

Abstract
A point mutation (I53A) in the core of Escherichia coli RNase H* is known to destabilize both the native conformation (ΔGUN) and the kinetic intermediate (ΔGUI) by 2 kcal/mole. Here, we have used native-state hydrogen deuterium exchange to ask how this destabilization is propagated throughout the molecule. Stability parameters were obtained for individual residues in I53A and compared with those from the wild-type protein. A destabilization of 2 kcal/mole was observed in residues in the core but was not detected in the periphery of the molecule. These results are consistent with the localized destabilization of the core observed in the early intermediate of the kinetic folding pathway, supporting the resemblance of this kinetic intermediate to the partially unfolded form detected in the native state at equilibrium. A thermodynamic cycle also shows no interaction between Ile 53 and a residue in the periphery. There is, however, an increase in the number of denaturant-independent exchange events in the periphery of I53A, showing that effects of the point mutation are communicated to regions outside the core, although perhaps not through changes in stability. In sum, this work shows that localized regions within a protein can be destabilized independently. Furthermore, it implies a correspondence between the kinetic intermediate and the equilibrium PUF, as the magnitude and localization of the destabilization are the same in both.
Keywords: Hydrogen exchange, protein stability, protein folding, amino acid substitution, mutation
The "native state" of a protein is comprised of the ensemble of conformations accessible to the protein under native conditions. Therefore, in addition to the majority of molecules that fold into the native conformation, the native state includes low populations of partially unfolded forms (PUFs) and even the unfolded conformation (the populations of each conformer weighted according to the Boltzman distribution). Intermediates transiently formed during the folding process (kinetic intermediates) should be represented within the ensemble. Traditional structural probes such as X-ray crystallography, circular dichroism (CD), and NMR do not report on these rare conformations, as the signal they detect arises from the majority of molecules in the native conformation. Native state hydrogen exchange (Bai et al. 1995), however, has successfully detected PUFs within the native state of several proteins including RNase H (Chamberlain et al. 1996), cytochrome c (Bai et al. 1995), and T4 lysozyme (Llinas et al. 1999). For both RNase H* and cytochrome c, these equilibrium PUFs resemble kinetic intermediates in the folding pathway (Englander 2000).
For all of these proteins, the stability is not distributed uniformly throughout the molecule, as would be expected for a completely cooperative, two-state system. Some regions are less stable than others, leading to partially unfolded molecules that are detected via hydrogen exchange through their unstructured regions (Chamberlain and Marqusee 2000; Englander 2000). The energetic coupling between these regions of differing stabilities is not well understood. Stabilizing the weakest region of cytochrome c via reduction was found to stabilize all PUFs, preserving their heirarchy (Xu et al. 1998). Here, we ask a complementary question: do changes in the most stable region of a protein affect the stability of less stable regions? If such changes have differential effects across the protein, then the resulting variant will have an altered stability profile or free energy landscape.
In these studies, we examined the free energy profile of a variant of E. coli RNase H* (the asterisk denotes a cysteine-free variant) possessing the mutation Ile 53 to Ala (I53A) (Fig. 1 ▶). Residue 53 is located in a region defined as the core in both equilibrium (Chamberlain et al. 1996) and kinetic studies (Raschke and Marqusee 1997). Previous studies have shown that this mutation destabilizes both the global unfolding and the kinetic intermediate by ∼2 kcal/mole (Raschke et al. 1999). These changes in stability are not a result of gross structural changes: we solved the crystal structure of I53A, which overlays well with the wt* structure determined previously (Goedken et al. 2000). We, therefore, turned to native-state hydrogen deuterium exchange to ask how this site-specific mutation affects the equilibrium energy landscape of RNase H. Is the destabilization localized to the core, as suggested by the refolding kinetics of I53A, or is it propagated to more distal regions of the molecule? We also address a possible coupling between the stabilities of the core and the periphery using a thermodynamic cycle between Ile 53 (in the core) and Ala 140 (in the periphery). Our evidence supports a 2 kcal/mole destabilization of the core that is not propagated to the periphery. This is consistent with the localized 2 kcal/mole destabilization of the core seen in the I53A kinetic intermediate.
Fig. 1.

Overlay of the E. coli RNase H* (Goedken et al. 2000) and I53A X-ray crystal structures. The wt* and I53A backbones are drawn as light and dark ribbons, respectively. I53 is the residue in black.
