

Abstract
EF-hand Ca2+-binding proteins participate in both modulation of Ca2+ signals and direct transduction of the ionic signal into downstream biochemical events. The range of biochemical functions of these proteins is correlated with differences in the way in which they respond to the binding of Ca2+. The EF-hand domains of calbindin D9k and calmodulin are homologous, yet they respond to the binding of calcium ions in a drastically different manner. A series of comparative analyses of their structures enabled the development of hypotheses about which residues in these proteins control the calcium-induced changes in conformation. To test our understanding of the relationship between protein sequence and structure, we specifically designed the F36G mutation of the EF-hand protein calbindin D9k to alter the packing of helices I and II in the apoprotein. The three-dimensional structure of apo F36G was determined in solution by nuclear magnetic resonance spectroscopy and showed that the design was successful. Surprisingly, significant structural perturbations also were found to extend far from the site of mutation. The observation of such long-range effects provides clear evidence that four-helix EF-hand domains should be treated as a single globally cooperative unit. A hypothetical mechanism for how the long-range effects are transmitted is described. Our results support the concept of energetic and structural coupling of the key residues that are crucial for a protein's fold and function.
Keywords: Calcium-binding protein, conformational change, EF-hand, mutagenesis, NMR spectroscopy, protein engineering
The EF-hand family of Ca2+-binding proteins (CaBPs) provides a rich framework for investigating fundamental relationships between protein sequence and biochemical function. The EF-hand motif is among the most common in animal cells (Henikoff et al. 1997); more than 1000 have been identified from their unique sequence signatures. These motifs are organized into structural units/domains containing two or more EF hands that form highly stable helical bundles (Nelson and Chazin 1998a). Despite the similarities in their sequences and structures, EF-hand proteins perform a diverse range of functions. There are two primary classes of EF-hand proteins: Ca2+ sensors, which transduce Ca2+ signals, and Ca2+ signal modulators, which modulate the shape and/or duration of Ca2+ signals or participate in Ca2+ homeostasis. Here, we describe research on representative sensor and signal modulator proteins, calmodulin (CaM) and calbindin D9k (calbindin), which is directed toward understanding how differences in amino acid sequence specify differences in how EF-hand domains respond to the binding of Ca2+ ions, the key biochemical function distinguishing various EF-hand proteins.
Calmodulin and calbindin are homologous proteins (i.e., ∼25% sequence identity) and have very similar structures in the absence of Ca2+ ions. However, there is a striking difference in the structures of CaM and calbindin in the presence of Ca2+. Both EF-hand domains of CaM undergo a Ca2+-induced change into an open conformation, characterized by an exposed hydrophobic patch (Kuboniwa et al. 1995; Finn et al. 1995; Zhang et al. 1995). Calbindin, on the other hand, remains in a closed conformation on Ca2+ binding that is similar to the conformation observed for the apo state (Skelton et al. 1994). This difference in the response to the binding of Ca2+ correlates well with the various roles of CaM and calbindin in the cell (Skelton et al. 1994). Understanding what structural factors and amino acid properties control Ca2+-induced conformational changes is a critical step toward understanding how sequence dictates EF-hand protein structure and function. Several protein design efforts today have similar objectives, for example, the determination of a few mutations to convert a protein from its native fold to a fundamentally different fold (Dalas et al. 1997).
Hypotheses about the relationship between sequence and structure at particular sites in EF-hand proteins can be tested by the design and analysis of specific mutant proteins. Our strategy for the design of mutations is based largely on detailed studies of EF-hand CaBP structure (Nelson and Chazin 1998b). Our thinking is guided by the strong evidence for long-range effects in EF-hand proteins, for example, as reflected in cooperativity in Ca2+ binding and site–site interactions (Mäler et al. 2000). This approach is predicated on the firm belief that reorganization of packing in and around the hydrophobic core drives the bulk of the conformational response to Ca2+ binding. These views are supported by, and fully consistent with, recent studies showing the importance of solvation energetics as a factor contributing to the response to the binding of Ca2+ by EF-hand proteins (Ababou and Desjarlais 2001).
Results
Design of F36G calbindin D9k
Factors affecting the stability of both the open and closed conformations contribute to the observed differences in the response to the binding of Ca2+ by EF-hand CaBPs. Hence, hypotheses and the corresponding calbindin mutant designs to understand how differences in amino acid sequence specify differences in the response to the binding of Ca2+ are based on two general goals: (1) alter the packing interactions of apo calbindin so that its closed conformation more closely resembles that of a prototypical Ca2+ sensor; and (2) stabilize a CaM-like open conformation for calbindin by relieving all predicted steric conflicts and making the hydrophobic residues that would become solvent exposed more easily solvated.
Phe 36 in calbindin was identified initially as a candidate for mutation from the sequence alignment of EF-hand CaBPs. The CaM sequence homolog of Phe 36 in the N-terminal domain (CaM-N) is Gly 40. In fact, a glycine is found at this sequence position in all known members of the CaM subfamily except for the N-terminal domain of calcium-dependent protein kinase and the C-terminal domain of squidulin, both of which have serine at this position.
