

Abstract
The three-dimensional structures of indinavir and three newly synthesized indinavir analogs in complex with a multi-drug-resistant variant (L63P, V82T, I84V) of HIV-1 protease were determined to ∼2.2 Åresolution. Two of the three analogs have only a single modification of indinavir, and their binding affinities to the variant HIV-1 protease are enhanced over that of indinavir. However, when both modifications were combined into a single compound, the binding affinity to the protease variant was reduced. On close examination, the structural rearrangements in the protease that occur in the tightest binding inhibitor complex are mutually exclusive with the structural rearrangements seen in the second tightest inhibitor complex. This occurs as adaptations in the S1 pocket of one monomer propagate through the dimer and affect the conformation of the S1 loop near P81 of the other monomer. Therefore, structural rearrangements that occur within the protease when it binds to an inhibitor with a single modification must be accounted for in the design of inhibitors with multiple modifications. This consideration is necessary to develop inhibitors that bind sufficiently tightly to drug-resistant variants of HIV-1 protease to potentially become the next generation of therapeutic agents.
Keywords: HIV protease, drug resistance, drug design, protein crystallography
The development of HIV protease inhibitors was one of the most successful structure-based drug design efforts to date. This effort was based on extensive investigations of the conformational and functional properties of the enzyme (Wlodawer and Erickson 1993). HIV protease is an aspartyl protease, responsible for post-translational processing of the viral gag-pol polyprotein gene products, yielding the structural proteins and enzymes necessary for the maturation of infectious viral particles (Debouck 1992). The active protease is a homodimer of 99 amino acid subunits bound together by noncovalent interactions (Wlodawer and Erickson 1993). The active site of the enzyme exists as a cleft at the interface between the two subunits. Two aspartic acid residues, one from each subunit, are responsible for enzymatic activity. The formation of the active site of aspartyl proteases, by the coming together of two identical subunits, is unique to retroviruses (Molla et al. 1998; Turner and Summers 1998; Wlodawer and Gustchina 2000).
All of the HIV protease inhibitors currently prescribed for treatment of HIV-infected patients are competitive inhibitors that bind to the active site. Their design was based on both the three-dimensional structure of the active site of the protease and the primary sequences of its natural substrates. Unfortunately, exposure to protease inhibitors often leads to drug-resistant mutations in the protease gene. The resulting protease is resistant to the inhibitor, yet still maintains at least a portion of its function in cleaving its natural substrates. Patients receiving inhibitor therapy often have an initial reduction in viral load, followed by a rebound caused by the development of drug-resistant mutations. Both in vivo and in vitro studies involving HIV and currently available drugs have demonstrated the generation of mutations that give rise to drug resistance (Schinazi et al. 1997; Boden and Markowitz 1998; Molla et al. 1998). As the population of heterogeneous HIV-1 viruses increase within a patient, most likely a combination of protease inhibitors will be necessary for effective treatment.
More than 200 crystal structures of HIV protease have been solved over the past several years (http://www-fbsc.ncifcrf.gov/HIVdb). Crystal structures of specific drug-resistant protease inhibitor complexes have been solved recently (Baldwin et al. 1995; Chen et al. 1995; Hong et al. 1996,1997,1998; Kervinen et al. 1996; Silva et al. 1996; Ala et al. 1997,1998; Swairjo et al. 1998). Comparing these mutant and wild-type protease inhibitor complexes allows an analysis of the mechanisms by which these mutations reduce drug binding ability. Additionally, investigations of the kinetics for these drug-resistant variant proteases provide a quantitative comparison of the mutational effects on substrate cleavage and inhibitor binding affinities (Gulnik et al. 1995; Lin et al. 1995; Pazhanisamy et al. 1996; Schock et al. 1996; Ermolieff et al. 1997; Wilson et al. 1997,1998; Klabe et al. 1998).
The protease inhibitor known as indinavir (Crixivan) (Chen et al. 1994) (Fig. 1a ▶) is often one of the first lines of treatment for patients infected with HIV (J. Sullivan and K. Luzuriaga, pers. comm.). Indinavir is a peptidomimetic; a peptide analog in which the scissile amide bond is replaced by a reduced moiety. This protease inhibitor has several large, mainly hydrophobic groups that interact with the hydrophobic P2–P2′ pockets in the active site of the protease (Wlodawer and Erickson 1993). Thus, nonpolar interactions contribute significantly to the inhibitory properties of indinavir to HIV protease, but these interactions are also often the sites at which drug-resistant mutations occur. Mutations at the active site such asV82T and I84V confer resistance to indinavir but several other mutations often occur simultaneously, combining a decrease in the affinity of the inhibitor to the protease and an increase in catalytic efficiency (Molla et al. 1998). The mutation L63P, located in “the hinge region” of the protease (Chen et al. 1995), frequently arises upon drug exposure and along with M46I has been shown to compensate for active site mutations, restoring the impaired enzyme's ability to catalyze the necessary cleavage reactions (Markowitz et al. 1995; Schock et al. 1996). In fact, in a comparison of catalytic efficiencies, the doubly substituted M46I/L63P demonstrated greater catalytic efficiency (110–360%) for each of the eight normal substrates than the wild-type enzyme (Schock et al. 1996). Thus, HIV-1 protease evolves to maintain activity while in the presence of inhibitors.
Fig. 1.

(a) A chemical schematic diagram of indinavir, with the protease substrate subsites labeled. (b) A schematic representation of the synthesis pathway for indinavir analogs.
A multi-drug resistant triple mutant variant of HIV-1 protease (3X-protease) having the substitutions L63P, V82T, I84V (Fig. 2 ▶) is the focus of this study. These mutations were chosen on the basis of their relative frequency of emergence reported in both in vivo and in vitro studies with various HIV protease inhibitors (Molla et al. 1998). Both structural investigations and activity analyses were used to characterize the differences in inhibitor binding and substrate cleavage between the drug-resistant and wild-type proteases. From the changes in structure, new inhibitors were designed. This work describes the X-ray crystal structure of this protease variant in complex with indinavir, as well as with a series of newly designed inhibitors. Analysis of the structures of these complexes, in combination with the relative binding activity of the inhibitors, elucidates why two independent modifications to indinavir are mutually exclusive and cannot be productively combined in a single compound. Understanding which functional groups can be productively combined to form a tight inhibitor is essential for designing the next generation of HIV-1 protease inhibitors.
Fig. 2.