Results
The E. coli RNase H* fold is conserved in I53A
Several studies, both high and low resolution, suggest that the mutation I53A does not alter the fold of RNase H. I53A forms an active RNase H with a circular dichroism (CD) spectrum similar to that of wt* (RNase H*, a cysteine free variant) (Raschke et al. 1999). A comparison of backbone chemical shifts between wt* and I53A shows only minor perturbations (I53A was assigned by HNCA—see Materials and Methods). The backbone protons on the surface of the protein overlay well (Fig. 2 ▶). Backbone protons with altered chemical shifts are localized in the core, as expected, because Ile 53 is a core residue with a large number of contacts. Finally, we used X-ray crystallography to demonstrate directly that the two proteins have the same structures. The structure of I53A was solved by molecular replacement (see Materials and Methods) to a resolution of 1.6 Å, yielding a final Rwork and Rfree of 20.7 and 25.7%, respectively. The overall rsym for the data (in space group P212121) was 6.2 %, the completeness of the data 95.2%, and the resolution range used in refinement 12.5–1.6 Å. An overlay of the backbone representations shows that I53A and wt* have the same fold (Fig. 1 ▶).
Fig. 2.

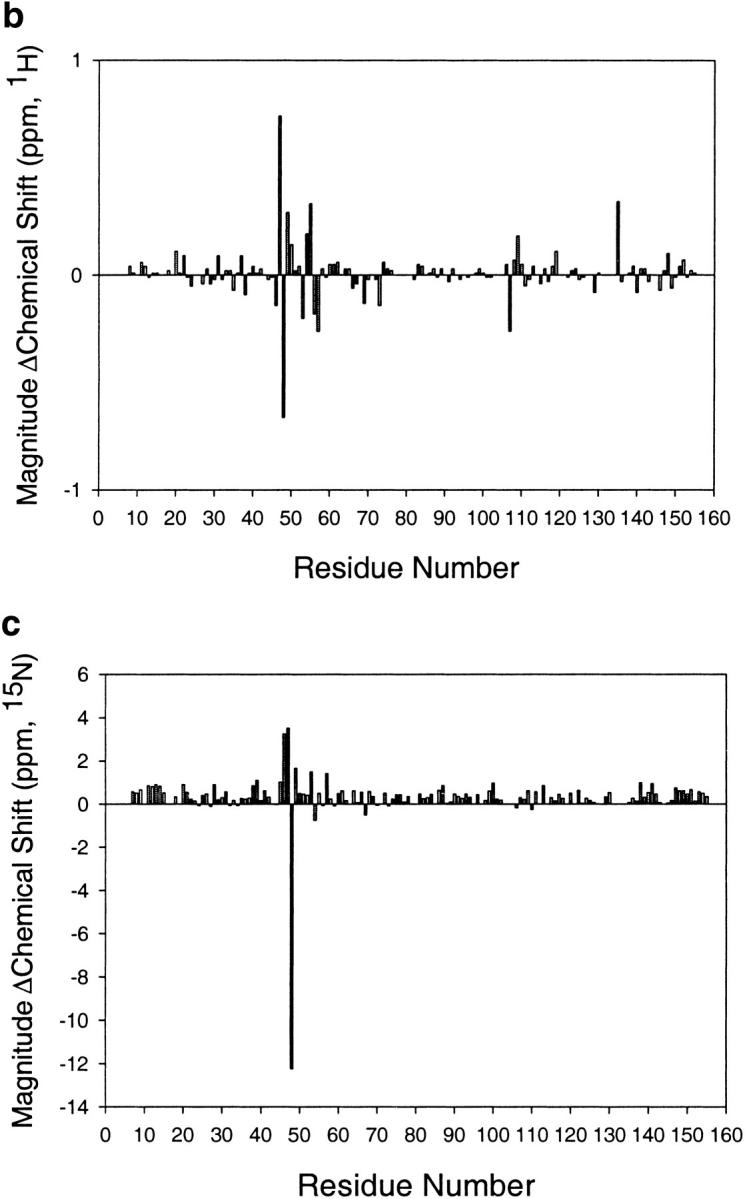
Comparison of E. coli RNase H* (Dabora et al. 1996) and I53A chemical shifts. (a) HSQC spectra overlayed. Spectra were taken in H2O at pH 5.5. I53A and wt* chemical shifts are drawn as peaks and boxes, respectively. (b,c) Histograms depicting the change in 1H or 15N chemical shift per residue upon mutating Ile 53 to Ala.
