

Abstract
Spontaneous formation of isoaspartyl residues (isoAsp) disrupts the structure and function of many normal proteins. Protein isoaspartyl methyltransferase (PIMT) reverts many isoAsp residues to aspartate as a protein repair process. We have determined the crystal structure of human protein isoaspartyl methyltransferase (HPIMT) complexed with adenosyl homocysteine (AdoHcy) to 1.6-Å resolution. The core structure has a nucleotide binding domain motif, which is structurally homologous with the N-terminal domain of the bacterial Thermotoga maritima PIMT. Highly conserved residues in PIMTs among different phyla are placed at positions critical to AdoHcy binding and orienting the isoAsp residue substrate for methylation. The AdoHcy is completely enclosed within the HPIMT and a conformational change must occur to allow exchange with adenosyl methionine (AdoMet). An ordered sequential enzyme mechanism is supported because C-terminal residues involved with AdoHcy binding also form the isoAsp peptide binding site, and a change of conformation to allow AdoHcy to escape would preclude peptide binding. Modeling experiments indicated isoAsp groups observed in some known protein crystal structures could bind to the HPIMT active site.
Keywords: Isoaspartyl, methyltransferase, deamidation, protein repair
Methyltransferases catalyze the transfer of methyl groups from adenosyl methionine (AdoMet) to a variety of specific substrate molecules including DNA, RNA, phospholipids, porphyrins, neurotransmitters, amino acids, peptides, and proteins. They produce methylated substrates and adenosyl homocysteine (AdoHcy). Even though they provide a diversity of cellular and physiological functions, recent structural studies of different methyltransferases reveal that they share common structural features and molecular mechanisms, e.g., chemotaxis receptor methyltransferase or CheRMT (Djordjevic and Stock 1997), Thermotoga maritima protein L-isoaspartate O-methyltransferase or TMPIMT (Skinner et al. 2000), adenine-N-6-DNA-methyltransferase (Labahn et al. 1994), glycine N-methyltransferase (Huang et al. 2000), HhaI DNA cytosine-C5-methyltransferase (Cheng et al. 1993), Rrna adenine N-6-methyltransferase (Schluckebier et al. 1999), catechol-O-methyltransferase or COMT (Vidgren et al. 1994), and HhaI cytosine-specific methyltransferase (O'Gara et al. 1999).
Protein methyltransferases are classified into two broad groups, i.e., those that methylate specific carboxyl groups and those that methylate specific nitrogen or sulfur atoms of proteins (Clarke 1992). Of the carboxyl protein methyltransferases, one of the most widely distributed in nature methylates isoAsp residues commonly found in short peptides and intact proteins.
Isoaspartyl residues in proteins arise spontaneously from deamidation of asparagines and isomerization of aspartates via the formation of transient succinimidyl intermediates as shown in Figure 1 ▶ (Geiger and Clark 1987; McFadden and Clarke 1987; Skinner et al. 2000). The succinimidyl intermediates can undergo racemization and hydrolysis to form D or L isomers of aspartate and isoaspartate in the peptide chain. An isoaspartyl residue (isoAsp) forms the main-chain backbone using the former side-chain carboxyl group instead of the α-carboxyl group, which then becomes the new side chain. IsoAsp formation is 30–50 times faster when the next residue is glycine, because the absence of a side chain favors succinimidyl group formation. Succinimide formation is also restricted by certain polypeptide backbone conformations. Therefore, not all asparagines and aspartates in proteins have the same propensities to convert to isoAsp.
Fig. 1.

Protein deamidation, isoAsp formation, and the repair cycle catalyzed by PIMT. 1. Protein deamidation/dehydration at the susceptible residue in the native protein. 2. Hydrolysis of the intermediate succinimide to form L-aspartate. 3. Hydrolysis of the intermediate succinimide to form L-isoaspartate. 4. O-methylation by PIMT. 5. Release of methanol forms the L-succinimide intermediate. (Reproduced from Skinner et al. (2000), with permission of Elsevier Science.)
The most probable physiological role of protein isoaspartyl methyltransferase (PIMT) is to provide a mechanism for converting isoAsp residues to normal L-aspartate residues (Clarke 1993). This conversion represents a complete restoration of modified aspartate residues and a partial restoration to a more native conformation of former asparagine residues. PIMT initiates this conversion by methylating the α-carboxyl group of the isoAsp residue to form an L-isoAsp-α-methyl ester (Johnson et al. 1987b). The L-Asp-succinimidyl group can form quickly again with the release of methanol. The newly formed L-Asp succinimidyl group can then either revert back to L-Asp or return to L-isoAsp. The constant presence of PIMT along with a high ratio of [AdoMet]/[AdoHcy] insures that the equilibrium favors the formation of L-Asp.
IsoAsp formation can cause disruption of the local and perhaps long-range three-dimensional structure of many proteins. When L-isoAsp formation occurs, the peptide backbone gains an extra methylene group and the new side-chain carboxyl group is one carbon atom shorter than before the isomerization. If deamidation of an Asn occurred, a negatively charged isoAsp would also replace the neutral Asn. These changes would likely disrupt molecular structural elements, intermolecular associations, hydrogen bonding networks, and enzyme active sites. Several crystal structures have been shown to contain one or more isoAsp residues in the folded proteins: ribonuclease A (Esposito et al. 2000), hen egg white lysozyme (Noguchi et al. 1998), ribonuclease U2 (Noguchi et al. 1995), porcine trypsin (Rester et al. 1999), and enolpyruvate transferase (Schonbrunn et al. 2000).
A number of studies have shown the critical protective role of PIMT in physiological systems. It was shown that calmodulin could be aged in vitro until it lost the ability to activate protein kinase (Johnson et al. 1987a). The loss of activity was found to be because of the conversion of asparagine residues to isoAsp residues by deamidation. Incubation of the modified calmodulin with bovine brain PIMT and an excess of AdoMet partially restored the activation ability of calmodulin.
PIMT-deficient mice have been produced by targeted disruption of the PIMT gene (Kim et al. 1997; Yamamoto et al. 1998). The PIMT-deficient mice died within 4 to 12 wk from progressive epileptic seizures. It was found that the brains of PIMT-deficient mice accumulated nine times the amount of isoAsp residues over that of brains from mice not deficient of PIMT. %PIMT has been shown to protect primary mouse cortical neurons from Bax-induced apoptosis (Huebscher et al. 1999). It was postulated that PIMT acts to maintain cytoskeletal organization in interphase cells, with tubulin being a specific target of maintenance by PIMT. Furthermore, PIMT is found to be associated with neurofibrillary tangles (NFTs) in Alzheimer's disease (Shimizu et al. 2000), suggesting that the formation of isoAsp in NFTs may play a pathological role in the neurodegeneration of Alzheimer's disease.
Mice immunized with the isoAsp form of a murine cytochrome C peptide have shown B- and T-cell immune responses, whereas the non-isoAsp peptide was found to be nonimmunogenic (Mamula et al. 1999). Similarly, mice immunized with an isoAsp peptide autoantigen of human systemic lupus erythematosus gave strong B- and T-cell responses but tolerated the non-isoAsp self-antigen. Thus, isoAsp protein accumulations may be involved with the development of autoimmune diseases for which PIMTs may be protective.