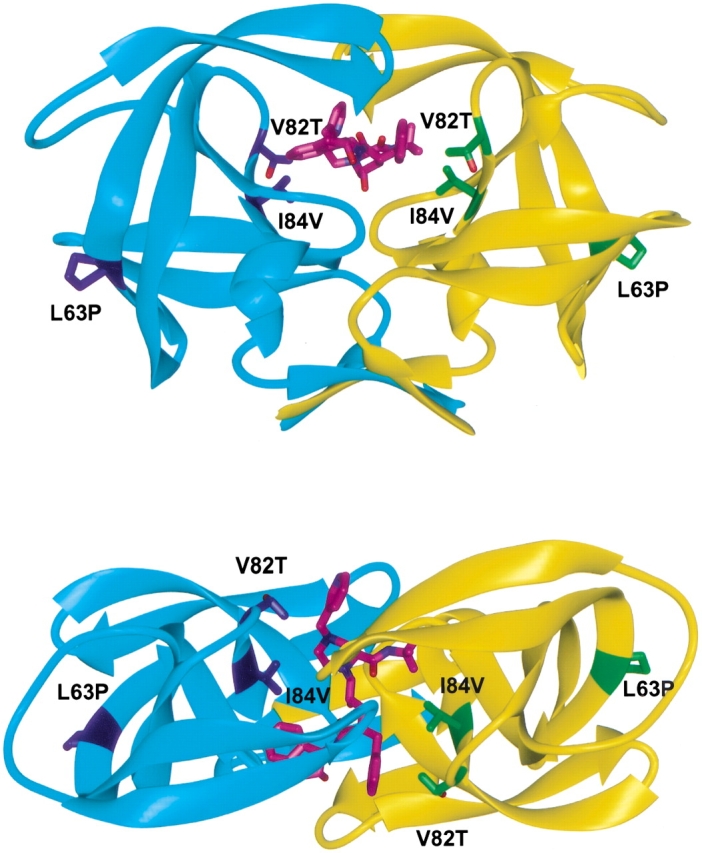
Ribbon diagrams of two views of the drug-resistant variant of HIV-1 protease dimer (in cyan and yellow) bound to indinavir (in magenta). The three modifications L63P, V82T, I84V are displayed and labeled in blue and green for each monomer, respectively. Figures were made with MIDAS (Ferrin et al. 1988).
Results and Discussion
General features
The multi-drug-resistant HIV protease variant (3X-protease) having the substitutions L63P, V82T, and I84V was complexed with four inhibitors and their crystal structures were determined and analyzed (Table 1). The 3X-protease inhibitor complexes displayed well-ordered electron density for the protease and all four ligands. The electron density for the mutated side chains was unambiguous. These included residues 82 and 84, which are located in the active site, and residue 63, located at the hinge region away from the inhibitor-binding site (Fig. 2 ▶). Because HIV protease is catalytically active as a homodimer, the mutations encoding two active site residues result in four amino acid changes in the active site where inhibitor/substrate bind. The change from a leucine to a proline at position 63 is not only frequently selected for during protease inhibitor drug therapy, but also commonly exists as a polymorphism found in viral isolates from untreated patients (Olsen et al. 1999). Thus, the change at position 63 is expected to have less impact on the inhibitor than those substitutions at positions 82 and 84.
Table 1.
Crystallographic statistics for complexes of the multi-drug-resistant variant (L63P/V82T/I84V) bound to indinavir and analogs
| Indinavir | 807-29-4 | XN1336-51 | XN1336-52 | |
| Resolution (Å) | 2.2 | 2.2 | 2.25 | 2.0 |
| Space group | P2l2l2l | P2l2l2l | P2l2l2l | P2l2l2l |
| a (Å) | 51.45 | 51.02 | 51.49 | 51.46 |
| b (Å) | 59.40 | 58.95 | 59.37 | 59.30 |
| c (Å) | 61.74 | 61.59 | 62.03 | 61.86 |
| Z | 4 | 4 | 4 | 4 |
| Rmerge (%) | 6.8 | 8.2 | 8.8 | 6.9 |
| Completeness (%) | 99.2 | 91.4 | 96.6 | 96.0 |
| Total number of reflections | 63726 | 31972 | 33172 | 95068 |
| Number of unique reflections | 10006 | 9067 | 9186 | 12820 |
| R-factor (%) | 19.4 | 18.3 | 18.4 | 17.7 |
| Rfree (%) | 23.1 | 23.0 | 25.0 | 21.1 |
| Number of crystallographic waters | 119 | 119 | 97 | 158 |
| Average of B-factors for the inhibitors (Å2) | 35.2 | 27.5 | 26.9 | 26.8 |
| RMSD in | ||||
| Bond lengths (Å) | 0.008 | 0.006 | 0.006 | 0.006 |
| Bond angles (°) | 1.4 | 1.3 | 1.3 | 1.2 |
| B-factors rmsd bonded (Å2) (main chain) | 0.9 | 1.4 | 1.5 | 1.0 |
| (side chain) | 1.1 | 2.4 | 2.5 | 1.4 |
Indinavir
The structure of the indinavir 3X-protease complex was refined to a resolution of 2.2 Å, with an R-factor of 19.5%. The average of the B-factors for the entire complex was 38.0 Å2. Indinavir was modeled in one conformation in the 3X-protease indinavir complex structure, similar to that of the wild-type protease indinavir complex (Chen et al. 1994). The electron density was strong for the entire indinavir molecule, in contrast with a wild-type HIV-2 protease indinavir complex, in which weaker density has been reported for the pyridine ring (Chen et al. 1994). In the present structure, the B-factors for the pyridine ring of indinavir, averaged 40.9 Å2, which is slightly higher than for the entire inhibitor, 35.2 Å2 (Table 1). However, in the wild-type HIV-2 protease indinavir complex the difference in B-factors was even larger, with the pyridine ring having an average of 54 Å2 relative to the average of 39 Å2 for the entire inhibitor (Chen et al. 1994). Although indinavir was modeled in a single conformation, additional difference density (Fo − Fc) was observed in proximity to the tertiary-butyl group. However, no sterically acceptable alternate conformations could be unambiguously modeled to account for this extra density. The average of the B-factors for the t-butyl moiety were 31.9 Å2, lower than the average for the entire inhibitor, thus providing support for the unique conformation modeled in that region of the molecule. Modeling of the nearby I47a (the monomers are distinguished from one another by referring to them as “a” and “b”) in a second conformation (Fig. 3a ▶) further ordered the electron density surrounding both I47a and the region around the t-butyl moiety. The added conformational variability at I47a may result from the cavity introduced into the region by the nearby protease mutation of I84aV.
Fig. 3.
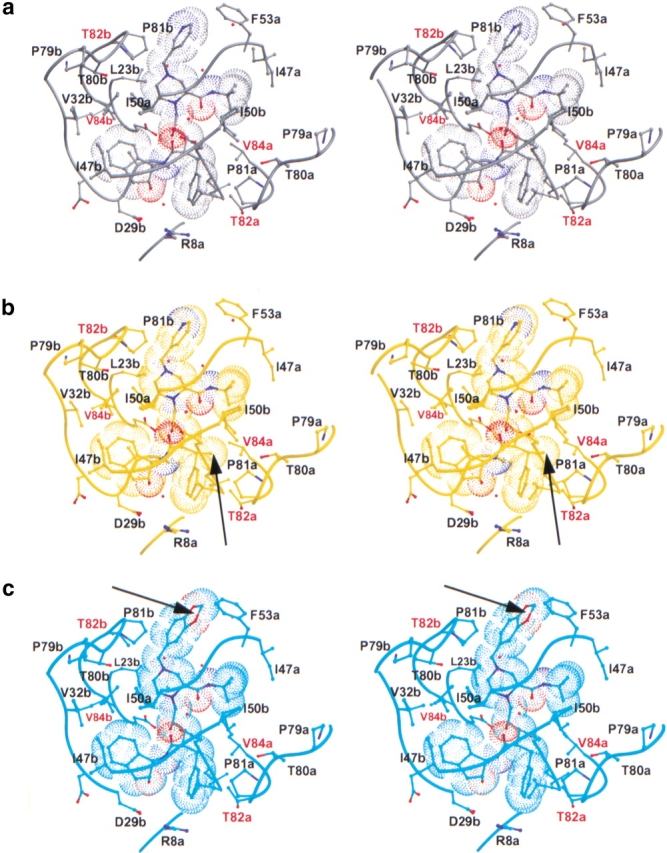
Stereo views of the variant HIV-1 protease inhibitor complexes showing all atoms within at least 3.5 Å of their respectively bound inhibitors. The side chains adjacent to the protease active site are labeled, with the sites of mutation labeled in red. Eighty percent of the van der Waals surface is shown around each inhibitor. Figures were made with MIDAS (Ferrin et al. 1988). (a) Indinavir complex (in gray). (b) XN1336–51 complex (in yellow). An arrow indicates the addition of an s-methyl group in the benzylic position relative to indinavir. (c) 807–29–4 complex (in cyan). An arrow indicates the methylenedioxyphenyl group, which has replaced the pyridine ring of indinavir. (d) XN1336–52 complex (in magenta). Arrows indicate both of the modifications relative to indinavir described in b and c.(e) All four complexes superimposed. An arrow indicates where in the protease the 807–29–4 complex varies from the rest of the set.