I53A is destabilized by 2 kcal/mole
Previous studies monitoring the CD signal as a function of urea (in H2O) have shown that the stability (ΔGUN) of I53A is decreased by ∼2 kcal/mole (versus wt*) (Raschke et al. 1999). We repeated these measurements under the conditions used in the native-state hydrogen exchange experiment (i.e., D2O and GdmCl) (Fig. 3 ▶, inset). Under these conditions, the stability of I53A in D2O and GdmCl is 7.6 kcal/mole (versus 10 kcal/mole for wt*).
Fig. 3.

Hydrogen-deuterium exchange for every probe can be described by the global unfolding event and/or a local fluctuation. Protons shown are: T9 (Strand 1) (open circles), F35 (Strand 3) (closed circles), E48 (Helix A) (closed squares), and D108 (Helix D) (closed triangles). Lines are fits to a fixed global unfolding event and a variable denaturant-independent exchange event. (Inset) I53A fraction folded (Ff) as a function of denaturant concentration, probed by circular dichroism at 222nm. I53A appears completely folded at all denaturant concentrations probed by native-state HX.
To determine how this ∼2 kcal/mole destabilization is distributed throughout the molecule, we carried out native-state hydrogen deuterium exchange on I53A and compared the results to wt* (Chamberlain et al. 1996). Exchange was monitored for individual amides at 10 different denaturant concentrations (from 0 to 0.8M GdmCl); I53A appears completely folded by CD under all these conditions (Fig. 3 ▶, inset). The hydrogen exchange profile for every backbone amide monitored could be fit to a single denaturant-dependent free energy (ΔGunf = 8.2 kcal/mole, m = 5.3 kcal mole−1 M−1), together with (for most residues) a variable denaturant-independent event (these "local fluctuations" are more abundant in I53A—see below) (Fig. 3 ▶). This single unfolding event is similar in free energy and m value to the global unfolding measured by CD under the same conditions (ΔGUN = 7.6 kcal/mole, m = 5.3 kcal mole−1 M−1). We, therefore, attribute the denaturant-dependent exchange observed in I53A to global unfolding. Although we did not detect a partially unfolded form (PUF) directly, we have evidence that a PUF is present in the I53A native state (see discussion). Nevertheless, by both hydrogen exchange and CD methods, the ΔG of global unfolding is decreased by ∼2 kcal/mole in I53A (compared with wt*).
New local fluctuations are prevalent in I53A
More residues were observed to undergo denaturant-independent exchange (local fluctuations) in I53A than in wt*. All sites that undergo local fluctuations in wt* also do so in I53A (if the site is monitored in both proteins), and the exchange rate, or ΔGFL, for these shared sites is unperturbed (Fig. 4B ▶). I53A contains additional sites where local fluctuations were observed. Strangely, all of these new fluctuations occur in strands I, II, III, V, and helix E, regions outside the core (Fig. 1 ▶), while the site of the mutation and the observed destabilization are in the core (Fig. 4A,C). Uncovering new local fluctuations is surprising, as the global unfolding of I53A occurs at a lower free energy. This should, if anything, mask any higher energy fluctuations, as the lowest stability exchange event dominates.
Fig. 4.

Comparison of denaturant-independent exchange events in I53A and wt*. (a) "Local fluctuations" mapped onto the crystal structure of I53A. Dark balls are amide protons that exchange by denaturant-independent events in both I53A and wt*; light balls represent new events in I53A (i.e., not seen in wt*). (b) Free energies of local fluctuations in wt* and I53A are compared. Only residues that exchange through a local fluctuation in both proteins are shown. A line with a slope of 1 is included to simulate the case where I53A ΔGFL = wt* ΔGFL. (c) Free energy of all local fluctuations in I53A (black bars) and wt* (gray bars).