The ability to repair damaged proteins in cells would be more energy efficient than to replace them by protein synthesis and recycling the replaced protein. Additionally, it would be important to repair sensitive proteins in cells that do not replenish them, e.g., erythrocytes or plant seeds. The same would be true for proteins in tissues requiring longevity, e.g., dentine, eye lens proteins, brain white matter, and other nerve cell proteins, all of which have been shown to contain some level of isoAsp residues (Clarke 1987).
The first PIMT structure to be determined (TMPIMT) was from the thermophilic bacterium, T. maritima (Skinner et al. 2000). Here we report the structure of human PIMT (HPIMT) to 1.6-Å resolution with AdoHcy bound. We have also modeled a peptide and fully folded proteins containing the isoAsp residue at the active site. These models represent how naturally formed isoAsp residues belonging to large protein structures could bind to HPIMT for methylation.
Results
There are two known isozymes of HPIMT attributable to differential mRNA splicing (Takeda et al. 1995). Isozyme 1 has one less amino acid at the C-terminal and ends with WK, whereas isozyme 2 ends with DEL, which is a known endoplasmic reticulum retention factor. The sequence for the present structure corresponds to isozyme 1. There is also a site of polymorphism at residue 119, which is 77% Ile and 23% Val (Ingrosso et al. 1989). DNA sequencing, as well as the high-resolution electron density map, indicated that the Val polymorph was present in this crystal structure. Enzyme assays using this highly purified recombinant HPIMT showed it was enzymatically active.
Vapor diffusion crystallization experiments with macroseeding produced diffraction quality single crystals as thin plates with dimensions of approximately 0.3 × 0.2 × 0.05 mm. Native HPIMT crystals diffracted to 1.5 Å. Unit cell parameters were a = 48.1 Å, b = 53.9 Å, c = 48.8 Å, and β = 116.15° for space group P21. The Matthews coefficient, assuming one molecule per asymmetric unit, was 2.4 Å3/dalton and the solvent content for these parameters was 45%. Summaries of the data collection and refinement statistics are given in Table 1.
Table 1.
Summary of data collection and refinement statistics for the crystal structure of HPIMT
| All data | Last shell | |
| Data collection: | ||
| Resolution range (Å) | 50–1.6 | 1.66–1.60 |
| Rsym (l) | 0.039 | 0.124 |
| Completeness (%) | 95.7 | 87.1 |
| Redundancy | 1.7 | 1.4 |
| No. of unique reflections | 28,488 | 2587 |
| <l/σ(l)> | 29.6 | |
| Refinement statistics: | ||
| Resolution range (Å) | 15–1.6 | |
| No. of refls. used in refinement | 27,830 | |
| No. of refls. used in working set | 26,446 | |
| No. of refls. in free R set | 1384 | |
| R factor (working set) | 0.180 | |
| R factor (test set) | 0.211 | |
| Rms deviations from ideal: | ||
| Bond lengths (Å) | 0.004 | |
| Bond angles (degrees) | 1.30 | |
| Dihedral angles (degrees) | 23.70 | |
| Improper angles (degrees) | 0.83 | |
| No. of water molecules | 279 | |
The overall architecture of the HPIMT structure is shown in Figure 2 ▶ as a Ribbons diagram (Carson 1997). The HPIMT structure is composed of an N-terminal three-helix bundle followed by an α/β open parallel β-sheet nucleotide-binding domain. Only the first and the last residues were not visible in the electron density map, presumably because of vibrational disorder. Residues near both termini had large temperature factors indicating high mobility. However, low-temperature factors throughout the remainder of the molecule suggest the surface shape is largely fixed. HPIMT has one cis proline, Pro 151, which is conserved in the TMPIMT structure as cis Pro 144.
Fig. 2.
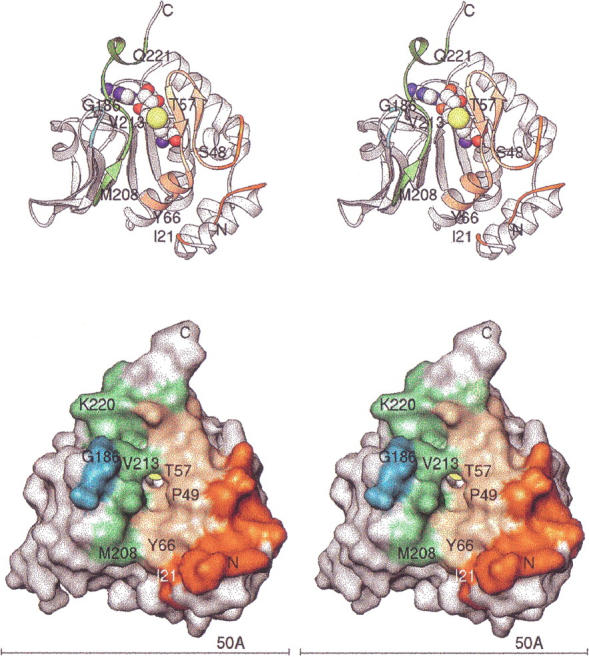
Architecture of HPIMT. (Top) Stereo ribbon diagram with key residues labeled. Residues forming the surface cleft for putative substrate interaction are colored. Yellow-orange and dark orange designate N-terminal residues; cyan and green designate residues of the C-terminal region. The AdoHcy is colored by atom type with sulfur yellow. (Bottom) Stereo view of the molecular surface (Connolly 1993) in the same orientation. The surface residues are colored as in the ribbon drawing. All molecular figures were prepared with Ribbons (Carson 1997; http://sgce.cbse.uab.edu/ribbons).
Figure 3 ▶ shows a stereo view of the AdoHcy binding site. Although sequence conservation for residues involved with AdoHcy binding is not strong (Figure 4 ▶), there are strong structural similarities among the known methyltransferase structures for this binding site. The glycine rich region centered on Gly 85 is highly conserved among PIMTs and also other types of methyltransferases. Gly 85 and Gly 87 are located beneath and make hydrophobic contacts with AdoHcy. The conserved Asp 109 Oδ1 and Oδ2 atoms make hydrogen bonds to the ribose ring oxygens O2 and O3. The conserved Asp 141 Oδ1 atom hydrogen bonds to the adenine N6. Conserved Gly 142 N, Thr 216 O, and N hydrogen bond to adenine atoms N1, N6, and N7, respectively. The adenine also has a parallel plane hydrophobic interaction with the His 110 side chain. The homocysteinyl carboxyl oxygens hydrogen bond to Ser 59 N, His 64 Nɛ2, and Ser 88 Oγ and N atoms. The homocysteinyl group also makes hydrophobic contacts with Ile 58, Ser 86, Gly 87, and Val 213.
Fig. 3.