The final model of the 3X-protease indinavir complex also included 119 water molecules and 5 acetate ions. A highly conserved water molecule tetrahedrally coordinates the tips of the protease flaps at the amide nitrogens of I50 in each monomer to the two oxygen atoms of the inhibitor (O1 and O3) (Figure 3a ▶). Indinavir binds to the active site in the same conformation seen in the wild-type protease indinavir complex (Chen et al. 1994) with the functional groups occupying similar subsites within the protease. Indinavir's pyridyl and piperidine groups occupy the S3 and S1 subsites, whereas the benzyl moiety occupies the S1′ subsite. The tertiary-butyl group and the indanol moiety occupy the S2 and S2′ subsites, respectively (Fig. 1a ▶). The catalytic aspartic acids of the enzyme, at the edge of the S1/S1′ subsites, are within hydrogen bonding distance of indinavir's O2 of the hydroxyethylene group. The binding constant from isothermal titration calorimetry is 38.4 times worse for indinavir to 3X-protease compared with its binding to wild-type HIV-1 protease (Table 2). The loss in binding is entirely due to a loss in the enthalpy of binding of 2.48 kcal/mole, presumably mainly because of the loss of van der Waals contacts at residues 82 and 84. The IC50 for indinavir inhibiting this 3X-protease is 0.47 μM (Table 3, Fig. 4 ▶).
Table 2.
Thermodynamic measurements from isothermal titration calorimetry collected at 20°C
| Enzyme | Inhibitor | Enthalpy (ΔH) (cal/mole) | Enthalpy (−TΔS) (cal/mole) | Free Energy (ΔG) (cal/mole) | Kd (M) |
| wt-protease | indinavir | 2.97e + 03 | −1.43e + 04 | −1.13e + 04 | 8.6e − 10 |
| 3X-protease | indinavir | 5.39e + 03 | −1.47e + 04 | −9.34e + 03 | 3.3e − 08 |
| 3X-protease | XN1336-51 | 6.79e + 03 | −1.63e + 04 | −9.53e + 03 | 2.4e − 08 |
| 3X-protease | 807-29-4 | 5.40e + 03 | −1.46e + 04 | −9.19e + 03 | 4.4e − 08 |
| 3X-protease | XN1336-52 | 7.06e + 03 | −1.57e + 04 | −8.65e + 03 | 1.2e − 07 |
Table 3.
Inhibition data for indinavir and analogs
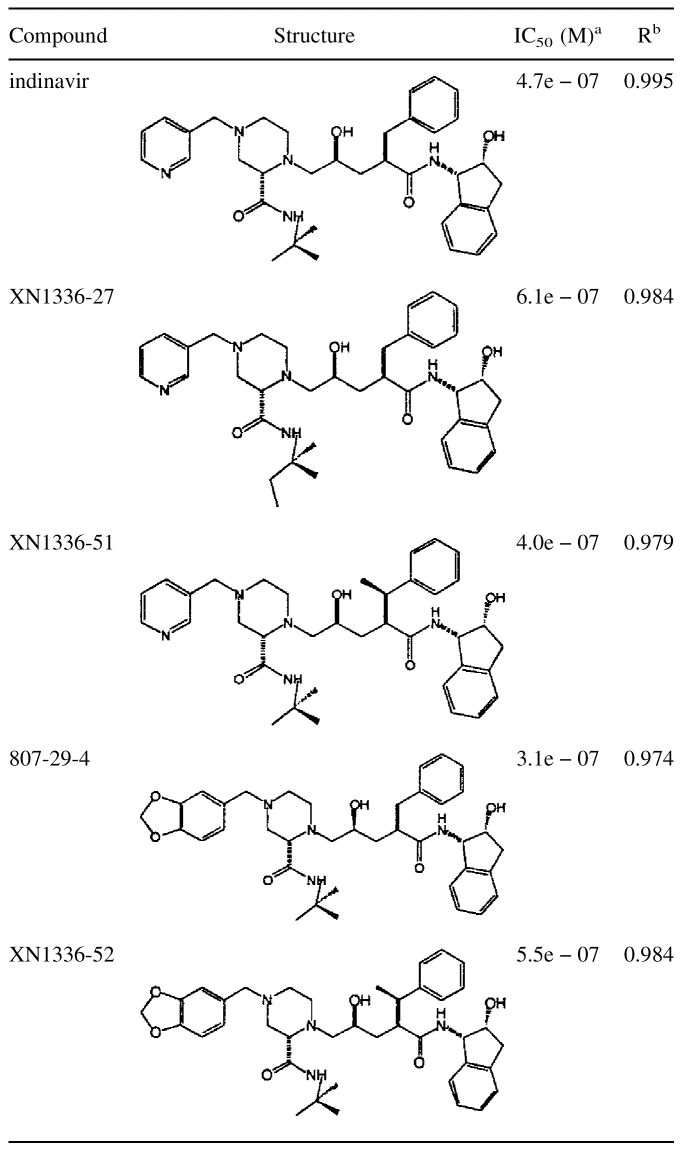
a IC50 values were determined by best linear fit of data points using Microsoft Excel.
b Square of the Pearson product moment correlation coefficient through data points.
Fig. 4.

Inhibition of 3X-protease by the four inhibitors in the presence of a substrate peptide, AcSQNYPVV-NH2. Percent activity is measured by the amount of remaining uncleaved peptide after incubation with protease and varying amounts of inhibitors. The IC50 is the concentration at which 50% of the activity is remaining.
XN1336–27
In an attempt to design an inhibitor that would fill the cavity caused by the protease mutation I84aV, a new analog was synthesized (XN1336–27) in which a methyl group was added to the t-butyl moiety of indinavir. Inhibition assays performed on this new indinavir analog unfortunately indicated a reduction in binding affinity compared with that of indinavir, with an IC50 of 0.61 μM (Table 3). Thus, this modification to indinavir was not further pursued.