No interaction is detected between residues 53 and 140
We created a thermodynamic cycle (Horovitz and Fersht 1990) of mutant proteins to ask if residues representing the periphery and the core interact to affect the overall stability of the fold. Ile 53 was chosen to represent the core, and Ala 140 (positioned in Helix E, with a large number of contacts to the strands) represents the periphery. Four variants were studied: wt*, I53A (in the core: Helix A), A140S (in the periphery: Helix E), and the double mutant (I53A/A140S). All variants were active, and have overlaying CD spectra (data not shown). The free energy of unfolding was determined for each variant. The difference in ΔGunf upon mutating a residue in the presence or absence of its partner yields the interaction energy (i.e., the difference of two vertical or horizontal sides of the cycle in Fig. 5 ▶). This cycle shows no interaction (ΔGint = −0.1 kcal/mole) between Ile 53 and Ala 140: A140S destabilizes the native state by 1 kcal/mole (with respect to wt*), and the same magnitude of destabilization is observed in the I53A background (Fig 5 ▶). Helix E contributes the same amount of stability to the native state regardless of the stability of the core.
Fig. 5.

Thermodynamic cycle addressing the possible interaction between Ile 53 and Ala 140. ΔGUNF for each variant was measured by circular dichroism @ 222 nm as a function of [urea]. The change in the ΔGUNF between variants (in kcal/mole) is reported along the arrows. The cycle shows no interaction between I53 and A140 in the native state.
Discussion
Traditional studies on the effect of site-specific mutations have addressed changes in the overall, global stability of proteins. More recent studies using hydrogen exchange have mapped the distribution of stability throughout the native state. Here, we combine the two approaches, using native-state hydrogen exchange to monitor the energetic effects of mutation on different regions of a protein. Previous amide exchange studies on variants of RNase T1 have shown that point mutations can affect both global and nonglobal exchange throughout the molecule (Huyghues-Despointes et al. 1999). The origin of the exchange not due to global unfolding, however, was not clear, as hydrogen exchange was not monitored as a function of denaturant. In our studies, we have examined the effects of a site-specific, destabilizing mutation on backbone amide exchange rates in E. coli RNase H. We monitor exchange as a function of [GdmCl], enabling us to distinguish denaturant-dependent unfolding events from denaturant independent "local fluctuations." The mutation, I53A, is located in the core, the most stable region of the molecule. Is the destabilization localized to the core, the region structured in the kinetic intermediate, or is it propagated to more peripheral regions of the protein? Our results suggest that, as in the kinetic intermediate, this destabilization event is localized to the core, and the change in stability of ∼2 kcal/mole is not propagated to more distal regions of the folded molecule (strands I, II, III, V, and Helix E). However, we do see a change in the local fluctuations in the periphery of the molecule.
Partially unfolded forms (PUFs) in the native state have been postulated to be identical to kinetic folding intermediates in both structure and stability (Englander 2000). Here, we compare the effects of a mutation in the PUF and the kinetic intermediate to determine if they are indeed the same intermediate. Previous native-state hydrogen exchange studies on wt* (Chamberlain et al. 1996) uncovered a partially unfolded form (PUF) of RNase H similar in both structure and stability to the kinetic intermediate (Raschke and Marqusee 1997). I53A folds through a similar kinetic intermediate, although it is highly destabilized (ΔΔGUI ∼2 kcal/mole) (Raschke et al. 1999). Therefore, kinetic studies on both proteins reveal an intermediate with a stability between that of the native and unfolded states. There is no reason that this intermediate should not be accessible by native-state hydrogen exchange on I53A, suggesting it is masked in our experiments. In fact, if the equilibrium PUF has the same stability as the kinetic intermediate, we would expect the local fluctuations to mask the PUF in I53A (see below).
Indeed, no subglobal exchange was detected in I53A; all the denaturant-dependent exchange can be described by one common free energy and m value (ΔGUNF = 8.2 kcal/mole, mUNF = 5.3 kcal mole−1 M−1). Exchange from the PUF (i.e., exchange from the unstructured periphery) is most likely masked (as outlined in Fig. 6 ▶). The lower energy global unfolding of I53A (blue line) offers only a narrow window of [GdmCl] in which exchange by a periphery with wt*-like stability and denaturant dependence (upper dotted line) could be detected. In this narrow window (0 to 0.3 M GdmCl) most residues in I53A exchange by local fluctuations. To illustrate this point, actual exchange data for Thr 9 (measured in I53A) are plotted as black circles in Figure 4 ▶. The PUF detected in wt* (upper dotted line) would be masked by the low-energy local fluctuation experienced by T9 at low [GdmCl], as the lowest energy event dominates hydrogen exchange. In sum, because I53A has been destabilized globally, and local fluctuations mask any higher energy exchange events at low [GdmCl], we cannot observe exchange from a periphery with wt*-like stability.