Structural analysis showed that this sequence difference has obvious implications for occupancy of an open conformation by calbindin. Gly 40 is completely exposed to solvent in Ca2+-loaded CaM-N, and correspondingly Phe 36 is solvent exposed in a model of calbindin in the open conformation (Fig. 1a ▶). The presence of phenylalanine instead of glycine at this position is predicted to destabilize the open conformation of calbindin, because of the much higher cost of solvating phenylalanine (Wimley et al. 1996).
Fig. 1.

Design of the F36G mutation. (a) Exposure of Phe 36 in the model of open calbindin D9k shown in CPK rendering. Phe 36 is highlighted in yellow. (b) Differences in apo state contacts made by Phe 36 and its CaM-N homolog Leu 39 in the interface between helices I and II. In calbindin (dark blue), Phe 36 interacts with Ile 9, whereas in CaM-N, Leu 39 interacts with Phe 12. The Protein Data Bank codes are 1CLB for apo calbindin D9k and 1CFC for apo CaM-N, and images were rendered with InsightII (MSI).
An important role for this site in stabilizing the closed conformation of calbindin also was revealed from the structural analyses. Inter-residue contact analyses (Nelson and Chazin 1998b) show that the closest CaM structural homolog of Phe 36 is not the sequence homolog Gly 40 but rather Leu 39, which is able to mimic most of the interactions of Phe 36 in the interface between helices II and IV. The structural identity is only partial, however, because Leu 39 does not mimic the contacts of Phe 36 in the interface between helices I and II (Fig. 1b ▶). The fact that our analysis anticipates important effects on both the closed and the open conformations suggests that this site has a particularly critical role in determining the response to Ca2+-binding in EF-hand proteins.
The F36G mutation has been produced and characterized to test our ability to rationally alter the interface between helices I and II of apo calbindin to make it more like CaM. There are two possible effects on conformation postulated to arise from this mutation. First, substituting a glycine for Phe 36 may shorten helix II. Glycine often functions as a helix breaker (Richardson and Richardson 1989), and, in fact, helix II of apo CaM-N ends at Leu 39 and not at Gly 40, whereas in calbindin it continues one residue further. Second, the substitution of glycine for Phe 36 also may alter the packing of helices I and II. The greater bulk of Phe 36 relative to its CaM-N structural homolog Leu 39 appears to force the C-terminal end of helix II away from helix I in the closed conformation of apo calbindin relative to apo CaM-N. Helices I and II in F36G are anticipated to be more closely packed than in the wild-type protein.
Characterization of F36G calbindin D9k
Initial biophysical characterization of F36G revealed that the effects of this mutation on the protein were unusual. Among the Phe → Gly mutations of calbindin (Julenius et al. 1998; Kragelund et al. 1998), F36G exhibits the smallest reductions in the stability of the apo state, the cooperativity of unfolding, and the Ca2+ affinities at 2 mM and 150 mM KCl (Table 1). These results imply that this site has some unique characteristics that allow it to better accommodate the mutation, presumably associated with the more peripheral location of F36 with respect to the protein core.
Table 1.
The effect of Phe→Gly mutations on stability and Ca2+-affinity of calbindin D9ka
| Apo stability | Cooperativity of unfolding | Ca2+-affinity | Ca2+-affinity | |
| δGNU(H2O) (kcal/mol) | A (kcal/mol • M) | δGtot (kcal/mol) 2 mM KC1 | δGtot (kcal/mol) 150 mM KC1 | |
| Wild-type | 6.29 ± 0.45 | 1.22 ± 0.10 | −22.30 | −17.60 |
| F36G | 2.92 ± 0.07 | 1.10 ± 0.02 | −20.75 | −16.00 |
| F10A | 1.72 ± 0.12 | 0.88 ± 0.05 | −19.24 | −15.49 |
| F66A | 1.58 ± 0.10 | 0.79 ± 0.05 | −20.36 | −14.40 |
a Stability data for F10A and F66A are taken from Julenius et al. (1998). Affinity data for wild-type, F10A, and F66A are taken from Kragelund et al. (1998). Similar values for wild-type were obtained in our control experiments.
An initial series of 1H nuclear magnetic resonance (NMR) analyses suggested that there were atypically large structural effects from the Phe 36 → Gly mutation. Nearly complete sequence-specific 1H NMR assignments were made for both the apo and Ca2+-loaded states of F36G. The resonance assignments were compared with the corresponding assignments for the wild-type protein, and chemical shift differences were found to be larger in magnitude and much more widely dispersed through the protein for the apo state. Thus, as anticipated in our design, the effect of the mutation was much greater on the apo state than the Ca2+-loaded state. One striking observation was that the degree of perturbation of chemical shifts induced by the Phe 36 → Gly mutation is far greater than in any other single-site mutant of calbindin studied to date.