XN1336–51
An alternative attempt at producing an inhibitor that would fill the cavity produced by the substitution I84aV involved adding an s-methyl group in the benzylic position to produce the indinavir analog XN1336–51. Thermodynamically, although the enthalpy of binding is somewhat worse than indinavir, the entropy has improved, thus improving the overall Kd (Table 2). Inhibition studies measure the IC50 for XN1336–51 as 0.40 μM (Table 3, Fig. 4 ▶). The 3X-protease XN1336–51 crystal structure was solved to determine how the added methyl group contributed to the improvement in inhibition (Fig. 3b ▶, arrow). The root mean square deviation (RMSD) between XN1336–51 and the indinavir 3X-protease complexes for all α carbons was 0.15 Å, indicating relatively little change in the protease backbone as a result of the added methyl group on XN1336–51. The added group does induce a 0.20 Å shift that can be seen throughout the entire inhibitor molecule, away from V84a compared with the position of indinavir (Fig. 5a ▶). In addition, a slight adjustment in the position of the side chain of V84a was observed, with a difference in χ1 of 12.7°. I47a of this inhibitor protease complex became more ordered as a result of the added methyl group and only one conformation of this side chain was necessary to fit the electron density. Thus, the methyl group added onto the inhibitor ordered the conformation of nearby groups in the active site.
Fig. 5.
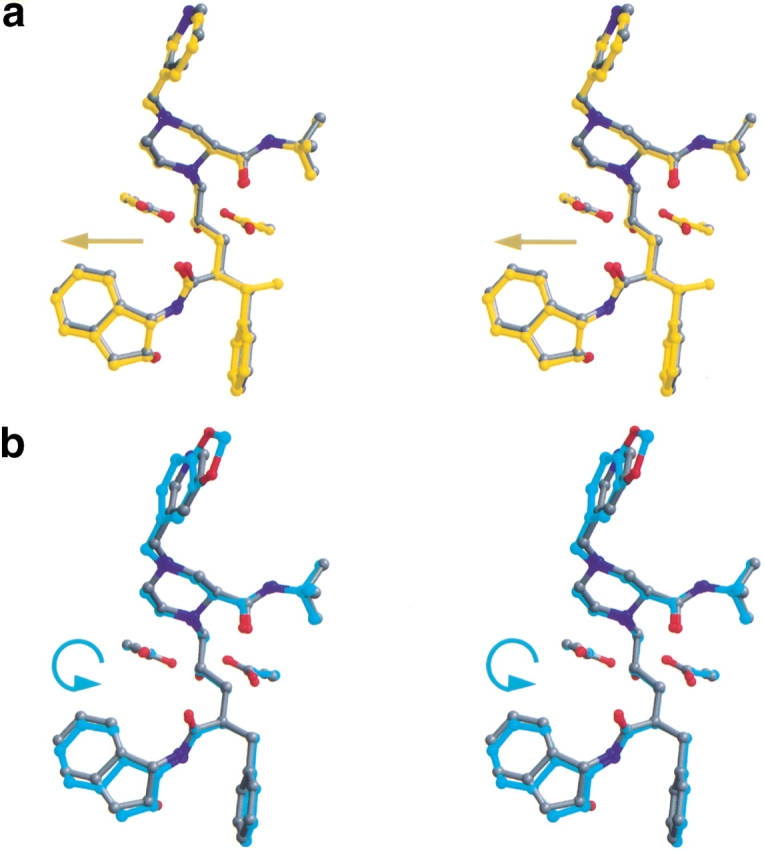
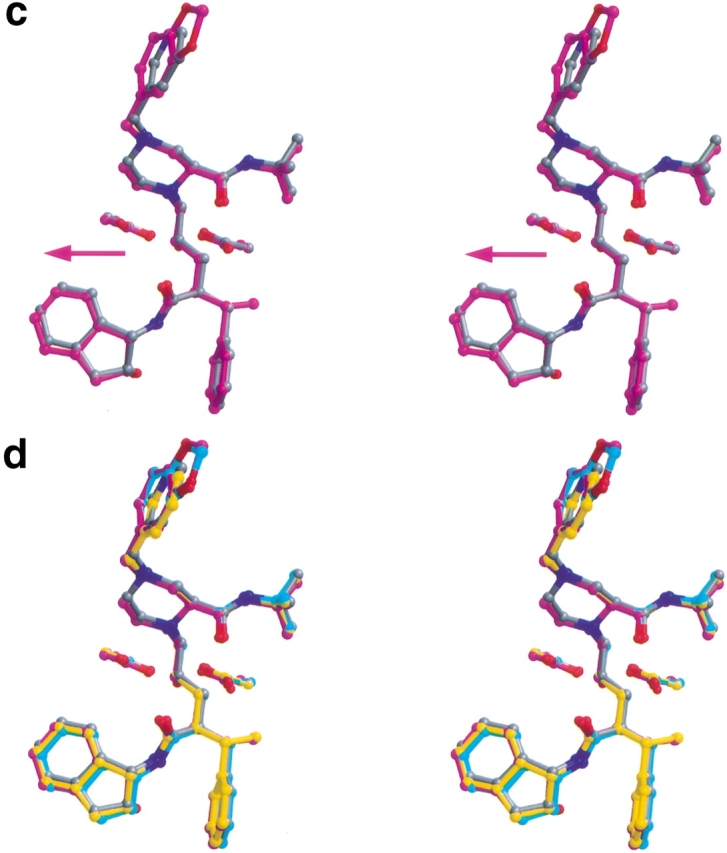
Stereo views overlapping each of the three indinavir analogs on indinavir (in gray). Also shown are the catalytic aspartic acids at residue 25 in each monomer of the protease. Figures were made with MIDAS (Ferrin et al. 1988). (a) XN1336–51 (in yellow). The arrow indicates the direction of the shift relative to indinavir (in gray). (b) 807–29–4 (in cyan). The arrow indicates the tilt of the inhibitor relative to indinavir (in gray). (c) XN1336–52 (magenta). The arrow indicates the direction of the shift relative to indinavir (in gray). (d) All four inhibitors superimposed.
807–29–4
Although the inhibitor's activity improved upon filling the cavity created by the protease mutation at position 84, further improvement was necessary to obtain a viable lead compound. An alternative strategy in the search for a better inhibitor involved replacing the pyridyl moiety. A library of functional groups at this position was randomly screened. Inhibition assays on the resulting compounds determined 807–29–4 to be the best inhibitor of this screen. In this analog, a methylenedioxyphenyl group has replaced the pyridine ring of indinavir. This modification resulted in an even tighter inhibitor, with an IC50 of 0.31 μM (Table 3), although the thermodynamics of binding in the absence of substrate did not show improvement over indinavir (Table 2).
