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6: 277–293. [DOI] [PubMed] [Google Scholar]
- Freedberg, D.I., Wang, Y.-X., Stahl, S.J., Kaufman, J.D., Wingfield, P.T., Kiso, Y., and Torchia, D.A. 1998. Flexibility and function in HIV protease: Dynamics of the HIV-1 protease bound to the asymmetric inhibitor kynostatin 272 (KNI-272). J. Am. Chem. Soc. 120: 7916–7923. [Google Scholar]
- Furfine, E.S., D'Souza, E., Ingold, K.J., Leban, J.J., Spector, T., and Porter, D.J. 1992. Two-step binding mechanism for HIV protease inhibitors. Biochemistry 31: 7886–7891. [DOI] [PubMed] [Google Scholar]
- Fushman, D., Tjandra, N., and Cowburn, D. 1998. Direct measurement of 15N chemical shift anisotropy in solution. J. Am. Chem. Soc. 120: 10947–10952. [Google Scholar]
- Garrett, D.S., Powers, R., Gronenborn, A.M., and Clore, G.M. 1991. A common sense approach to peak picking in two-, three-, and four-dimensional double- and triple-resonance heteronuclear magnetic resonance spectroscopy. J. Magn. Reson. 95: 214–220. [DOI] [PubMed] [Google Scholar]
- Grzesiek, S. and Bax, A. 1993. The importance of not saturating H2O in protein NMR. Application to sensitivity enhancement and NOE measurements. J. Am. Chem. Soc. 115: 12593–12594. [Google Scholar]
- Harte, W.E.J., Swaminathan, S., Mansuri, M.M., Martin, J.C., Rosenberg, I.E., and Beveridge, D.L. 1990. Domain communication in the dynamical structure of human immunodeficiency virus 1 protease. Proc. Natl. Acad. Sci. 87: 8864–8868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishima, R., Freedberg, D.I., Wang, Y.-X., Louis, J.M., and Torchia, D.A. 1999. Flap opening and dimer-interface flexibility in the free and inhibitor bound HIV protease and their implications for function. Structure 7: 1047–1055. [DOI] [PubMed] [Google Scholar]
- Ishima, R. and Torchia, D.A. 2000. Protein dynamics from NMR. Nat. Struct. Biol. 7: 740–743. [DOI] [PubMed] [Google Scholar]
- Kamath, U. and Shriver, J.W. 1989. Characterization of thermotropic state changes in myosin subfragment-1 and heavy meromyosin by UV difference spectroscopy. J. Biol. Chem. 264: 5586–5592. [PubMed] [Google Scholar]
- Kay, L.E. 1998. Protein dynamics from NMR. Nat. Struct. Biol. 5 Suppl: 512–517. [DOI] [PubMed] [Google Scholar]
- Kay, L.E., Nicholson, L.K., Delaglio, F., Bax, A., and Torchia, D.A. 1992. Pulse sequences for removal of the effects of cross correlation between dipolar and chemical-shift anisotropy relaxation mechanisms on the measurement of heteronuclear T1 and T2 values in proteins. J. Magn. Reson. 97: 359–375. [Google Scholar]
- Kroenke, C.D., Rance, M., and Palmer, A.G. 1999. Variability of the 15N chemical shift anisotropy in Escherichia coli ribonuclease H in solution. J. Am. Chem. Soc. 121: 10119–10125. [Google Scholar]
- Lapatto, R., Blundell, T., Hemmings, A., Overington, J., Wilderspin, A., Wood, S., Merson, J.R., Whittle, P.J., Danley, D.E., Geoghegan, K.F., et al. 1989. X-ray analysis of HIV-1 proteinase at 2.7 Å resolution confirms structural homology among retroviral enzymes. Nature 342: 299–302. [DOI] [PubMed] [Google Scholar]
- Laskowski, R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Cryst. 26: 283–291. [Google Scholar]
- Lipari, G. and Szabo, A. 1982a. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity. J. Am. Chem. Soc. 104: 4546–4559. [Google Scholar]
- Lipari, G. and Szabo, A. 1982b. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 2. Analysis of experimental results. J. Am. Chem. Soc. 104: 4559–4570. [Google Scholar]
- Liu, H., Muller-Plathe, F., and van Gunsteren, W.F. 1996. A combined quantum/classical molecular dynamics study of the catalytic mechanism of HIV protease. J. Mol. Biol. 261: 454–469. [DOI] [PubMed] [Google Scholar]
- Mildner, A.M., Rothrock, D.J., Leone, J.W., Bannow, C.A., Lull, J.M., Reardon, I.M., Sarcich, J.L., Howe, W.J., Tomich, C.S., Smith, C.W., et al. 1994. The HIV-1 protease as enzyme and substrate: Mutagenesis of autolysis sites and generation of a stable mutant with retained kinetic properties. Biochemistry 33: 9405–9413. [DOI] [PubMed] [Google Scholar]
- Nicholson, L.K., Yamazaki, T., Torchia, D.A., Grzesiek, S., Bax, A., Kaufman, J.D., Stahl, S.J., Wingfield, P.T., Lam, P.Y.S., Jadhav, P.K., et al. 1995. Flexibility and function in HIV-1 protease. Nat. Struct. Biol. 2: 274–280. [DOI] [PubMed] [Google Scholar]
- Pillai, B., Kannan, K.K., and Hosur, M.V. 2001. 1.9 Å X-ray study shows closed flap conformation in crystals of untethered HIV-1 PR. Proteins 43: 57–64. [DOI] [PubMed] [Google Scholar]
- Press, W.H., Flannery, B.P., Teukolsky, S.A., and Vetterling, W.T. 1988. Numerical recipes in C. Cambridge University Press, Cambridge, UK.