The three-dimensional structure of apo F36G calbindin D9k was determined using standard 1H and 15N-1H NMR methods (Akke et al. 1995; Skelton et al. 1995; Mäler et al. 2000). The final ensemble of 24 conformers representing apo F36G is shown in Figure 2 ▶. The precision and accuracy of the structure is of the same order as that of the wild-type structure (Skelton et al. 1995), as judged by the comparable number of restraints, similar root-mean-square deviations, similar values of the restraint violation and total molecular energies, and backbone conformational analysis (which showed that most residues occupy the most favorable regions of φ/ξ space) (Table 2).
Fig. 2.

Structure of apo F36G calbindin D9k. Ensemble of 24 conformers representing the apo F36G structure. N, Cα, and C backbone atoms are displayed with the N-terminal EF-hand in light green, the C-terminal EF-hand in dark green, and the linker between the EF-hands in gray. Prepared using InsightII (MSI).
Table 2.
Comparison of structural statistics of apo F36G and wild-type calbindin D9k
| Wild-type | F36G | |
| Restraints | ||
| Total NOE restraints | 994 | 1042 |
| Intraresidue | 144 | 186 |
| Sequential (i, i+1) | 277 | 269 |
| Medium range a (i, i+2−i, i+4) | 268 | 289 |
| Long range (>i, i+4) | 305 | 298 |
| Hydrogen bond restraints | 32 | 18 |
| Total dihedral restraints | 122 | 115 |
| φ | 45 | 45 |
| ξ | 52 | 42 |
| κ 1 | 25 | 28 |
| Avg. RMSDs from mean structure (Å) | ||
| All atoms | 1.59 | 1.30 |
| All backbone atoms | 1.17 | 0.671 |
| All atoms in helices b | 1.04 | 1.08 |
| All backbone atoms in helices b | 0.55 | 0.41 |
| Amber energies (kcal/mol) | ||
| Avg. E rst ± RMSD | 4.25 ± 1.05 | 3.65 ± 0.44 |
| Avg. E tot ± RMSD | −1007.1 ± 13.4 | −1050.4 ± 10.7 |
| Procheck | ||
| Residues in most favored regions | 83.0% | 89.4% |
| Residues in additional allowed regions | 14.1% | 8.7% |
a Medium range restraints are defined to be between protons 2–4 residues apart in the sequence.
b Helical elements were defined on the basis of phi and psi angles and the presence of (i, i+3) or (i, i+4) hydrogen bonds (Kördel et al. 1993). The helices in wild-type and F36G, respectively, were as follows: I-2–15, 2–16; II-24–36, 25–35; III-46–54, 45–55; IV-62–74, 62–74.
Comparison of apo F36G to the wild-type protein and apo CaM-N
The structure of the mutant retains the global fold of the wild-type protein, consisting primarily of four helices, with a short β-sheet between the two Ca2+-binding loops. An extensive set of comparisons has been made between apo F36G, the wild-type protein and CaM-N, and these are summarized in Figure 3a ▶. Overall, the extent of the differences between F36G and the wild-type protein are greater than anticipated for a single-site mutation and far larger than any effect observed directly in structures from previous studies of calbindin. The differences between the F36G and wild-type structures are supported by the fact that 548 of the 1042 nuclear overhauser effect (NOE)-derived distance restraints used to calculate the F36G structure are not on the list of 994 restraints used to calculate the wild-type calbindin structure.
Fig. 3.
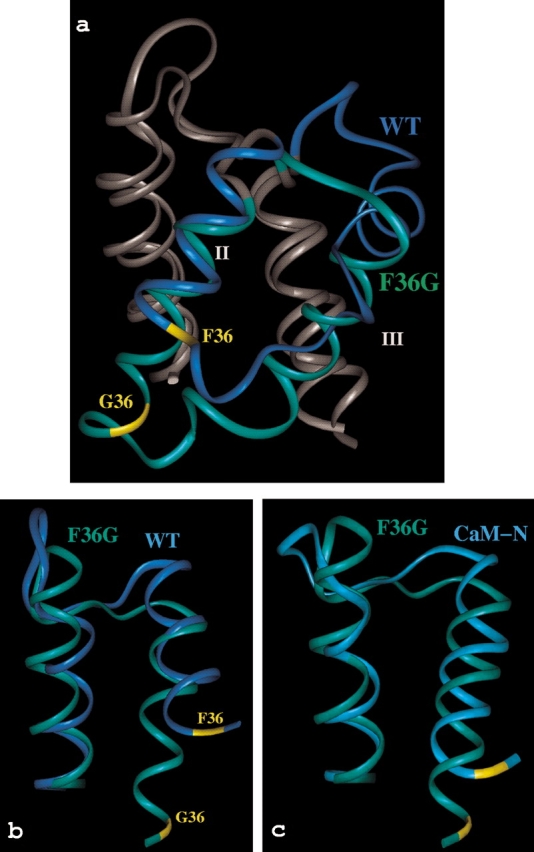
Comparison of apo F36G to wild-type calbindin D9k and CaM-N. (a) Overview of the repositioning of helices II and III of calbindin (dark blue) by the F36G mutation (green). (b) Differences in the packing of helices I and II in F36G and wild-type calbindin. (c) Differences in the packing of helices I and II in F36G (green) and CaM-N (light blue). Best-fit superpositions were made using the backbone atoms of helices I and IV. Images were rendered using InsightII (MSI).