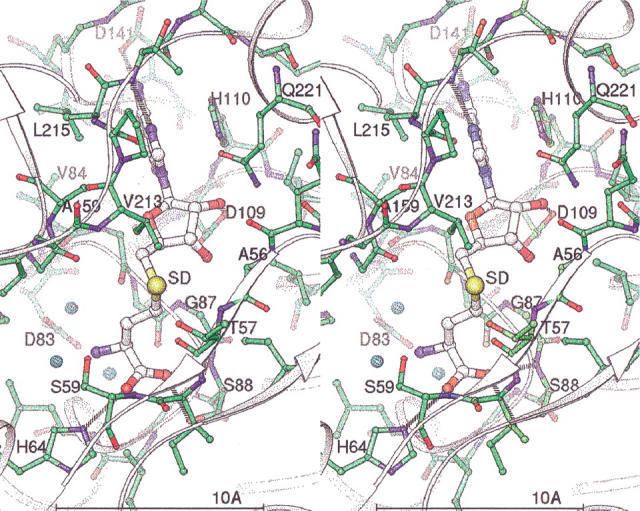
Stereo view of the AdoHcy binding site. Key residues and atoms are labeled. Atoms are colored by type, with green carbon/bonds for the protein and white carbon/larger bonds for AdoHcy. Three tightly bound waters are shown as small cyan spheres. Hydrogen bonds between the protein and AdoHcy are shown as dashed lines.
Fig. 4.

Multiple sequence alignment of PIMTs. Eukaryotic and noneukaryotic sequences were aligned using the program PILEUP from the GCG Wisconsin Package. The ALSCRIPT program (Barton 1993) was used to prepare the figure. Sequence numbering and the secondary structure designations correspond to the HPIMT sequence (pimt-human). The secondary structure labels correspond to those used for the TMPIMT structure (Skinner et al. 2000). Conservation analysis was based only on eukaryotic residues. Residues completely conserved among these eukaryote sequences have a black background. Residues conserved in all but one of these eukaryotic sequences have a gray background. Noneukaryotic residues that share the eukaryotic residue conservation are indicated the same way. The sequences used were pimt-wheat: Triticum aestivum, pimt-arath: Arabidopsis thaliana, pimt-caeel: Caenorhabditis elegans, pimt-drome: Drosophilia melanogaster, pimt-rat: Rattus norvegicus, pimt-mouse: Mus musculus, pimt-bovin: Bos taurus, pimt-human: human, pimt-metja: Methanococcus jannaschii, pimt-metth: Methanobacterium thermoautotrophicum, pimt-pyrho: Pyrococcus horikoshii, pimt-aerpe: Aeropyrum pernix, pimt-helpj: Helicobacter pylori j99, pimt-helpy: Helicobacter pylori, pimt-pseae: Pseudomonas aeruginosa, pimt-ecoli: Escherichia coli, pimt-rhime: Rhizobium meliloti, and pimt-thema: Thermotoga maritima.
Discussion
Figure 5a ▶ is a Ribbons diagram showing the superposition of HPIMT onto TMPIMT. The HPIMT structure lacks the large C-terminal domain found in the TMPIMT structure. However, between HPIMT and TMPIMT there is structural conservation of the core sheet topology, including the last antiparallel pair of β strands that differ from all other known methyltransferase structures (Skinner et al. 2000). The structures diverge significantly at HPIMT residue 217. In the TMPIMT structure, a large C-terminal domain is positioned away from the active site. In the human enzyme, a short C-terminal region winds back to contribute to AdoMet/AdoHcy nucleotide binding. The C-terminal main chain passes over the nucleotide binding site, and Gln 221 Nɛ2 hydrogen bonds to AdoHcy O2. C-terminal residues beyond TMPIMT Gly 210 do not pass back over the nucleotide-binding site and do not directly interact with AdoHcy. Figure 5b ▶ compares the proposed binding site surfaces of HPIMT and TMPIMT.
Fig. 5.
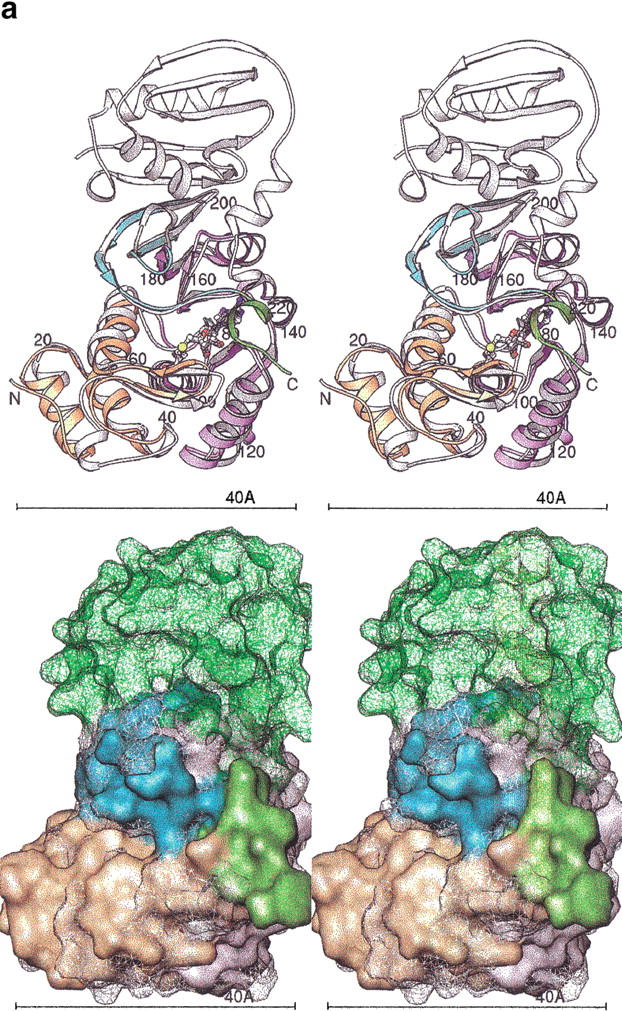
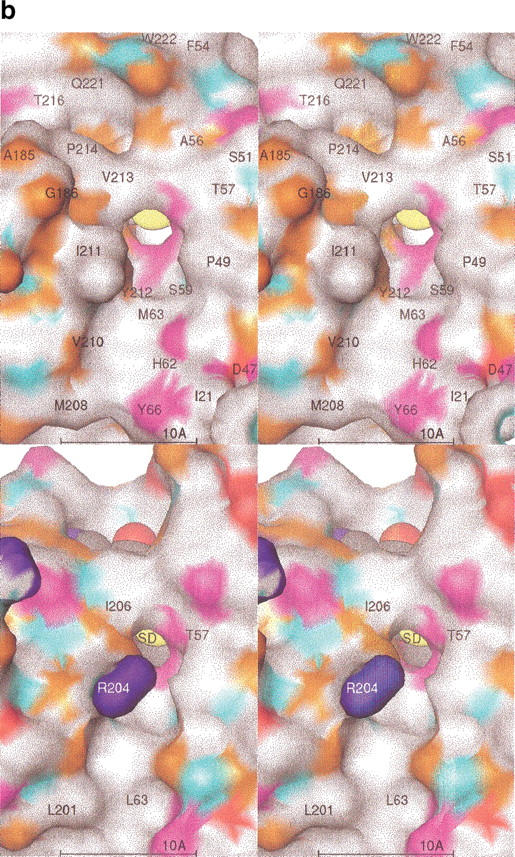
(a) Superposition of HPIMT and TMPIMT structures. (Top) Stereo ribbons diagrams. The human structure is colored by its sequence domains: yellow-orange is the N-terminal, lavender is the conserved MT core, cyan are the sheets with the unique topology in PIMTs, and green is the divergent C-terminal region. The termini and every 20 residues are labeled. The AdoHcy molecule is colored by atom type. The superposed TMPIMT backbone is white and its AdoHcy gray. (Bottom) Stereo molecular surfaces. The human structure is solid and colored as above. The TMPIMT surface is shown as a mesh, colored white for the common catalytic domain and green for its divergent and unique C-terminal domain. (b) PIMT comparison of substrate-binding surface. The molecular surfaces are colored by chemical property: white, hydrophobic; red, negative; blue, positive; orange, H-bond acceptor; cyan, H-bond donor; and magenta, polar. Key residues are labeled. The AdoHcy molecule is shown as spheres colored by atom type. (Top) HPIMT. (Bottom) TMPIMT.