The RMSD for the superposition of the crystal structure of the 3X-protease 807–29–4 complex and the 3X-protease indinavir complex was 0.20 Å for all α carbons, with the main deviations in the loop involving active site residues 80b-82b in the S1 subsite (Fig. 3c,e ▶). This region of the S1 loop has moved away from the inhibitor an average of 0.56 Å relative to the corresponding region in the protease-3X indinavir complex. Similar variations in this region have been observed in other protease inhibitor structures (Baldwin et al. 1995; Ala et al. 1998). In this case, the adaptation in the protease as a result of replacing the pyridyl moiety paralleled the movement of the inhibitor. The modification in 807–29–4 resulted in a “tilt” in its position relative to the position of indinavir in their respective protease complexes (Fig. 5b ▶). The inhibitor 807–29–4 in this complex has pushed away from the protease flaps with the modified region of the inhibitor closer to P81b. The indanol group at the opposite end of the inhibitor has also been pushed away from the S2′ binding pocket. An additional adaptation by the protease in its complex with 807–29–4 was seen at L23b (Fig. 3c ▶), the side chain of L23b has rotated away from the inhibitor piperidine ring, possibly because of the tilt in the ligand, which has shifted the piperidine ring towards L23b. A further adjustment in the protease occurred at the tip of the flap of monomer B. The side chain of I50b assumed two conformations, with the CD1 atom of the second conformation directed towards the space between the inhibitor's t-butyl group and protease residue V84a, as an attempt to fill the void created by the I84aV mutation (Fig. 3c ▶). Although there was some deviation in the position of I50b in the other inhibitor protease complexes, the side chain rotamers were basically the same. Flexibility at I50 has also been seen in the quadruple-mutant protease (M46I,L63P,V82T,I84V) indinavir complex (Chen et al. 1995). In addition, in the 3X-protease 807–29–4 complex, the side chain of I50a has relocated to fill the pocket created by the I84bV mutation on the opposite side of the active site, near the piperidine ring of the inhibitor. Thus, the flexibility of both backbone and side chains in the flap region of the protease allows it to adapt in this complex as well as in related complexes. The precise nature of the adaptability depends upon the exact protease variant and the inhibitor bound.
XN1336–52
Because the two beneficial modifications made to indinavir occurred in different regions of the compound, both modifications were combined into a single compound, with the hope of obtaining a synergistic effect. However, both the inhibition (Table 3, Fig. 4 ▶) and the thermodynamic data (Table 2) for this new indinavir analog, XN1336–52, show it to be a less effective inhibitor than indinavir. Superposition of the crystal structure of the 3X-protease complex and the doubly modified XN1336–52 with the two singly modified inhibitor analog 3X-protease complexes can be seen in Fig. 5d ▶. The RMSDs were 0.15 Å and 0.20 Å for the superposition of all α carbon atoms of the 3X-protease XN1336–52 and 3X-protease XN1336–51 complexes and the 3X-protease XN1336–52 and 3X-protease 807–29–4 complexes, respectively. This observation indicates that the complex with the doubly modified analog is more similar to the complex containing the inhibitor analog with the smaller methyl group added (XN1336–51) than it is to the complex containing the larger modification (807–29–4). Thus, the s-methyl group addition in the benzylic position of the inhibitor appears to have more impact on structural adaptations in the protease than does the substitution of the methylenedioxyphenyl group for the inhibitor's pyridine ring (Fig. 3d,e ▶).
Comparing complexes
Examination of the substituted residues in the active site of the 3X-protease indinavir complex reveals both similarities and differences with related structures. In two previously solved protease indinavir complexes, wild-type and quadruple-mutant (M46I,L63P,V82T,I84V) (Chen et al. 1994,1995), the conformations of residue 82a in the S1′ loop were the same as those in the triple-mutant complexes. The CG1 atom of residue 82a makes van der Waals contacts with the benzyl ring of indinavir. In the quadruple-mutant protease indinavir complex and in the present 3X-protease indinavir complex, threonine has been substituted for the isosteric valine at that position. In these mutant protease structures, the side chain oxygen faces away from the active site, thus reducing its inhibitory effect. On the other hand, the corresponding side chain at position 82b in the S1 loop of the 3X-protease inhibitor complex is rotated 96° about the χ1 bond relative to that in the wild-type structure. (The position of the side chain of T82b in the quadruple-mutant protease indinavir complex was similar to that seen for V82b in the wild-type complex.) Thus, in the present 3X-protease indinavir complex, the oxygen of the side chain of T82b (Fig. 3a ▶) also points away from the inhibitor, unlike that seen in the related indinavir structures. The conformations of T82a and T82b in all of the complexes of the 3X-protease indinavir and indinavir analogs were basically the same (Fig. 3 ▶).
In contrast with V82T, the protease mutation I84V physically changes the volume of the S1 and S1′ subsites within the active site. Each inhibitor occupies a slightly different position in the active site, causing slight shifts in the position of V84a in the S1 subsite, although the side chain rotamers remain the same. On the other hand, for each of the 3X-protease inhibitor complexes, V84b in the S1 subsite had two conformations differing by a rotation about χ1 of 109°–122° (Fig. 3 ▶). Multiple conformations at I84 have previously been characterized for the wild-type protease inhibitor complex, presumably because of the relatively larger S1 pocket (Baldwin et al. 1995).
Thus far, all of the differences described here primarily involve local adaptations in the protease. However, these adaptations do not take into account any long-range adjustments (beyond the RMSDs) that the protease may make between the various inhibitor complexes. These differences can arise as a result of subtle changes in the positions of the inhibitor molecules bound in the active site. A set of double difference plots (Fig. 6 ▶) showing the differences in domain to domain distances for the various structures provides information that is otherwise not readily apparent. Figure 6a ▶ shows the α carbon difference distance matrix for the comparison between the 3X-protease XN1336–51 complex and the 3X-protease indinavir complex. This plot shows little variation between the two complexes. In contrast, large structural differences can be seen in the double difference plot comparing the 3X-protease 807–29–4 complex and the indinavir complex (Fig. 6b ▶). However, in the double difference plot between the XN1336–52 protease complex and the 3X-protease indinavir complex (Fig. 6c ▶) little change is once again observed even though, as in 807–29–4, a methylenedioxyphenyl group is part of the inhibitor. In fact, as shown in Figure 6d ▶, the 3X-protease XN1336–51 complex and the 3X-protease XN1336–52 complex are virtually identical. This indicates that the presence of an added methyl group in the S1′ pocket of the active site has a stronger impact on the structure of the protease inhibitor complex than does the methylenedioxyphenyl moiety.
Fig. 6.

Double difference distance plots for the various inhibitor complexes of HIV-1 protease. The value plotted is Dij = dij (first complex) − dij (second complex), where dij indicates the distance between the α carbons i and j in a particular complex. Contours in the plots show whether the respective distances in the two complexes being compared are closer or further apart. Black indicates a difference of <−0.6 Å, red a difference of between −0.59 and −0.3 Å, blue a difference of between 0.3 and 0.59 Å, and yellow a difference of >0.6 Å. (a) Drug-resistant variant HIV-1 protease complexes bound with indinavir and XN1336–51. (b) Drug-resistant variant HIV-1 protease complexes bound with indinavir and 807–29–4. (c) Drug-resistant variant HIV-1 protease complexes bound with indinavir and XN1336–52. (d) Drug-resistant variant HIV-1 protease complexes bound with XN1336–51 and XN1336–52.
These findings demonstrate that a small change in the central active site S1′ pocket results in more significant adaptations in the protease than a larger modification occurring in the S1/S3 subsite. However, 807–29–4 was the tightest binding analog tested here when competing with substrate, with an IC50 of 0.31 μM compared with 0.47 μM for indinavir. Therefore, the protease modifications observed in the S1/S3 subsite, around residue P81b, were likely advantageous for better inhibition, and the lack of such an adaptation in the 3X-protease XN1336–52 complex resulted in a weaker inhibitor.