Our results support the hypothesis that the phenylalanine at position 36 in calbindin contributes to the difference in the packing of helices I and II in wild-type apo calbindin relative to apo CaM-N. We find that the Phe → Gly mutation does repack the helix I/II interface, making it more similar to the interface in CaM-N, particularly near the site of mutation (Fig. 3b,c ▶). However, the transformation of this interface is not complete as the similarity to CaM-N is not propagated throughout the entire length of the two helices. For instance, the contacts involving Tyr 13 and Phe 10 in helix I, and residues in helix II in the wild-type protein, are largely conserved in the mutant. The conservation of the interactions between Tyr 13, Leu 23, and Leu 31 is particularly obvious (Fig. 4 ▶). This set of side-chain contacts appears to serve as an anchor in the hydrophobic core, which prevents repacking along the entire length of the helices. These results for F36G suggest that mutations at these additional sites may be required to achieve complete repacking of the entire helix I/II interface.
Fig. 4.

Conservation of contacts at the top of the I/II interface in F36G. The interaction of Tyr 13, Leu 23, and Leu 31 is shown for (a) wild-type calbindin and (b) F36G. The distances between the centers of geometry of Tyr 13 and Le 23 in F36G and wild-type calbindin D9k are 6.38 Å and 7.09 Å, respectively. The corresponding distances between Tyr 13 and Leu 31 are 5.13 Å and 4.45 Å, and the distances between Leu 23 and Leu 31 are 5.94 Å and 5.00 Å, respectively. Superpositions and rendering were as described for Fig. 3 ▶.
The most surprising effect of the Phe → Gly mutation is the drastic repositioning of helix III, which is closer and more parallel to helix II than in the wild-type protein (Fig. 3a ▶). This difference is a reflection of a substantially greater number of helix II/III side-chain contacts in F36G than either calbindin or CaM-N. The N terminus of helix III is also more closely packed to the C terminus of helix IV, whereas these two helices are further apart at the opposite ends. Overall, the mutation appears to result in a more intimate packing of helix III into the rest of the globular EF-hand domain.
Discussion
The unexpected repositioning of helix III in apo F36G shows that we cannot yet predict which interactions in calbindin D9k are relatively plastic and which will persist despite perturbations in nearby regions. Despite the dramatic structural alterations, the effects on the stability and Ca2+ affinity are modest. Moreover, the effects for F36G are smaller than those observed for other Phe → Gly mutants, which do not show significant structural alterations. We opt not to speculate here on the origin of these observations because deeper insights are anticipated from calbindin D9k protein design/engineering studies in progress in our laboratory.
Why were structural effects from the mutation transmitted specifically to helix III? Helix III is the shortest and the least well packed of the four helices in typical EF-hand domains. Moreover, in the S100 EF-hand proteins, the binding of Ca2+ results in structural effects restricted almost exclusively to helix III (Drohat et al. 1998; Matsumura et al. 1998; Smith and Shaw 1998). Thus, helix III appears to be unique, having a greater potential to occupy various packing arrangements than the other three helices in the EF-hand domain. This observation fits with the recently introduced concept of mutations causing redistributions between pre-existing conformational substates (Sinha and Nussinov 2001). Regardless of the specific mechanisms for the observed long-range effects, our results clearly indicate that the four-helix domain comprising a pair of EF hands can behave as a globally cooperative structural unit and that this unit should be treated as an integrated structural/functional entity.
The observed effect of the F36G mutation on relatively distant regions of the calbindin structure is consistent with the idea that the evolutionarily conserved identities of residues at distant sequence positions may be coupled. The work of Lockless and Ranganathan suggests that such coupling could be achieved through a pathway of key residues, which are crucial for the fold and function of the protein (Lockless and Ranganathan 1999). Phe 36 in calbindin D9k appears to be one such key residue.
What is the pathway leading to the structural perturbations so far from the site of mutation in calbindin? One plausible explanation for the shift in helix III is that the translation and rotation of helix II with respect to helix I transmits effects to Ca2+ binding loop I, and because the hydrogen bonds between the two Ca2+ binding loops are preserved, the effect is transmitted in turn to Ca2+ binding loop II. This Ca2+ binding loop includes the last two residues at the C terminus of helix III (Asp 54, Lys 55), so the structural changes transmitted from the C-terminal end of helix II through to Ca2+ binding loop II results in an effect on the local environment of helix III at the opposite side of the protein. It is conceivable that the Ca2+-induced conformational changes in S100 proteins noted above also may use this coupling pathway. If this view of the coupling pathway is accurate, then key residues, in fact, can be quite sparsely distributed along a pathway as long as there are stable and conserved secondary structure elements to relay the information/effects between the key residues.