The N-terminal domain up to residue 74 is a small helical bundle with a small β-sheet-hairpin loop that forms one side of the proposed active site. COMT (Vidgren et al. 1994) and CheR (Djordjevic and Stock 1997) methyltransferases have structurally similar domains, but the domains have different orientations with respect to the central sheet domain. A cleft is formed between the N-terminal domain and the C-terminal end of the central sheet domain. This cleft is the putative binding site of isoAsp substrates based on an analogy with other MT structures. At the bottom of the cleft is an opening in which it is presumed the transferable methyl group would sit if AdoMet were bound, based on an analogy with the COMT structure.
The protein substrate recognition surface has been proposed as formed by the sides of the cleft (Skinner et al. 2000). The highly conserved residues lining the cleft, i.e., 57–59 and 212–216, then would form the majority of the protein substrate recognition residues. These residues participate in an extensive hydrogen-bonding network across the cleft involving solvent waters. The waters are held with classical tetrahedral hydrogen bonds having distances between 2.7 and 3.0 Å. A search was made for shorter bonds with square bipyramidal bonding geometry to locate a suspected Mg+2 binding site. None of the isolated electron density met these criteria and none were designated as Mg+2 sites. A Mg+2 binding site had been suspected by analogy with the COMT structure also and the necessity of having Mg+2 ions present for crystallization.
The COMT structure was determined with AdoMet bound and with an inhibitor bound at an analogous active site (Vidgren et al. 1994). In the COMT structure, the transferable methyl group does protrude through a similar opening toward the inhibitor, which is bound in the cleft. The methyl group is located at a tetrahedral position on the δ-sulfur.
Based on similarities to the DNA amino-methyltransferases (Malone et al. 1995), SN2 is the proposed reaction mechanism for transfer of the methyl group from AdoMet to the isoAsp carboxyl group (Ho et al. 1991). The reaction has been proposed (Skinner et al. 2000; Johnson and Aswad 1993) to follow an ordered sequence of (1) nucleophilic attack by the carboxyl oxygen of the isoAsp group at the AdoMet methyl group, (2) release of the methylated isoAsp residue, (3) conformational change of the HPIMT C-terminal domain to allow release of the AdoHcy product, (4) binding of new AdoMet substrate, (5) reestablishment of the original conformation of the C-terminus, and (6) binding of new isoAsp substrate. The ordered sequential mechanism is probable because the C-terminal residues provide structure to the active site cleft for isoAsp and adjacent residues to bind and also covers the AdoHcy. A conformational change must occur for these C-terminal residues to allow the AdoHcy to escape and AdoMet to enter. The conformational change would disrupt isoAsp binding.
A number of mechanisms are plausible that could open a path for AdoHcy and AdoMet exchange. One is that C-terminal residues covering the cofactor site move or become disordered on methyl transfer from AdoMet. Another mechanism that has been suggested for TMPIMT (Skinner et al. 2000) involves movements of residues in the βa-βb β-sheet, which also cover the cofactor binding site. Interestingly, there are four residues in the refined HPIMT structure that have unusually small Cα main-chain bond angles. These are Ile 58 (99.7°), Pro 174 (103.4°), Val 213 (103.6°), and Leu 215 (97.8°). Pro 174 aligns with Glu 164 in TMPIMT, and both are located at sharp turns, which could act as hinges for movement of a C-terminal structural unit covering the cofactor site. Ile 58 is at the end of the βa-βb sheet that covers the cofactor site in both structures. Val 213 and Leu 215 are directly over the cofactor site as well. These residues may have small bond angles as a result of structural stresses brought about by cofactor binding. One or more of these stresses may be relieved by conformational changes that then allow exchange of AdoHcy and AdoMet.
Any proposed mechanism for the sequence of events following isoAsp methylation requires an initiating signal. One possible signal is the disappearance of the formal positive charge on the δ-sulfur of the AdoMet when the methyl group leaves. Simultaneously, the negative charge on the isoAsp carboxyl group is neutralized. The δ-sulfur position is surrounded by a number of carbonyl oxygens, which could serve to stabilize the formal positive charge of AdoMet on the δ-sulfur atom. Gly 85 contributes one of those carbonyl oxygens and is highly conserved across all families of PIMT. Gly 158 is another contributor and also is highly conserved among the eukaryotic PIMTs. Changes of the charge configuration (both on the AdoMet and the isoAsp side chain) may be the signal that causes movements of the C-terminal residues or βa-βb sheet residues to effect cofactor exchange and perhaps peptide exchange. Although this structure is stable with AdoHcy present in the crystal, either the ternary complex or under other physiological conditions it may not be.
An alternative signal may be the Val 213 side chain positioned to interact with the transferable methyl group. As the methyl group moves from the AdoMet to the isoAsp peptide, Val 213 could loose an anchoring hydrophobic interaction or rotate away, thereby allowing a change of conformation of the downstream C-terminal residues holding the AdoHcy. Val 213 is highly conserved among the PIMTs.
Another aid to the enzymatic reaction would be a mechanism for orienting and positioning the isoAsp carboxyl group to receive the methyl group. Ideally the nucleophilic oxygen should approach the methyl group along the axis of the S-to-methyl bond to form a methyl ester bond of approximately 110° on the oxygen and have a ∼1.43 Å CH3-O bond distance. Thr57 is a highly conserved residue in a position to hydrogen bond with the incoming isoAsp carboxyl oxygens and perhaps play a role in orienting the attacking carbonyl oxygen.
A comparison of the conformations of the AdoMet from the COMT structure and AdoHcy from the HPIMT structure indicated small conformational changes between the two. Therefore, the demethylation of AdoMet may not create any significant conformational changes of the AdoHcy, which could affect the HPIMT contacts or conformation.