The details of the IC50s and the thermodynamics of the four inhibitors vary upon comparison of their relative binding. This is not completely unexpected, as although the two methods are measuring similar properties, they are not identical. The IC50s (Table 3, Fig. 4 ▶) of these competitive inhibitors are determined as described in the methods by blocking the cleavage of a peptide AcSQNYPVV-NH2 by the 3X-protease. Thus, the inhibitors compete with the binding of substrates and with product release. This competition increases IC50s. The thermodynamic measurements (Table 2), on the other hand, are made in the absence of substrates and are measuring the direct binding of the inhibitors to the 3X-protease. As the absolute binding of these various inhibitors is fairly similar, subtle changes due to possible product inhibition could account for the change in the relative inhibition and binding between the two techniques. Nevertheless, both techniques verify the nonadditivity of the two functional groups when combined in the XN1336–52 compound.
Conclusions
The emergence of drug-resistant mutations in HIV protease has caused significant difficulties in the treatment of HIV infection. Not only does the continuation of presently existing protease inhibitor therapies become more difficult, but more recently, physicians have basically no effective treatment options if the initial HIV infection is caused by an already drug-resistant variant of the virus. The possibility of widespread transmission of these drug-resistant viruses emphasizes the need for developing drugs that will effectively inhibit newly emerging proteases.
This study focused on a multiple drug-resistant variant of HIV protease having amino acid substitutions L63P, V82T, and I84V. Both in vitro and in vivo studies involving a variety of protease inhibitors have shown that these substitutions commonly emerge (Condra et al. 1995; Molla et al. 1998). Solving the crystal structure of this protease with the inhibitor indinavir led to the design and analysis of indinavir analogs. Modifications made to different regions of the indinavir molecule resulted in similar improvements in inhibition, despite very different structural adaptations in the protease. The addition of an s-methyl group at the benzylic position of indinavir in one analog, XN1336–51, led to an increase in van der Waals interactions between the inhibitor and nearby atoms in the active site of the protease. In another analog, 807–29–4, substitution of the pyridine ring of indinavir with a methylenedioxyphenyl group resulted in structural rearrangements that propagated throughout the protease dimer.
Combining these two modifications into a single indinavir analog, XN1336–52, however, resulted in a compound with decreased inhibition and binding affinity relative to indinavir. The protease was also unable to adapt and assume the structurally beneficial rearrangements seen in either of its complexes with inhibitors containing the individual modifications alone. The challenge remains to develop an inhibitor that optimally combines the advantageous aspects of both analogs. Perhaps such an inhibitor would be a non-indinavir type molecule, having a novel scaffold that both fills the active site pocket formed by the I84V mutation and allows the movements within the protease seen around residue P81 in the S1 loop of the 807–29–4 complex, hopefully preserving the higher affinity.
Materials and methods
Protease gene construction
The 3X-protease (L63P, V82T, I84V) gene was constructed using standard site-directed mutagenesis of a synthetic protease variant. The N-terminal encoding sequence of HXB2 was replaced by a synthetic gene sequence having the (neutral) polymorphisms V3I, K14R, and S37N. The protease variant also included an additional substitution of Q7K to prevent autoproteolysis (Rose et al. 1993).
Protease expression and purification
The gene encoding HIV protease was cloned into the plasmid pXC34 (ATCC), which contains a λ PL promoter (Cheng and Patterson 1992). The protease was expressed by heat induction in Escherichia coli TAP 106 cells using this plasmid. Cells from 12 L of fermentation were lysed and the protein was purified from inclusion bodies (Hui et al. 1993). The inclusion body centrifugation pellet was dissolved in 50% acetic acid followed by another round of centrifugation to remove impurities. Size exclusion chromatography was used to separate high molecular weight proteins from the desired protease. This was carried out on a 2.1-L Sephadex G-75 superfine (Sigma Chemical) column equilibrated with 50% acetic acid. The protein was refolded in 10 mM formic acid (Todd et al. 1998). A final purification was performed with a Pharmacia Superdex 75 FPLC column equilibrated with 0.05 M sodium acetate at pH 5.5, 5% ethylene glycol, 10% glycerol, and 5 mM DTT.
Synthesis of inhibitors
The synthesis and preparation of the protease inhibitors used in this study are shown in Figure 1b ▶. The penultimate piperazines (compound I in Fig. 1b ▶) were prepared according to the method published in US patent 5,436,067, assigned to Merck & Co., Inc. T-amylamine was substituted for t-butylamine in the preparation of the piperazine amide for synthesis of the precursor to XN1336–27. In the cases of the precursors to XN1336–51 and XN1336–52, (S)-3-phenylbutyric acid (purchased from Fluka) replaced hydrocinnamic acid for the preparation of compound I (Fig. 1b ▶). Preparation of the final products was accomplished by reductive alkylation of compound I by either pyridine-3-carboxaldehyde (XN1336–27, XN1336–51) or piperonal (807–29–4, XN1336–52), in the presence of sodium triacetoxyborohydride, as described (Abdel-Magid et al. 1996). The final products were purified by preparative thin layer chromatography using silica plates and 10% methanol in methylene chloride as the eluent. Structures and purities were confirmed by proton and carbon NMR and mass spectroscopy, with no evidence of hydration.
Crystallization and data collection
Crystals were set up using a three- to fivefold molar excess of inhibitor to protease, which ensures ubiquitous binding. The final concentration of protease was approximately 2 mg/mL in 0.05 M sodium acetate at pH 5.5, 5% ethylene glycol, 10% glycerol, 5 mM DTT. Equal volumes of the inhibitor–protein mixture and the reservoir solution were combined to set up hanging drops of 5 μL. The reservoir solution consisted of 126 mM phosphate buffer at pH 6.2, 63 mM sodium citrate, and ammonium sulfate in a range of 27–33% (Silva et al. 1996). Crystals were grown at ambient temperature and were evident within 24–72 hours. The data collection took place at room temperature on an R-AXIS-IV imaging plate system. The data were reduced and scaled using the programs DENZO and SCALEPACK (Otwinowski 1993), respectively. Crystals of both the indinavir and the indinavir analog protease complexes were of the P212121 space group, with one dimer per asymmetric unit.
Refinement
The program CNS [Crystallography and NMR System (Brunger et al. 1998)] was used to refine the structures. The wild-type HIV-1 protease indinavir complex (1HSG) (Chen et al. 1994) served as a model for solving the 3X-protease indinavir structure by molecular replacement. The 3X-protease indinavir complex was, in turn, used as a model for refining the corresponding 3X-protease XN1336–51 structure. The model used for refinement of the subsequent indinavir analog 3X-protease 807–29–4 complex was a high-resolution, well-refined substrate–protease structure recently solved in our laboratory (M. Prabu-Jeyabalan, E. Nalivaika, and C.A. Schiffer, unpubl.), having the same P212121 space group. The structure for 3X-protease 807–29–4 complex was then used as the model to refine the structure of the 3X-protease XN1336–52 complex. For each structure, an initial rigid body refinement was performed at 4Å resolution with difference Fourier electron density maps (Fo − Fc and 2Fo − Fc) subsequently computed. The program CHAIN (Sack 1988) was used for model building. Positional and B-factor refinements were carried out, and the difference Fourier electron density map (Fo − Fc) unambiguously revealed the positions of the inhibitors. The resolution limits were gradually extended in equal steps to accomplish the highest possible resolution. Solvent molecules were added manually at positions indicated by the electron density. For cross-validation, R-free values were monitored and simulated annealed omit maps were used to decrease model bias. The stereochemical parameters of the final structures were analyzed using the programs PROCHECK (Laskowski et al. 1993) and WHATIF (Vriend 1990; Hooft et al. 1996). There were no outliers in the Ramachandran maps. The crystallographic and refinement statistics are shown in Table 1. Complexes were superimposed using the α-carbon backbone of the relatively immobile (Rose et al. 1998) terminal domain of all these structures (residues 1–9 and 86–99) with the program MIDAS (Ferrin et al. 1988). The coordinates have been deposited in the protein databank, 1K6C, 1K6P, 1K6T, and 1K6V for the 3X-protease indinavir, 807–29–4, XN 1336–51 and XN1336–52 complexes, respectively.