Protein chemists continue to make progress toward understanding the individual components that specify protein structure. However, we do not yet understand these factors and, in particular, how they work together to produce a folded and functional protein. One way to increase our understanding of these components and how they interact is to formulate hypotheses about how specific amino acids in a protein contribute to an observed property or structural feature, and then design mutations to test these hypotheses. The rational design of site-specific mutants as we have shown here is an essential step in attempts to uncover the physical explanations for how a protein's sequence determines its structure and function.
Materials and methods
Sequence alignments
The information about the natural sequence variations among EF-hand CaBPs is very valuable for mutant design. For example, residues that are invariant in the sensor class of EF-hand proteins and differ from the corresponding residues in signal modulator proteins are likely to be important in determining differences in the response to the binding of Ca2+. Sequence information is most useful if it is linked with information about the structure and function of the proteins. The EF-Hand Calcium-Binding Proteins Data Library (Nelson 1999) (http://structbio.vanderbilt.edu/chazin/cabp_database/) was constructed to facilitate these sorts of interconnections among various types of information. The sequence information and appropriate annotations for our designs were taken from this resource.
The alignment of representative sequences originally were extracted from multiple sequence alignments constructed with CLUSTALW (Thompson et al. 1994), using the Web interface provided by the Network Protein Sequence Analysis group at Pôle Bio-Informatique Lyonnais (http://pbil.ibcp.fr/). All available sequences of the proteins in the subfamily were aligned using the default settings at the NPSA Web site (BLOSUM weight matrix, gap opening penalty of 1, gap extension penalty of 0.5, hydrophilic gaps on, hydrophilic residues = GPSNDQERK, residue-specific gap penalties on).
Mutant production and characterization
The F36G mutant was produced using cassette mutagenesis, overexpressed in Escherichia coli, and purified (Linse et al. 1987; Johansson et al. 1990). This mutant also contained the mutation of Pro 43 to methionine. Biophysical characterization of the F36G mutant therefore was interpreted by comparison to the corresponding data for P43M calbindin D9k.
Ca2+ affinities of F36G were measured by the competitive chelator method, using Quin-2 as a competitive chelator (Linse et al. 1987; Kragelund et al. 1998). Titrations were made in 2 mM Tris-HCl at pH 7.5. The absorbance of Quin-2 was followed at 263 nm during the titration of CaCl2 into the solution. The macroscopic Ca2+-binding constants were obtained from nonlinear least squares fits to the absorbance at 263 nm as a function of CaCl2 concentration.
The stability of apo F36G was determined from urea denaturation monitored by circular dichroism (Julenius et al. 1998). Protein (2 μM) was added to solutions of 9.8 M urea, in 1 mM potassium phosphate buffer at pH 7.0, in the presence of 0.5 mM EGTA. Data were collected at 25°C. The ellipticity at 222 nm was measured for each urea concentration in the titration, with more points collected near the transition point identified in a preliminary titration. The signal due to buffer was subtracted from the data points, which then were fit to the two-state unfolding model described in (Julenius et al. 1998).
NMR spectroscopy
NMR spectra were collected using standard methods (Cavanagh et al. 1996) following protocols previously described for calbindin D9k (Akke et al. 1995; Skelton et al. 1995; Nelson and Chazin 1999). Spectra were collected using Bruker AMX-500 or DMX-750 spectrometers on samples with a protein concentration of ∼2.5 mM at pH 6.0 and 27°C, in the presence of a trace amount of EDTA and again in the presence of 5.5 mM CaCl2. The data were processed with FELIX (MSI).
Sequence-specific 1H NMR assignments were made from two-dimensional COSY, TOCSY, and NOE spectroscopy (NOESY) spectra collected in H2O. The NOESY spectra were collected with a 200-msec mixing time. The following additional spectra were collected for structure calculations of apo F36G: two-dimensional homonuclear NOESY collected in H2O with a 45-msec mixing time; two-dimensional homonuclear NOESY collected in D2O with a 200-msec mixing time; two-dimensional 15N-1H HSQC spectrum and three-dimensional 15N TOCSY-HSQC spectrum; three-dimensional 15N NOESY-HSQC spectrum collected with a 130-msec mixing time; three-dimensional HNHA spectrum; three-dimensional HNHB.
Sequence-specific assignments were primarily made by comparison to the wild-type assignments (Skelton et al. 1990). The GENXPK program was used to facilitate this process (Gippert 1995). Backbone assignments were confirmed by sequential connectivities in the NOESY spectra, and side-chain assignments were confirmed in the COSY spectrum, following standard methods such as those described for the N56A calbindin D9k mutant (Wimberly et al. 1995). The chemical shift assignments have been deposited under accession code 5207.