When Human PIMT binds peptide substrates, the residue before and the two residues after the isoAsp, i.e., R−1, R+1, R+2, appear to have the most affect on binding affinity, whereas residues R−2 and R+3 have a weaker effect, according to Km measurements (Lowenson and Clarke 1991). There also appears to be preference for bulky hydrophobic groups preceding the isoAsp (R−1) and for a neutral or positive group following the isoAsp (R+1, R+2, R+3). A putative site for the R−1 site is a depression 10–15 Å away from the δ-sulfur site. Pro 61, His 62, Met 63, and Tyr 66 surround this area and make nonbonded contacts with Met 208 and Val 210. These residues are well conserved, especially among the eukaryotes (see Fig. 4 ▶). The preference for non-negative residues at R+1, R+2, and R+3 is not as clearly explained by the structure. There is no clear abundance of negative residues in the probable region where these residues would bind. The preference may be because of a disruption of AdoMet binding and/or conformation when negatively charged side chains are present in the shallow groove of this site. A clearer explanation of this phenomenon will require crystal structures of complexes of PIMTs with isoAsp peptides.
A number of protein structures have recently been reported in the Protein Data Bank (PDB) that contain isoAsp residues, i.e., ribonuclease A 1DY5 (Esposito et al. 2000), hen egg white lysozyme 1AT6 (Noguchi et al. 1998), porcine trypsin 1C9P (Rester et al. 1999), enolpyruvate transferase 1EYN (Schonbrunn et al. 2000), and ribonuclease U2 1RTU (Noguchi et al. 1995). The highest resolution structure, the ribonuclease A 1DY5 provided a starting point for full protein modeling studies. IsoAsp 67 and the three residues on each side were extracted from a loop in the 1DY5 coordinate file. This heptapeptide was visually docked via a rigid body transformation into the HPIMT active site, placing the isoAsp side chain in the channel leading to the AdoHcy sulfur atom. Complementary hydrogen bonds with the protein established the preferred direction of the bound peptide. The residue preceding the isoAsp residue, Lys 66, was converted to Trp, with a favorable rotamer chosen to fill the hydrophobic depression below the active site. There was no obvious way to model the polypeptide conformation after the isoAsp. It was left as observed in the 1DY5 structure. This peptide complex was energy-minimized with CNS (Brunger et al. 1998), tightly restraining the protein/AdoHcy coordinates. Figure 6 ▶ shows this complex model along with the AdoMet substituted for the AdoHcy and with the methyl group positioned for transfer.
Fig. 6.
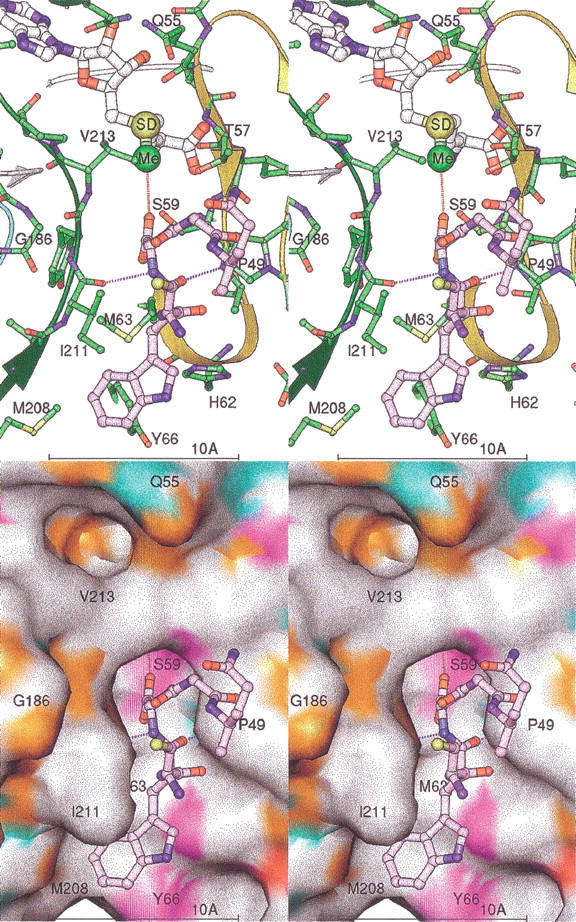
Stereo views of the isoAsp peptide-binding model. The isoAsp-containing peptide is shown in lavender. Only five residues are shown for clarity. The protein ribbon is colored as in Fig. 2 ▶. Key residues of the protein are labeled. Dashed lines mark the hydrogen bonding of the protein to the peptide (lengths ∼2.9 Å). The AdoMet model is shown with the SD and activated methyl group (Me) as larger spheres. A dashed line marks the path of methyl transfer, with a distance of ∼3.1 Å. The surface is colored by chemical property as in Fig. 5b ▶.
To model the binding of whole proteins to HPIMT, the isoAsp-containing proteins from the PDB were juxtaposed to our model complex. Most of the isoAsp residues in these structures are found in loops on the surface of the proteins. The N, CA, C, CB, and CG atoms of each isoAsp were used to least-square fit against the corresponding atoms in the model described above. This resulted in considerable steric clashes with the HPIMT in most cases. Each protein/HPIMT complex model was adjusted visually in a crude docking experiment. Two points in the backbone were chosen, one or two amino acids before and after each isoAsp. Main-chain bonds were broken at these points, leaving the isoAsp loop stationary. The rest of the protein was then transformed by a single rotation to visually minimize any clash with HPIMT. Figure 7 ▶ illustrates the model docking results. The AdoMet of the HPIMT structure is shown for reference.
Fig. 7.