Protease inhibition assays
Inhibition of the triple-mutant protease by the various inhibitors was determined. Activity assays were performed at 37°C with the substrate AcSQNYPVV-NH2 (Sigma). Constant concentrations of protease, 0.05 mg/mL, and substrate, 492 μM, were used for all experiments. Inhibitor concentrations were varied from 0.0–0.80 μM. For each concentration of inhibitor, aliquots of the reaction mixture were removed at 15-min intervals, quenched with equal volumes of cold trichloroacetic acid, and assayed by reverse phase HPLC (Moore et al. 1989). The percent of activity remaining for each inhibitor concentration was plotted and IC50s were obtained (Fig. 4 ▶).
Thermodynamic binding assays
An isothermal titration calorimeter, a VP-ITC (MicroCal Inc.), was used to measure binding energies of inhibitors. Twenty to twenty-four 10–12-μl injections of 0.2 mM inhibitor were made into 29 μM wild-type HIV-1 protease and 22.7 μM 3X-protease HIV-1 protease at 20°C. The buffer in which both proteins and the inhibitor were suspended was 10 mM sodium acetate, 2.0% DMSO, and 2mM TCEP at pH 5.0. Heats of dilution were subtracted from the corresponding heats of reaction to obtain the heat due solely to the binding of the ligand to the enzyme. Data was processed and analyzed using the MicroCal Origin software package.
Acknowledgments
This work was supported in part by grants from the University of Massachusetts Medical School Center for AIDS Research, the American Foundation for AIDS Research, and Sepracor, Inc. N.M. King was also supported by NIH F32-GM62993–02 postdoctoral fellowship. Some of the indinavir was obtained through the AIDS Research and Reference Reagent Program, NIAID, NIH:Indinavir. We also thank Kendall Knight and Anthony Carruthers for helpful discussions and Claire Baldwin for editorial advice.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.25502.
References
- Abdel-Magid, A.F., Carson, K.G., Harris, B.D., Maryanoff, C.A., and Shah, R.D. 1996. Reductive amination of aldehydes and ketones with sodium triacetoxyborohydride. Studies on direct and indirect reductive amination procedures. J. Org. Chem. 61 3849–3862. [DOI] [PubMed] [Google Scholar]
- Ala, P.J., Huston, E.E., Klabe, R.M., McCabe, D.D., Duke, J.L., Rizzo, C.J., Korant, B.D., DeLoskey, R.J., Lam, P.Y., Hodge, C.N., and Chang, C.H. 1997. Molecular basis of HIV-1 protease drug resistance: Structural analysis of mutant proteases complexed with cyclic urea inhibitors. Biochemistry 36 1573–1580. [DOI] [PubMed] [Google Scholar]
- Ala, P.J., Huston, E.E., Klabe, R.M., Jadhav, P.K., Lam, P.Y., and Chang, C.H. 1998. Counteracting HIV-1 protease drug resistance: Structural analysis of mutant proteases complexed with XV638 and SD146, cyclic urea amides with broad specificities. Biochemistry 3715042–15049. [DOI] [PubMed] [Google Scholar]
- Baldwin, E.T., Bhat, T.N., Liu, B., Pattabiraman, N., and Erickson, J.W. 1995. Structural basis of drug resistance for the V82A mutant of HIV-1 proteinase. Struc. Biol. 2 244–249. [DOI] [PubMed] [Google Scholar]
- Boden, D. and Markowitz, M. 1998. Minireview: Resistance to human immunodeficiency virus type 1 protease inhibitors. Antimicrob. Agents Chemother. 42 2775–2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., Read, R.J., Rice, L.M., Simonson, T., and Warren, G.L. 1998. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D54 905–921. [DOI] [PubMed] [Google Scholar]
- Chen, Z., Li, Y., Chen, E., Hall, D.L., Darke, P.L., Culberson, C., Shafer, J.A., and Kuo, L.C. 1994. Crystal structure at 1.9 Angstrom resolution of human immunodeficiency virus (HIV) II protease complexed with L-735,524, and orally bioavailable inhibitor of the HIV proteases. J. Biol. Chem. 269 26344–26348. [PubMed] [Google Scholar]
- Chen, Z., Schock, H.B., Hall, D., Chen, E., and Kuo, L.C. 1995. Three-dimensional structure of a mutant HIV-1 protease displaying cross-resistance to all protease inhibitors in clinical trials. J. Biol. Chem. 270 21433–21436. [DOI] [PubMed] [Google Scholar]
- Cheng, X. and Patterson, T.A. 1992. Construction and use of l PL promoter vectors for direct cloning and high level expression of PCR amplified DNA coding sequences. Nucleic Acids Res. 20 4591–4598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condra, J.H., Schleif, W.A., Blahy, O.M., Gabryelski, L.J., Graham, D.J., Quintero, J.C., Rhodes, A., Robbins, H.L., Roth, E., Shivaprakash, M., Titus, D., Yang, T., Teppler, H., Squires, K.E., Deutsch, P.J., and Emini, E. 1995. In vivo emergence of HIV-1 variants resistant to multiple protease inhibitors. Nature 374 569–571. [DOI] [PubMed] [Google Scholar]
- Debouck, C. 1992. The HIV-1 protease as a therapeutic target for AIDS. AIDS Res. Hum. Retroviruses 8 153–164. [DOI] [PubMed] [Google Scholar]
- Ermolieff, J., Lin, X., and Tang, J. 1997. Kinetic properties of Saquinavir resistant mutants of human immunodeficiency virus type 1 protease and their implications in drug resistance in vivo. Biochemistry 36 12364–12370. [DOI] [PubMed] [Google Scholar]
- Ferrin, T.E., Huang, C.C., Jarvis, L.E., and Langridge, R. 1988. The MIDAS display system. J. Mol. Graphics. 6 13–27. [Google Scholar]
- Gulnik, S.V., Suvorov, L.I., Liu, B., Yu, B., Anderson, B., Mitsuya, H., and Erickson, J.W. 1995. Kinetic characterization and cross-resistance patterns of HIV-1 protease mutants selected under drug pressure. Biochemistry 34 9282–9287. [DOI] [PubMed] [Google Scholar]
- Hong, L., Treharne, A., Hartsuck, J.A., Foundling, S., and Tang, J. 1996. Crystal structures of complexes of a peptidic inhibitor with wild type and two mutant HIV-1 proteases. Biochemistry 35 10627–10633. [DOI] [PubMed] [Google Scholar]
- Hong, L., Zhang, X.J., Foundling, S., Hartsuck, J.A., and Tang, J. 1997. Structure of a G48H mutant of HIV-1 protease explains how glycine-48 replacements produce mutants resistant to inhibitor drugs. FEBS Lett. 420 11–16. [DOI] [PubMed] [Google Scholar]