Structure determination
Determination of the three-dimensional structure of apo F36G followed previously developed protocols (Mäler et al. 2000). Distance restraints were derived from the intensity of NOEs, assigning each NOE to a distance bin. The bins were calibrated separately for each NOESY spectrum. The φ torsion angle restraints were derived from 3JHN-Hα coupling constants calculated from the ratio of the intensity of the diagonal and crosspeaks in the HNHA spectrum. Additional φ and also ξ restraints were assigned based on a combination of the α proton chemical shift index (CSI) and the presence of medium-range NOEs characteristic of helical conformation. If the HNHA, CSI, and NOE data all indicated the presence of helix, and characteristic (i, i + 4) NOEs were identified, then restraints were set to φ = −40° to −90° and ξ = 0° to −80°. The φ angle for residues in which the 3JHN-Hα was greater than 8 Hz was restrained to −80° to −160°. Side-chain χ1 angles were restrained based on the relative magnitude of 3JNHβ derived from the HNHB spectrum in conjunction with the relative intensities of the NOEs between the amide proton and the two β-protons and between the α-proton and the two β-protons (Barsukov and Lian 1993). Eighteen hydrogen bond restraints for nine hydrogen bonds in the helical regions of the protein were assigned on the basis of slow exchange of the amide proton and NOEs characteristic of α-helix (Skelton et al. 1995).
Stereospecific assignments of 15 β-protons initially were made on the basis of the HNHB data and NOEs. The β-protons of one of proline and the side-chain amide protons of three Asn and Gln residues could be stereospecifically assigned based on NOEs (Kline et al. 1989). Two of the three sets of valine methyl groups were stereospecifically assigned based on the relative intensity of the NOEs between the two methyl groups and the amide proton (Zuiderweg et al. 1985). Additional stereospecific assignments were made using the GLOMSA program (Güntert et al. 1991). This enabled stereospecific assignment of five more β protons, the δ protons of Pro 37, and the methyl groups of Leu 31.
Fifty starting structures were generated from the restraints by distance geometry, using the DIANA program (Güntert et al. 1991). The structures then were refined by simulated annealing, using the AMBER program (Pearlman et al. 1995). Restraint violations in the structures were analyzed and resolved after each round of calculations. Once the structures coalesced into a coherent family, the members of this preliminary family were used to identify further restraints from the list of ambiguous NOEs, filtering on the basis of the distances in the then-current structures (Sastry et al. 1998).
A final ensemble of 24 structures was selected by first ordering the structures on the basis of increasing restraint violation energies. Structures that had a total AMBER energy or a specific term of the force field greater than two standard deviations above the mean were carefully scrutinized for potential exclusion from the final ensemble. The minimum number of structures required to adequately represent the conformational space allowed by the data was 22, as determined using the FINDFAM program (Smith 1999).The number of structures in the final ensemble was selected to be similar to that used for previous calbindin D9k structures to facilitate comparison.
The quality of the final family of structures was assessed with PROCHECK-NMR (Laskowski et al. 1996); 89.4% of the residues fall within the most favored regions of φ/ξ space, and a further 8.7% fall within additionally allowed regions. No residues fall consistently in disallowed regions. The coordinates have been deposited under accession code 1KCY.
Structural analyses
Comparisons of the apo F36G structure to the wild-type apo calbindin D9k and apo CaM-N structures were made using the methods described previously (Nelson and Chazin 1998b). This included analysis of interhelical angles calculated with the INTERHLX program (kindly provided by K. Yap and M. Ikura, University of Toronto), inter-residue contacts calculated with CHARMM (Brooks et al. 1983), and distance difference matrices calculated with DISCOM (Gippert 1995). These comparisons guided extensive graphics-based comparisons using InsightII (Molecular Simulations Inc.). Structures were always superimposed on helices I and IV for graphics-based comparisons, because the bias-independent methods showed that this interhelical interface is similar in all of the structures.
Acknowledgments
We thank Drs. Karin Julenius, Bryan Finn, and Sara Linse for assistance with stability and Ca2+ affinity measurements, Drs. John Chung, Lena Mäler, and Randal R. Ketchem for assistance with NMR experiments and structure calculations, MSI for providing software, Professor R.J.P. Williams and members of the Chazin lab for helpful discussions, the Scripps Metalloprotein Structure and Design Group for their interest and support in pursuing this research, and Drs. Anders Malmendal and Laura Mizoue for critical reading of the article. This research was supported by an operating grant from the National Institutes of Health (GM 40120 to W.J.C.) and a graduate fellowship from the National Science Foundation (to M.R.N.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Note added in proof
A highly complementary study of the Leu39 → Phe mutation in the N-terminal domain of calmodulin has recently been published by Ababou et al. 2001 (Biochemistry 40: 12719–12726).