Fig. 6.

Free energy of exchange versus denaturant concentration of: wt* global unfolding (black line), I53A global unfolding (blue line), unfolding of the strands in the wt* PUF (black dotted line), predicted unfolding by a destabilized PUF (red dotted line), and actual exchange for residue T9 in I53A (filled circles).
Furthermore, if the periphery were destabilized and exchange from the PUF occurred at a lower stability in I53A than in wt*, we would be able to detect it. The lower dotted line in Figure 6 ▶ models exchange from the periphery at a free energy of 2 kcal/mole less than in wt* (the same magnitude of destabilization as the core). In this case, exchange by the PUF should dominate over a large window of detection, and would, in fact, be seen in our experiments.
In sum, we propose that the PUF goes undetected in I53A due to the increased number of local fluctuations. As we see no exchange by a PUF in I53A, we can rule out the possibility that the periphery is destabilized by 2 kcal/mole. We cannot rule out that this region has been slightly destabilized, as a small (up to 1 kcal/mole) destabilization would be impossible to detect in these experiments. However, if we assume the PUF exists in the I53A native state, then the periphery is not destabilized to the extent of the core, which demonstrates that the destabilization is not evenly distributed throughout the molecule.
The proposed energy diagrams for wt* and I53A are diagrammed in Figure 7 ▶. Using the logic outlined above, we placed exchange by the I53A periphery at the same ΔGHX as in wt*. This results in a destabilization of the core (ΔGU-PUF) consistent with the destabilization of the kinetic intermediate (ΔGUI). Our model is therefore consistent with both the presence and stability of the kinetic intermediate, and the masking of the PUF by the local fluctuations in the I53A native-state hydrogen exchange experiments. This work provides a link between the kinetic intermediate and the equilibrium PUF, as the specific effects, that is, the magnitude and localization of the destabilization due to the I53A mutation, are the same in both.
Fig. 7.

Predicted energy diagram for I53A compared with wt*. I53A global exchange is directly measured, and exchange by the periphery is modeled at the ΔGHX observed in wt*.
Despite the fact that the destabilization in I53A is not observed in the unfolding free energy of the periphery and appears localized to the core, we do observe changes in the exchange properties of the periphery. We detect a multitude of new local fluctuations in this region. The significance of theses fluctuations is unclear because the biophysical mechanism of local fluctuations is still not known. "Solvent penetration," "local unfolding," and "ensemble" models have all been proposed to explain seemingly "denaturant-independent" exchange (Miller and Dill 1995; Wooll et al. 2000). In the first two models exchange occurs without a major change in surface area exposed from a minor amount of solvent exposure or small amplitude motions. The third model (and aspects of the second model), however, poses that exchange could occur via large excursions from the native fold. Our data cannot differentiate these. Temperature factors measured in the crystal structure of the I53A backbone are higher than in wt*, implying a more dynamic overall fold. However, these B factors are higher all over the molecule, and retain the same pattern as wt* (data not shown), while the new local fluctuations are specifically located in the periphery of the molecule. Furthermore, local fluctuations that are conserved in both proteins (which are located in the core) exchange with the same rate in I53A and wt*, implying the change in denaturant-independent exchange is localized to the periphery. Further studies on the dynamic nature of I53A may lead to a better understanding of the local fluctuations, and why they have drastically increased in number.
We have observed a site-specific modification in the core of a protein, the effect of which is not equally distributed throughout the entire folded molecule, demonstrating that localized regions within a protein can be destabilized independently. Although the core is destabilized in I53A, it remains the most stable region of the folded protein, as in wt*. Other studies on RNase H have shown that a stabilizing mutation in the periphery of the molecule (D10A) increases the ΔGunf of all regions of the molecule, preserving the hierarchy of stability (Goedken and Marqusee 2001). Further studies must be done to elucidate if the hierarchy of these regions can change, and if the cooperativity of stability within a protein depends on the preservation of this hierarchy. These studies can aid us in understanding how localized events (such as mutation or ligand binding) that change stability are communicated throughout a protein in the absence of a structural change.