- Rick, S.W., Erickson, J.W., and Burt, S.K. 1998. Reaction path and free energy calculations of the transition between alternate conformations of the HIV-1 protease. Proteins 32: 7–16. [DOI] [PubMed] [Google Scholar]
- Rodriguez, E.J., Debouck, C., Deckman, I.C., Abu-Soud, H., Raushel, F.M., and Meek, T.D. 1993. Inhibitor binding to the Phe53Trp mutant of HIV-1 protease promotes conformational changes detectable by spectrofluorometry. Biochemistry 32: 3557–3563. [DOI] [PubMed] [Google Scholar]
- Rose, J.R., Salto, R., and Craik, C.S. 1993. Regulation of autoproteolysis of the HIV-1 and HIV-2 proteases with engineered amino acid substitutions. J. Biol. Chem. 268: 11939–11945. [PubMed] [Google Scholar]
- Rose, R.B., Craik, C.S., and Stroud, R.M. 1998. Domain flexibility in retroviral proteases: Structural implications for drug resistant mutations. Biochemistry 37: 2607–2621. [DOI] [PubMed] [Google Scholar]
- Ross, A., Czisch, M., and King, G.C. 1997. Systematic errors associated with the CPMG pulse sequence and their effect on motional analysis of biomolecules. J. Magn. Reson. 124: 355–365. [Google Scholar]
- Scott, W.R.P. and Schiffer, C.A. 2000. Curling of flap tips in HIV-1 protease as a mechanism for substrate entry and tolerance of drug resistance. Structure 8: 1259–1265. [DOI] [PubMed] [Google Scholar]
- Shafer, R.W., Stevenson, D., and Chan, B. 1999. Human immunodeficiency virus reverse transcriptase and protease sequence database. Nucleic Acids Res. 27: 348–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinelli, S., Liu, Q.Z., Alzari, P.M., Hirel, P.H., and Poljak, R.J. 1991. The three-dimensional structure of the aspartyl protease from the HIV-1 isolate BRU. Biochimie 73: 1391–1396. [DOI] [PubMed] [Google Scholar]
- Tjandra, N., Feller, S.E., Pastor, R.W., and Bax, A. 1995. Rotational diffusion anisotropy of human ubiquitin from 15N NMR relaxation. J. Am. Chem. Soc. 117: 12562–12566. [Google Scholar]
- Tjandra, N., Wingfield, P., Stahl, S., and Bax, A. 1996. Anisotropic rotational diffusion of perdeuterated HIV protease from 15N NMR relaxation measurements at two magnetic fields. J. Biomol. NMR 8: 273–284. [DOI] [PubMed] [Google Scholar]
- Wang, Y.-X., Jacob, J., Cordier, F., Wingfield, P., Stahl, S.J., Lee-Huang, S., Torchia, D., Grzesiek, S., and Bax, A. 1999. Measurement of 3hJNC` connectivities across hydrogen bonds in a 30 kD protein. J. Biomol. NMR 14: 181–184. [DOI] [PubMed] [Google Scholar]
- Wishart, D., S. and Sykes, B., D. 1994. The 13C Chemical-Shift Index: A simple method for the identification of protein secondary structure using 13C chemical-shift data. J. Biomol. NMR 4: 171–180. [DOI] [PubMed] [Google Scholar]
- Wlodawer, A. and Erickson, J.W. 1993. Structure-based inhibitors of HIV-1 protease. Annu. Rev. Biochem. 62: 543–585. [DOI] [PubMed] [Google Scholar]
- Wlodawer, A., Miller, M., Jaskolski, M., Sathyanarayana, B.K., Baldwin, E., Weber, I.T., Selk, L.M., Clawson, M., Schneider, J., and Kent, S.B.H. 1989. Conserved folding in retroviral proteases: Crystal structure of a synthetic HIV-1 protease. Science 245: 616–621. [DOI] [PubMed] [Google Scholar]
- Yamazaki, T., Hinck, A.P., Wang, Y.X., Nicholson, L.K., Torchia, D.A., Wingfield, P., Stahl, S.J., Kaufman, J.D., Chang, C.H., Domaille, P.J., et al. 1996. Three-dimensional solution structure of the HIV-1 protease complexed with DMP323, a novel cyclic urea-type inhibitor, determined by nuclear magnetic resonance spectroscopy. Protein Sci. 5: 495–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- York, D.M., Darden, T.A., Pedersen, L.G., and Anderson, M.W. 1993. Molecular dynamics simulation of HIV-1 protease in a crystalline environment and in solution. Biochemistry 32: 1443–1453. [DOI] [PubMed] [Google Scholar]
- Zutshi, R. and Chmielewski, J. 2000. Targeting the dimerization interface for irreversible inhibition of HIV- 1 protease. Bioorg. Med. Chem. Lett. 10: 1901–1903. [DOI] [PubMed] [Google Scholar]
- Zutshi, R., Franciskovich, J., Shultz, M., Schweitzer, B., Bishop, P., Wilson, M., and Chmielewski, J. 1997. Targeting the dimerization interface of HIV-1 protease: Inhibition with cross-linked interfacial peptides. J. Am. Chem. Soc. 119: 4841–4845. [Google Scholar]