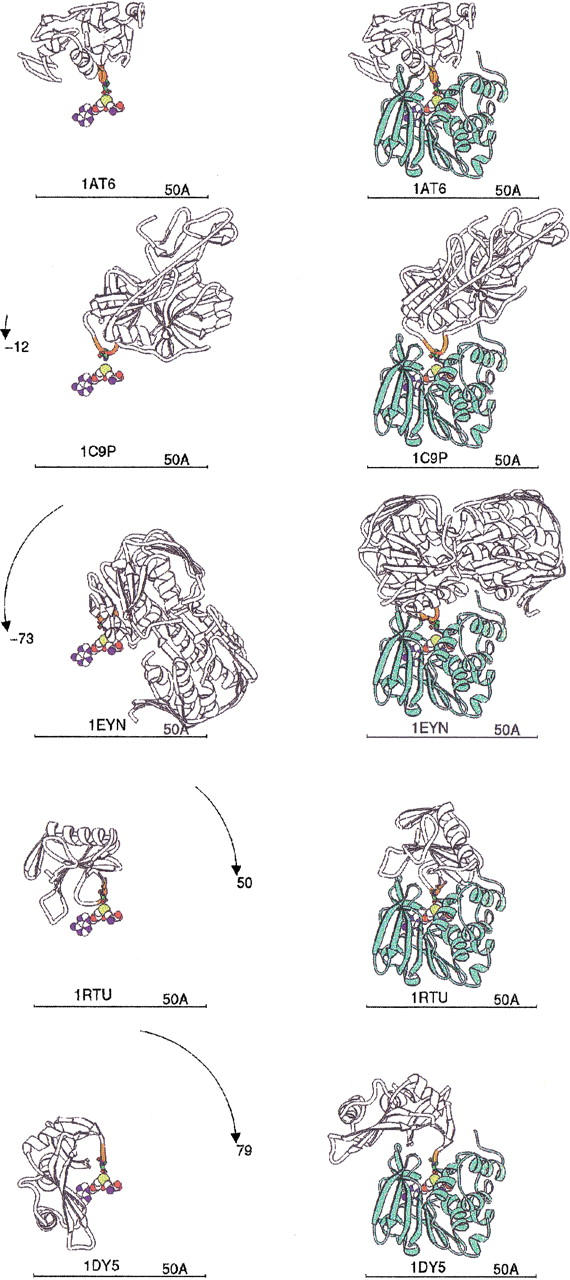
IsoAsp-containing protein binding models. Each PDB file containing isoAsp is shown as a white ribbon with the five residues centered on isoAsp colored orange, and the isoAsp is shown as a stick figure. Each protein is labeled by its PDB code. The AdoMet as bound to human PIMT provides the reference point, shown as spheres colored by atom type. (Left) PDB proteins aligned to superpose their isoAsp residue with the isoAsp modeled into the HPIMT site (Fig. 6 ▶). The rotation to be applied is shown on the arc. (Right) PDB isoAsp proteins docked into HPIMT by a single rotation about the isoAsp loop as explained in the text. HPIMT is shown as a light green ribbon. PDB codes 1AT6: hen egg white lysozyme; 1C9P: porcine trypsin; 1EYN: enoylpyruvate transferase; 1RTU: Ustilago sphaerogena ribonuclease U2; 1DY5: bovine pancreatic ribonuclease.
The maximum hinge rotation required was nearly 80° for the 1DY5 structure. No rotation was required for 1AT6 and hardly any for 1C9P. Only 1EYN still has considerable steric hindrances remaining, not easily removable by energy minimization. These modeling experiments indicate conformational changes may often be required, if possible, to enable HPIMT repair of many proteins having isoAsp residues. Alternatively, if those changes are not possible, the native protein may not be repairable. However, it also appears that some proteins can be repaired without experiencing major conformational changes, and we suggest such proteins could be good substrates for HPIMT given that residues adjacent to the isoAsp group are conducive to the binding site.
These modeling conclusions are supported by experiments in which bovine seminal ribonuclease containing an isoAsp residue was used as a model substrate for bovine brain PIMT (Galletti et al. 1988). Methyltransferase activity could not be detected for native folded ribonuclease. However, peptides containing the isosapartyl residue and denatured monomeric ribonuclease were substrates. The conclusion of those experiments was that the tertiary structure of the bovine seminal ribonuclease hindered the methyl esterification of the isoAsp residues. In our modeling studies, the highly homologous bovine pancreatic ribonuclease A required the most unfolding (79°) to allow the isoAsp residue to enter the active site of HPIMT. Furthermore, lysozyme required no unfolding in the modeling experiment, and native folded lysozyme is known to be a good substrate for human erythrocyte PIMT (Lowenson and Clarke 1991).
Materials and methods
Expression and purification of recombinant enzyme
The coding sequence for HPIMT was amplified from a human fetal brain cDNA clone using PCR and engineered into Escherichia coli expression vector pET21 (Novagen) using standard recombinant DNA techniques. The resulting plasmid was sequenced using automated DNA sequencing. For protein expression, the plasmid was transformed into E. coli strain BL21(DE3) competent cells and the transformants were selected on LB agar plate containing 50-μg/mL ampicillin. Single colonies were grown overnight at 37°C in LB medium containing ampicillin. Fresh LB medium containing 50-μg/mL ampicillin was inoculated with 1:100 dilution of overnight culture and grown with constant shaking (250 rpm) at 37°C. When the absorbance of the culture at 600 nm reached 0.6, isopropyl-D-thiogalactoside (IPTG) was added to a final concentration of 1 mM, and the culture was grown for 4 h at 37°C with constant shaking. The culture was harvested by centrifugation at 4000 rpm for 15 min and the pellet was frozen at −80°C until use.
The cell pellet was suspended in a lysis buffer composed of 50-mM Tris HCl at pH 8.0, 1-mM EDTA, 100-mM NaCl, 1-mM DTT, 1-mM benzamidine hydrochloride, 1-mM PMSF, and 1 μg/mL each of leupeptin and bestatin hydrochloride. Cells were lysed by incubation with lysozyme (final concentration of 0.1 mg/mL) at 4°C for 40 min. The lysate was then subjected to sonication for disrupting the DNA and decreasing the viscosity. The insoluble cellular debris was removed by centrifugation at 30,000 rpm for 30 min. Solid ammonium sulfate was added to the supernatant to achieve 70% saturation, and the suspension was stirred at 4°C for 30 min. The precipitated protein was isolated by centrifugation at 30,000 rpm for 30 min. The pellet was suspended in and dialyzed against the column buffer comprised of 10-mM Bis Tris HCl at pH 7.2, 1.5-mM MgCl2, and 2-mM DTT. The dialyzate was applied to a Q-Sepharose FF column (34 mL) equilibrated with the column buffer. After sample application, the column was thoroughly washed with the same buffer and the bound protein was eluted using a linear gradient of NaCl concentration (0–0.3 M in 400 mL of column buffer).
Collected fractions were analyzed by SDS-PAGE. Nearly homogeneous protein that migrated at 20 kD on SDS-PAGE, eluted at ∼0.15–0.2 M NaCl concentration. These fractions were pooled and dialyzed against MonoQ buffer consisting of 10-mM Bis Tris HCl at pH 7.0, 1.5-mM MgCl2, and 2-mM DTT and further chromatographed on a MonoQ HR10/10 column (Pharmacia) equilibrated with MonoQ buffer. After washing the unbound protein from the column, HPIMT was eluted with a linear gradient of 0–0.25 M NaCl concentration in 10 volumes of the MonoQ buffer. The recombinant protein eluted from this column in a single peak and migrated as a single band on SDS-PAGE. Fractions containing the protein were pooled and analyzed by N-terminal sequence analysis. Pooled fractions were dialyzed against MonoQ buffer and concentrated by ultrafiltration on Amicon membrane YM10 to a final concentration of 6.6 mg/mL. A sample of protein was analyzed by size exclusion chromatography on a Superdex 75 column developed in MonoQ buffer, and the molecular weight of the eluted protein was found to be approximately 20 kD. Thus the recombinant protein, like native PIMTs, existed as a monomer.