Materials and methods
Protein expression and purification
I53A, constructed by site-directed mutagenesis of E. coli RNase H* (Raschke et al. 1999), was expressed in E. coli BL21 (DE3) pLysS cells in M9 media and labeled with 15N (ammonium chloride) or both 15N and 13C (D-glucose). Protein was soluble and purified as previously described (Dabora and Marqusee 1994), with the following exceptions: a cation-exchange column (Pharmacia SOURCE 15 S) replaced the size-exclusion column, and protein was dialyzed against 200 mM ammonium bicarbonate before lyophilizing.
Crystal growth, X-ray diffraction, and refinement
Crystals of I53A were grown by the hanging-drop vapor diffusion method in conditions similar to those used for wt* (20 mM HEPES, pH 7.5, 10% PEG-3350, 4 mg/mL of protein) (Goedken and Marqusee 2001) and by microseeding with E. coli RNase H*. Crystals formed in a few days, and were incubated for ∼1 min in cryoprotectant (20% MPD, 20 mM HEPES pH 8.0, 10% PEG-3350) and flash-frozen in liquid nitrogen. X-ray diffraction data were collected at the Advanced Light Source (Beamline 5.0.2) at the Lawrence Berkeley National Laboratory. DENZO and SCALEPACK (Otwinowski and Minor 1997) were used to integrate and scale the data. The overall rsym for the data (in space group P212121) was 6.2%, the completeness of the data 95.2%, and the resolution range used in refinement 12.5–1.6 Å. I53A (PDB accession number 1JXB) was solved by molecular replacement using the E. coli RNase H* structure (PDB accession number 1F21) (Goedken et al. 2000) and AMoRe (Navaza 1994). REFMAC (Murshudov et al. 1997) and ARP (Lamzin and Wilson 1993) were used for automated refinement of the I53A model, and to build water into the structure. Manual rebuilding between REFMAC cycles was done in O (Jones et al. 1991) on 2Fo–Fc and Fo–Fc maps created in MAPMAN. The final Rwork and Rfree are 25.7 and 20.7%, respectively.
Stability determination by circular dichroism
CD measurements were taken in an Aviv 62DS spectropolarimeter with a Peltier temperature-controlled sample holder using a 1-cm cuvette. The CD signal at 222 nm as a function of denaturant was monitored, and data were fit using a two-state assumption (described previously; Santoro and Bolen 1988), yielding ΔGUN = 7.6 kcal/mole, m = 5.3 kcal mole−1 M−1.
Assignments
15N-13C-labeled I53A was dissolved to a 1 mM concentration in 100 mM d3-NaAc, in 90% H2O/10% D2O at pH 5.5. Direct overlaps were assigned by comparing 1H-15N heteronuclear single-quantum coherence (HSQC) spectra in H2O. Assignment of the backbone was completed using 1H-15N-13C HNCA. Data were collected at 25°C on a Bruker 500-MHz spectrometer, and processed using either NMRPipe (Delaglio et al. 1995) or FELIX (Molecular Simulations).
Hydrogen-deuterium exchange
Lyophilized 15N-labeled I53A was brought up in 100 mM NaAc, H2O, and an appropriate concentration of GdmHCl. Samples contained 0.7–1.0 mM protein and were buffer exchanged into 100 mM d3-NaAc, D2O, and deuterated GdmHCl (pD 5.5, or pDread 5.1). Twenty-minute HSQC spectra were collected immediately upon buffer exchange for 3 h continuously, and then periodically for up to 3 mo for 10 samples (0 to 0.8 M GdmHCl). 1D spectra in the 1H dimension were used to determine the consistency of the protein concentration over time. To ascertain that exchange takes place by the EX2 mechanism, a sample was prepared in 0.8 M GdmHCl at pD 5.2 (pDread 4.8), and the observed rates of exchange were compared to the 0.8 M GdmHCl sample at pD 5.5.
HSQC spectra were processed using FELIX 97.0 (Molecular Simulations). Maximal peak intensities as a function of time were fit to single exponentials (kobs). The free energy of exchange for individual amides at each denaturant concentration was obtained by the following equation: ΔGHX = −RTln(kobs/krc), where krc is the rate of exchange for residues in random coil (Bai et al. 1993). Free energy (ΔGHX) as a function of denaturant concentration was then fit to obtain the free energy of local fluctuation (ΔGfl), using a fixed unfolding event: 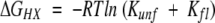 , where
, where 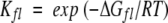 and
and 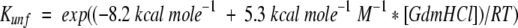 .
.