- Hong, L., Hartsuck, J.A., Foundling, S., Ermolieff, J., and Tang, J. 1998. Active site mobility in human immunodificiency virus, type 1, protease as demonstrated by crystal structure of A28S mutant. Protein Sci. 7 300–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooft, R.W.W., Vriend, G., Sander, C., and Abola, E.E. 1996. Errors in protein structures. Nature 381 272. [DOI] [PubMed] [Google Scholar]
- Hui, J.O., A.G., T., Reardon, I.M., Lull, J.M., Brunner, D.P., Tomich, C.C., and Heinrikson, R.L. 1993. Large scale purification and refolding of HIV-1 protease from Escherichia coli inclusion bodies. J. Prot. Chem. 12 323–327. [DOI] [PubMed] [Google Scholar]
- Kervinen, N., Thanki, N., Zdanov, A., Tino, J., Barrish, J., Lin, P., Colonno, F., Riccardi, K., Samanta, H., and Wlodawer, A. 1996. Structural analysis of the native and drug-resistant HIV-1 proteinases complexed with an aminodiol inhibitor. Protein Pept. Lett. 3 399. [Google Scholar]
- Klabe, R.M., Bacheler, L.T., Ala, P.J., Erickson-Viitanen, S., and Meek, J.L. 1998. Resistance to HIV protease inhibitors: a comparison of enzyme inhibition and antiviral potency. Biochemistry 37 8735–8742. [DOI] [PubMed] [Google Scholar]
- Laskowski, R.A., Mac Arthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK. A program to check the stereochemical quality of protein structures. J. Appl. Cryst. 26 283–291. [Google Scholar]
- Lin, Y., Lin, X., Hong, L., Foundling, S., Heinrikson, R.L., Thaisrivongs, S., Laalamanit, W., Raterman, D., Shah, M., Dunn, B.M., and Tang, J. 1995. Effect of point mutations on the kinetics and inhibition of human immunodeficiency virus type 1 protease: Relationship to drug resistance. Biochemistry 34 1143–1152. [DOI] [PubMed] [Google Scholar]
- Markowitz, M., Mo, H., Kempf, D.J., Norbeck, D.W., Bhat, T.N., Erickson, J.W., and Ho, D.D. 1995. Selection and analysis of human immunodeficiency virus type 1 variants with increased resistance to ABT-538, a novel protease inhibitor. J. Virol. 69 701–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molla, A., Granneman, G.R., Sun, E., and Kempf, D.J. 1998. Recent developments in HIV protease inhibitor therapy. Antiviral Res. 39 1–23. [DOI] [PubMed] [Google Scholar]
- Moore, M.L., Bryan, W.M., Fakhoury, S.A., Magaard, V.W., Huffman, W.F., Dayton, B.D., Meek, T.D., Hyland, L., Dreyer, G.B., Metcalf, B.W., Strickler, J.E., Gorniak, J.G., and Debouck, C. 1989. Peptide substrates and inhibitors of the HIV-1 protease. Biochem. Biophys. Res. Commun. 159 420–425. [DOI] [PubMed] [Google Scholar]
- Olsen, D.B., Stahlhut, M.W., Rutkowski, C.A., Schock, H.B., vanOlden, A.L., and Kuo, L.C. 1999. Non-active site changes elicit broad-based cross-resistance of the HIV-1 protease to inhibitors. J. Biol. Chem. 274 23699–23701. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. 1993. Oscillation data reduction program. In Data collection and processing (ed. L. Sawyer, N. Isaacs, and S. Bailey), pp. 56–62. SERC Daresburg Laboratory, UK.
- Pazhanisamy, S., Stuver, C.M., Cullinan, A.B., Margolin, N., Rao, B.G., and Livingston, D.J. 1996. Kinetic characterization of human immunodeficiency virus type-1 protease-resistant variants. J. Biol. Chem 271 17979–17985. [DOI] [PubMed] [Google Scholar]
- Rose, J.R., Salto, R., and Craik, C.S. 1993. Regulation of autoproteolysis of the HIV-1 and HIV-2 proteases with engineered amino acid substitutions. J. Biol. Chem. 268 11939–11945. [PubMed] [Google Scholar]
- Rose, R.B., Craik, C.S., and Stroud, R.M. 1998. Domain flexibility in retroviral proteases: structural implications for drug resistant mutations. Biochemistry 37 2607–2621. [DOI] [PubMed] [Google Scholar]
- Sack, J.S. 1988. CHAIN—A crystallographic modeling program. J. Mol. Graphics 6 224–225. [DOI] [PubMed] [Google Scholar]
- Schinazi, R.F., Larder, B.A., and Mellors, J.W. 1997. Mutations in retroviral genes associated with drug resistance. Int. Antiviral News 5 129–142. [Google Scholar]
- Schock, H.B., Garsky, V.M., and Kuo, L.C. 1996. Mutational anatomy of an HIV protease variant conferring cross resistance to protease inhibitors in clinical trials. J. Biol. Chem. 271 31957–31963. [DOI] [PubMed] [Google Scholar]
- Silva, A.M., Cachau, R.E., Sham, H.L., and Erickson, J.W. 1996. Inhibition and catalytic mechanism of HIV-1 aspartic protease. J. Mol. Biol. 255 321–346. [DOI] [PubMed] [Google Scholar]
- Swairjo, M.A., Towler, E.M., Debouck, C., and Abdel-Meguid, S.S. 1998. Structural role of the 30's loop in determining inhibitor specificity of the human immunodeficiency virus protease. Biochemistry 37 10928–10936. [DOI] [PubMed] [Google Scholar]
- Todd, M.J., Semo, N., and Freire, E. 1998. The structural stability of the HIV-1 protease. J. Mol. Biol. 283 475–488. [DOI] [PubMed] [Google Scholar]
- Turner, B.G. and Summers, M.F. 1998. Structural biology of HIV. J. Molec. Biol. 285 1–32. [DOI] [PubMed] [Google Scholar]
- Vriend, G. 1990. What IF: A molecular modelling and drug design program. J. Mol. Graph. 8 52–56. [DOI] [PubMed] [Google Scholar]
- Wilson, S.I., Phylip, L.H., Mills, J.S., Gulnik, S.V., Erickson, J.W., Dunn, B.M., and Kay, J. 1997. Escape mutants of HIV-1 proteinase: Enzymic efficiency and susceptibilty to inhibition. Biochim. Biophys. Acta 1339 113–125. [DOI] [PubMed] [Google Scholar]
- Wilson, S.I., Phylip, L.H., Gulnik, S.V., Mills, J.S., Bur, D., Dunn, B.M., and Kay, J. 1998. Sensitivity to inhibition and catalytic efficiency of HIV proteinase mutants. Plenum Press, New York. [DOI] [PubMed]
- Wlodawer, A. and Erickson, J.W. 1993. Structure-based inhibitors of HIV-1 protease. Ann. Rev. Biochem. 62 543–585. [DOI] [PubMed] [Google Scholar]
- Wlodawer, A. and Gustchina, A. 2000. Structural and biochemical studies of retroviral proteases. Biochim. Biophys. Acta 1477 16–34. [DOI] [PubMed] [Google Scholar]