Assay of enzymatic activity
The purified protein was analyzed for PIMT activity in an in vitro assay. The assay solution contained 0.1-M Tris HCl at pH 7.5, 100–300 μg of γ-globulin (Sigma), 100-μM AdoMet (3H, 0.5 μCi/mL) (Sigma), and 1 μg of purified protein in 100-μL total volume. The reaction mixture was incubated at 30°C for 5 min. The reaction was stopped by adding 800 μL of chilled 7% TCA, and the mixture was incubated on ice for 5 min. The precipitated protein was isolated by centrifugation at 14,000 rpm for 30 min at 4°C. To the pellet, 100 μL of formic acid and 800 μL of TCA was added, and the mixture was incubated on ice for 5 min followed by centrifugation at 14,000 rpm for 30 min at 4°C. The pellet was washed another time with 800 μL of TCA and finally dissolved in 75 μL of a resuspension buffer containing 0.2-M NaOH, 5% Triton X-100, and 1.3% methanol and mixed with 925 μL of scintillation fluid and counted in a liquid scintillation counter. Control assays received no enzyme.
Crystallization of HPIMT
A two-stage process grew initial diffraction grade crystals of HPIMT. Crystals were obtained by the hanging drop method using 6 mg/mL of HPIMT in buffer mixed in equal proportions with 5% PEG 8000 in sodium cacodylate buffer at pH 6.5 containing 15 mM magnesium acetate and 2.6 mM AdoHcy. Crystals appeared overnight at 4°C but were often multinucleated. More useful crystals were produced using pieces of the initial crystals as macroseeds in hanging droplets set up as before but with half the protein concentration. The seeds grew into large rectangular or triangular plates in 1 day.
X-ray diffraction experiments
HPIMT crystals could be successfully flash frozen to −170°C after soaking in a 4°C holding/cryoprotectant solution consisting of 20% PEG 8000, 30% glycerol, 100-mM sodium cacodylate, 15-mM magnesium acetate, and 2.6-mM AdoHcy at pH 6.5. X-ray diffraction data were collected using a fine-focus Rigaku RU300 copper rotating anode X-ray generator operating at 50 kV and 100 mA with Yale-type double-focusing mirrors and a Rigaku Raxis-IV dual-image plate system (Molecular Structure Corporation). The X-rays were prefiltered for β radiation with Ni foil 0.00015" thick and passed thorough a 0.5-mm collimator. The crystal-to-detector distance was 100 mm. The oscillation angle for each frame was 1°, and exposure duration was 45 min per frame. The diffraction data were integrated using DENZO and scaled with SCALEPACK (Otwinowski and Minor 1997).
Initial phases were determined by the molecular replacement method using the cross-rotation and translation programs of AMORE (Navaza 1994). The search model was constructed from residues 5 to 224 of the T. maritima PIMT. Conserved residues determined by sequence alignment were left unchanged, glycines were left as glycine, and all others were changed to alanine. The AdoHcy was left out of the search model. Data in the range of 20 to 4.5 Å were used for the cross-rotation search and gave a Patterson correlation coefficient of 0.201 compared to 0.177 for the next peak. After translation function and rigid body refinement, the starting R-factor was 0.532. QUANTA was used to manually build in missing loops and the rest of the C-terminus missing in the search model.
Iterative cycles of model refinement, manual rebuilding using QUANTA, and adding waters with WATERPICK (CNS; Brunger et al. 1998) were performed using high-resolution data until the process converged. We found it necessary to remove constraints imposed by CNS on the sugar ring dihedrals to complete the refinement and remove significant difference density from around the AdoHcy. Otherwise, the default parameter files of CNS forced a bias toward the sugar puckers found in RNA/DNA. The resulting conformation of the AdoHcy ribose ring has a 1` exo conformation.
Acknowledgments
We thank Ana Teodoro for helpful discussions and expert assistance using ALSCRIPT for the sequence alignment figure. We thank Randy Mann for his efforts toward maintaining the laboratory diffraction equipment. Atomic coordinates for HPIMT have been deposited with the Protein Data Bank under the code IDL5.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.37802.
References
- Barton, G.J. 1993. ALSCRIPT A tool to format multiple sequence alignments. Protein Eng. 6: 37–40. [DOI] [PubMed] [Google Scholar]
- Brunger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunsleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., Read, R.J., Rice, L.M., Simonson, T., and Warren, G.L. 1998. Crystallography and NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D 54905–921. [DOI] [PubMed] [Google Scholar]
- Carson, M., 1997. Ribbons. In Methods of Enzymology, Vol. 277 (eds. R.M. Sweet and C.W. Carter) pp. 493–505, Academic Press, New York, N. Y.
- Cheng, X., Kumar, S., Posfai, J., Pflugrath, J.W., and Roberts, R.J. 1993. Crystal structure of the HhaI DNA methyltransferase complexed with S-adenosyl-L-methionine. Cell 74299–307. [DOI] [PubMed] [Google Scholar]
- Clarke, S. 1987. Propensity for spontaneous succinimide formation from aspartyl and asparaginyl residues in cellular proteins. Int. J. Pept. Protein Res. 30808–821. [DOI] [PubMed] [Google Scholar]
- Clarke, S. 1992. The biological functions of protein methylation. In Fundamentals of medical cell biology. (ed. E.E. Bittar), Vol. 3B, pp. 413–436. JAI Press, Greenwich, CT.