Abbreviations
CaBP, Ca2+-binding protein
CaM, calmodulin
calbindin, calbindin D9k
NMR, nuclear magnetic resonance
CSI, chemical shift index
NOE, nuclear Overhauser effect
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.33302
References
- Ababou, A. and Desjarlais, J.R. 2001. Solvation energetics and conformational change in EF-hand proteins. Protein Sci. 10 301–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akke, M., Forsén, S., and Chazin, W.J. 1995. Solution structure of (Cd2+)1-calbindin D9k reveals details of the stepwise structural changes along the apo → (Ca2+)II1 → (Ca2+)I,II2 binding pathway. J. Mol. Biol. 252 102–121. [DOI] [PubMed] [Google Scholar]
- Barsukov, I.L. and Lian, L.-Y. 1993. Structure determination from NMR data I. Analysis of NMR data. In NMR of macromolecules: A practical approach (ed. G.C.K. Roberts), pp. 315–357. Oxford University Press, Oxford, UK.
- Brooks, B.R., Bruccoleri, R.E., Olafson, B.D., States, D.J., Swaminathan, S., and Karplus, M. 1983. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 4 187–217. [Google Scholar]
- Cavanagh, J., Fairbrother, W.J., Palmer, A.G.I., and Skelton, N.J. 1996. Protein NMR spectroscopy: Principles and practice. Academic Press, San Diego.
- Dalas, S., Balasubramanian, S., and Regan, L. 1997. Protein alchemy: Changing β-sheet to α-helix. Nat. Struct. Biol. 4 548–552. [DOI] [PubMed] [Google Scholar]
- Drohat, A.C., Baldisseri, D.M., Rustandi, R.R., and Weber, D.J. 1998. Solution structure of calcium-bound rat S100B(ββ) as determined by nuclear magnetic resonance spectroscopy. Biochemistry 37 2729–2740. [DOI] [PubMed] [Google Scholar]
- Finn, B.E., Evenäs, J., Drakenberg, T., Waltho, J.P., Thulin, E., and Forsén, S. 1995. Calcium-induced structural changes and domain autonomy in calmodulin. Nat. Struct. Biol. 2 777–783. [DOI] [PubMed] [Google Scholar]
- Gippert, G. 1995. "New computational methods for 3D NMR data analysis and protein structure determination in high-dimensional internal coordinate space." Ph.D. thesis, The Scripps Research Institute, La Jolla, CA.
- Güntert, P., Braun, W., and Wüthrich, K. 1991. Efficient computation of three-dimensional protein structures in solution from nuclear magnetic resonance data using the program DIANA and the supporting programs CALIBA, HABAS and GLOMSA. J. Mol. Biol. 217 517–530. [DOI] [PubMed] [Google Scholar]
- Henikoff, S., Greene, E.A., Pietrokovski, S., Bork, P., Attwood, T.K., and Hood, L. 1997. Gene families: The taxonomy of protein paralogs and chimeras. Science 278 609–614. [DOI] [PubMed] [Google Scholar]
- Johansson, C., Brodin, P., Grundstrom, T., Forsén, S., and Drakenberg, T. 1990. Mutation of the pseudo-EF hand of calbindin D9k into a normal EF hand. Eur. J. Biochem. 187 455–460. [DOI] [PubMed] [Google Scholar]
- Julenius, K., Thulin, E., Linse, S., and Finn, B.E. 1998. Hydrophobic core substitutions in calbindin D9k: Effects on stability and structure. Biochemistry 37 8915–8925. [DOI] [PubMed] [Google Scholar]
- Kline, A.D., Braun, W., and Wüthrich, K. 1989. Determination of the complete three-dimensional structure of the α-amylase inhibitor tendemistat in aqueous solution by nuclear magnetic resonance and distance geometry. J. Mol. Biol. 104 675–724. [DOI] [PubMed] [Google Scholar]
- Kördel, J., Skelton, N., Akke, M., and Chazin, W.J. 1993. High resolution solution structure of calcium-loaded calbindin D9k. J. Mol. Biol. 231 711–734. [DOI] [PubMed] [Google Scholar]
- Kragelund, B.B., Jonsson, M., Bifulco, G., Chazin, W.J, Nilsson, H., Finn, B.E, and Linse, S. 1998. Hydrophobic core substitution in calbindin D9k: Effects on Ca2+ binding and dissociation. Biochemistry 37 8926–8937. [DOI] [PubMed] [Google Scholar]
- Kuboniwa, H., Tjandra, N., Grzesiek, S., Ren, H., Klee, C.B., and Bax, A. 1995. Solution structure of calcium-free calmodulin. Nat. Struct. Biol. 2 768–776. [DOI] [PubMed] [Google Scholar]
- Laskowski, R.A., Rullmannn, J.A., MacArthur, M.W., Kaptein, R., and Thornton, J.M. 1996. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J Biomol. NMR 8 477–486. [DOI] [PubMed] [Google Scholar]
- Linse, S., Brodin, P., Drakenberg, T., Thulin, E., Sellers, P., Elmden, K., Grundstrom, T., and Forsén, S. 1987. Structure-function relationships in EF-hand Ca2+-binding proteins. Protein engineering and biophysical studies of calbindin D9k. Biochemistry 26 6723–6735. [DOI] [PubMed] [Google Scholar]
- Lockless, S. and Ranganathan, R. 1999. Evolutionarily conserved pathways of energetic connectivity in protein families. Science 286 295–299. [DOI] [PubMed] [Google Scholar]
- Mäler, L., Blankenship, J., Rance, M., and Chazin, W.J. 2000. Site–site communication in the EF-hand Ca2+-binding protein calbindin D9k. Nat. Struct. Biol. 7 245–250. [DOI] [PubMed] [Google Scholar]
- Matsumura, H., Shiba, T., Inoue, T., Harada, S., and Kai, Y. 1998. A novel mode of target recognition suggested by the 2.0 A structure of holo S100B from bovine brain. Structure 6 233–241. [DOI] [PubMed] [Google Scholar]
- Nelson, M.R. 1999. "A rational approach to understanding the relationship of sequence, structure and function in the EF-hand calcium-binding proteins." Ph.D. thesis, The Scripps Research Institute, La Jolla, CA.