- Clarke, S. 1993. Protein methylation. Curr. Opin. Cell Biol. 5977–983. [DOI] [PubMed] [Google Scholar]
- Connolly, M.L. 1993. The molecular surface package. J. Mol. Graph. 11139–141. [DOI] [PubMed] [Google Scholar]
- Djordjevic, S. and Stock, A.M. 1997. Crystal structure of the chemotaxis receptor methyltransferase CheR suggests a conserved structural motif for binding S-adenosylmethionine. Structure 5545–558. [DOI] [PubMed] [Google Scholar]
- Esposito, L., Vitagliano, L., Sica, F., Sorrentino, G., Zagari, A., and Mazzarella, L. 2000. The ultra-high resolution crystal structure of ribonuclease A containing an isoAsp residue at position 67. J. Mol. Biol. 297713–732. [DOI] [PubMed] [Google Scholar]
- Galletti, P., Ciardiello, A., Ingrosso, D., Di Donato, A., and D'Alessio, G. 1988. Repair of isopeptide bonds by protein carboxyl O-methyltransferase: Seminal ribonuclease as a model system. Biochemistry 271752–1757. [DOI] [PubMed] [Google Scholar]
- Geiger, T. and Clarke, S. 1987. Deamidation, isomerization, and racemization at asparaginyl and aspartyl residues in peptides. Succinimide-linked reactions that contribute to protein degradation. J. Biol. Chem. 262785–794. [PubMed] [Google Scholar]
- Ho, D.K., Wu, J.C., Santi, D.V., and Floss, H.G. 1991. Stereochemical studies of the C-methylation of deoxycytidine catalyzed by HhaI methylase and the N-methylation of deoxyadenosine catalyzed by EcoRI methylase. Arch. Biochem. Biophys. 284264–269. [DOI] [PubMed] [Google Scholar]
- Huang, Y., Komoto, J., Konishi, K., Takata, Y., Ogawa, H., Gomi, T., Fujioka, F., and Takusagawa, F. 2000. Mechanisms for auto-inhibition and forced product release in glycine N-methyltransferase: Crystal structures of wild-type, mutant R175K and S-adenosylhomocysteine-bound R175K enzymes. J. Mol. Biol. 298149–162. [DOI] [PubMed] [Google Scholar]
- Huebscher, K.J., Lee, J., Rovelli, G., Ludin, B., Matus, A., Stauffer, D., and Furst, P. 1999. Protein isoaspartyl methyltransferase protects from Bax-induced apoptosis. Gene 240333–341. [DOI] [PubMed] [Google Scholar]
- Ingrosso, D., Fowler, A.V., Bleibaum, J., and Clarke, S. 1989. Sequence of the D-aspartyl/L-isoAsp protein methyltransferase from human erythrocytes. J. Biol. Chem. 26420131–20139. [PubMed] [Google Scholar]
- Johnson, B.A. and Aswad, D.W. 1993. Kinetic properties of bovine brain protein L-isoaspartyl methyltransferase determined using a synthetic isoAsp peptide substrate. Neurochem. Res. 1887–94. [DOI] [PubMed] [Google Scholar]
- Johnson, B.A., Langmack, E.L., and Aswad, D.W. 1987a. Partial repair of deamidation-damaged calmodulin by protein carboxyl methyltransferase. J. Biol. Chem. 26212283–12287. [PubMed] [Google Scholar]
- Johnson, B.A., Murray, E.D. Jr., Clarke, S., Glass, D.B., and Aswad, D.W. 1987b. Protein carboxyl methyltransferase facilitates conversion of atypical L-isoAsp peptides to normal L-aspartyl peptides. J. Biol. Chem. 2621159–1165. [PubMed] [Google Scholar]
- Kim, E., Lowenson, J.D., MacLaren, D.C., Clarke, S., and Young, S.G. 1997. Deficiency of a protein-repair enzyme results in the accumulation of altered proteins, retardation of growth, and fatal seizures in mice. Proc. Natl. Acad. Sci. 946132–6137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labahn, J., Granzin, J., Schluckebier, G., Robinson, D.P., Jack, W.E., Schildkraut, I., and Saenger, W. 1994. Three-dimensional structure of the adenine specific DNA methyltransferase M. TaqI in complex with the cofactor S-adenosylmethionine. Proc. Nat. Acad. Sci. 9110957–10961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowenson, J.D. and Clarke, S. 1991. Structural elements affecting the recognition of L-isoAsp residues by the L-isoAsp protein methyltransferase. Implications for the repair hypothesis. J. Biol. Chem. 26619396–19406. [PubMed] [Google Scholar]
- McFadden, P.N. and Clarke, S. 1987. Conversion of isoAsp peptides to normal peptides: Implications for the cellular repair of damaged proteins. Proc. Natl. Acad. Sci. 842595–2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone, T., Blumenthal, R.M., and Cheng, X. 1995. Structure-guided analysis reveals nine sequence motifs conserved among DNA amino-methyltransferases, and suggests a catalytic mechanism for these enzymes. J. Mol. Biol. 253618–632. [DOI] [PubMed] [Google Scholar]
- Mamula, M.J., Gee, R.J., Elliott, J.I., Sette, A., Southwood, S., Jones, P.-J., and Blier, P.R., 1999. Isoaspartyl post-translational modification triggers autoimmune responses to self-proteins. J. Biol. Chem. 27422321–22327. [DOI] [PubMed] [Google Scholar]
- Noguchi, S., Satow, Y., Uchida, T., Sasaki, C., and Matsuzaki, T. 1995. Crystal structure of Ustilago sphaerogena ribonuclease U2 at 1.8 Å resolution. Biochemistry 3415583–15589. [DOI] [PubMed] [Google Scholar]
- Noguchi, S., Miyawaki, K., and Satow, Y. 1998. Succinimide and isoAspartate residues in the crystal structures of hen egg-white lysozyme complexed with tri-N-acetylchitotriose. J. Mol. Biol. 278231–238. [DOI] [PubMed] [Google Scholar]
- O'Gara, M., Zhang, X., Roberts, R.J., and Cheng, X. 1999. Structure of a binary complex of HhaI methyltransferase with S-adenosyl-L-methionine formed in the presence of a short nonspecific DNA oligonucleotide. J. Mol. Biol. 287201–209. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. In Methods of enzymology (eds. R.M. Sweet and C.W. Carter), Vol. 276 , pp. 307–326, Academic Press, New York. [DOI] [PubMed]
- Rester, U., Bode, W., Moser, M., Parry, M.A., Huber, R., and Auerswald, E. 1999. Structure of the complex of the antistasin-type inhibitor Bdellastasin with trypsin and modeling of the Bdellastasin-microplasmin system. J. Mol. Biol. 29393–106. [DOI] [PubMed] [Google Scholar]
- Schonbrunn, E., Eschenburg, S., Luger, K., Kabsch, W., and Amrhein, N. 2000. Structural basis for the interaction of the fluorescence probe 8-anilino-1-naphthalene sulfonate (Ans) with the antibiotic target Mura. Proc. Nat. Acad. Sci. 976345–6349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schluckebier, G., Zhong, P., Stewart, K.D., Kavanaugh, T.J., and Abad-Zapatero, C. 1999. The 2.2 Å Structure of the Rrna Methyltransferase Ermc` and its complexes with cofactor and cofactor analogs: Implications for the reaction mechanism. J. Mol. Biol. 289277–291. [DOI] [PubMed] [Google Scholar]
- Shimizu, T., Watanabe, A., Ogawara, M., Mori, H., and Shirasawa, T. 2000. Isoaspartate formation and neurodegeneration in Alzheimer's disease. Arch. Biochem. Biophys. 381225–234. [DOI] [PubMed] [Google Scholar]
- Skinner, M.M., Puvathingal, J.M., Walter, R.L., and Friedman, A.M. 2000. Crystal structure of protein isoaspartyl methyltransferase: A catalyst for protein repair. Structure 81189–1201. [DOI] [PubMed] [Google Scholar]
- Takeda, R., Mizobuchi, M., Murao, K., Sato, M., and Takahara, J. 1995. Characterization of three cDNAs encoding two isozymes of an isoAsp protein carboxyl methyltransferase from human erythroid leukemia cells. J. Biochem. 117683–685. [DOI] [PubMed] [Google Scholar]
- Vidgren, J., Svensson, L.A., and Liljas, A. 1994. Crystal structure of catechol O-methyltransferase. Nature 368354–358. [DOI] [PubMed] [Google Scholar]
- Yamamoto, A., Takagi, H., Kitamura, D., Tatsuoka, H., Nakano, H., Kawano, H., Kuroyanagi, H., Yahagi, Y., Kobayashi, S., Koizumi, K., Sakai, T., Saito, K., Chiba, T., Kawamura, K., Suzuki, K., Watanabe, T., Mori, H., and Sihrasawa, T. 1998. Deficiency in protein L-isoaspartyl methyltransferase results in a fatal progressive epilepsy. J. Neurosci. 182063–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]