- Nelson, M.R. and Chazin, W.J. 1998a. Structures of EF-hand proteins: Diversity in the organization, packing and response to Ca2+ binding. Biometals 11 297–318. [DOI] [PubMed] [Google Scholar]
- ———. 1998b. An interaction-based analysis of calcium-induced conformational changes in Ca2+ sensor proteins. Protein Sci. 7 270–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearlman, D.A., Case, D.A., Caldwell, J.C., Seibel, G.L., Singh, U.C., Weiner, P., and Kollman, P.A. 1995. AMBER. University of California, San Francisco.
- Richardson, J.S. and Richardson, D.C. 1989. Principles and patterns of protein conformation. In Prediction of protein structure and principles of protein conformation (ed. G. Fassman), pp. 1–98. Plenum Press, New York.
- Sastry, M., Ketchem, R.R., Crescenzi, O., Weber, C., Lubienski, M.J., Hidaka, H., and Chazin, W.J. 1998. The three-dimensional structure of Ca2+-bound calcyclin: Implications for Ca2+-signal transduction by S100 proteins. Structure 6 223–231. [DOI] [PubMed] [Google Scholar]
- Sinha, N. and Nussinov, R. 2001. Point mutations and sequence variability in proteins: Redistribution of preexisting populations. Proc. Natl. Acad. Sci. 98 3139–3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skelton, N.J., Forsén, S., and Chazin, W.J. 1990. 1H NMR resonance assignments, secondary structure, and global fold of apo bovine calbindin. Biochemistry 29 5752–5761. [DOI] [PubMed] [Google Scholar]
- Skelton, N.J., Kördel, J., Akke, M., Forsén, S., and Chazin, W.J. 1994. Signal transduction versus buffering activity in Ca2+-binding proteins. Nat. Struct. Biol. 1 239–245. [DOI] [PubMed] [Google Scholar]
- Skelton, N.J., Kördel, J., and Chazin, W.J. 1995. Determination of the solution structure of apo calbindin D9k by NMR spectroscopy. J. Mol. Biol. 249 441–462. [DOI] [PubMed] [Google Scholar]
- Smith, J. 1999. "NMR and computational methods applied to the 3-dimensional structure determination of DNA and ligand-DNA complexes in solution." Ph.D. thesis, The Scripps Research Institute, La Jolla, CA.
- Smith, S.P. and Shaw, G.S. 1998. A novel calcium-sensitive switch revealed by the structure of human S100B in the calcium-bound form. Structure 6 211–222. [DOI] [PubMed] [Google Scholar]
- Thompson, J.D., Higgins, D.G., and Gibson, T.J. 1994. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimberly, B., Thulin, E., and Chazin, W.J. 1995. Characterization of the N-terminal half-saturated state of calbindin D9k: NMR studies of the N56A mutant. Protein Sci. 4 1045–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimley, W., Creamer, T., and White, S. 1996. Solvation energies of amino acid sidechains and backbone in a family of host-guest pentapeptides. Biochemistry 35 5109–5124. [DOI] [PubMed] [Google Scholar]
- Zhang, M., Tanaka, T., and Ikura, M. 1995. Calcium-induced conformational transition revealed by the solution structure of apo calmodulin. Nat. Struct. Biol. 2 758–767. [DOI] [PubMed] [Google Scholar]
- Zuiderweg, E.R.P., Boelens, R., and Kaptein, R. 1985. Stereospecific assignments of 1H-NMR methyl lines and conformation of valyl residues in the lac repressor headpiece. Biopolymers 24 601–611. [Google Scholar]