Acknowledgments
We thank Jeffrey Pelton for help with the NMR assignments; Eric Goedken for the crystallography; Dave King for mass spectrometry, Julie Hollien, Eric Nicholson, and Chiwook Park for critical reading of the manuscript and scientific discussion; and the Marqusee lab for support and advice. This work was funded by NIH Grant GM50945.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.37202.
References
- Bai, Y., Milne, J.S., Mayne, L., and Englander, S.W. 1993. Primary structure effects on peptide group hydrogen exchange. Proteins Struct. Funct. Genet. 17 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai, Y., Sosnick, T.R., Mayne, L., and Englander, S.W. 1995. Protein folding intermediates: Native-state hydrogen exchange. Science 269 192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain, A.K., Handel, T.M., and Marqusee, S. 1996. Detection of rare partially folded molecules in equilibrium with the native conformation of RNaseH. Nat. Struct. Biol. 3 782–787. [DOI] [PubMed] [Google Scholar]
- Chamberlain, A.K. and Marqusee, S. 2000. Comparison of equilibrium and kinetic approaches for determining protein folding mechanisms. Adv. Protein Chem. 53 283–328. [DOI] [PubMed] [Google Scholar]
- Dabora, J.M. and Marqusee, S. 1994. Equilibrium unfolding of Escherichia coli ribonuclease H: Characterization of a partially folded state. Protein Sci. 3 1401–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabora, J.M., Pelton, J.G., and Marqusee, S. 1996. Structure of the acid state of Escherichia coli ribonuclease HI. Biochemistry 35 11951–11958. [DOI] [PubMed] [Google Scholar]
- Delaglio, F., Grzesiek, S., Vuister, G.W., Zhu, G., Pfeifer, J., and Bax, A. 1995. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6 277–293. [DOI] [PubMed] [Google Scholar]
- Englander, S.W. 2000. Protein folding intermediates and pathways studied by hydrogen exchange. Annu. Rev. Biophys. Biomol. Struct. 29 213–238. [DOI] [PubMed] [Google Scholar]
- Goedken, E.R., Keck, J.L., Berger, J.M., and Marqusee, S. 2000. Divalent metal cofactor binding in the kinetic folding trajectory of Escherichia coli ribonuclease HI. Protein Sci. 9 1914–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedken, E.R. and Marqusee, S. 2001. Co-crystal of Escherichia coli RNase HI with Mn2+ ions reveals two divalent metals bound in the active site. J. Biol. Chem. 276 7266–7271. [DOI] [PubMed] [Google Scholar]
- Horovitz, A. and Fersht, A.R. 1990. Strategy for analysing the co-operativity of intramolecular interactions in peptides and proteins. J. Mol. Biol. 214 613–617. [DOI] [PubMed] [Google Scholar]
- Huyghues-Despointes, B.M., Langhorst, U., Steyaert, J., Pace, C.N., and Scholtz, J.M. 1999. Hydrogen-exchange stabilities of RNase T1 and variants with buried and solvent-exposed Ala → Gly mutations in the helix. Biochemistry 38 16481–16490. [DOI] [PubMed] [Google Scholar]
- Jones, T., Zhou, J., Cowan, S., and Kjeldgaard, M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. 47 110–119. [DOI] [PubMed] [Google Scholar]
- Lamzin, V. and Wilson, K. 1993. Automated refinement of protein models. Acta Crystallogr. 49 129–147. [DOI] [PubMed] [Google Scholar]
- Llinas, M., Gillespie, B., Dahlquist, F.W., and Marqusee, S. 1999. The energetics of T4 lysozyme reveal a hierarchy of conformations. Nat. Struct. Biol. 6 1072–1078. [DOI] [PubMed] [Google Scholar]
- Miller, D.W. and Dill, K.A. 1995. A statistical mechanical model for hydrogen exchange in globular proteins. Protein Sci. 4 1860–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov, G., Vagin, A., and Dodson, E. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. 49 240–255. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276 307–326. [DOI] [PubMed] [Google Scholar]
- Raschke, T.M., Kho, J., and Marqusee, S. 1999. Confirmation of the hierarchical folding of RNase H: A protein engineering study. Nat. Struct. Biol. 6 825–831. [DOI] [PubMed] [Google Scholar]
- Raschke, T.M. and Marqusee, S. 1997. The kinetic folding intermediate of ribonuclease H resembles the acid molten globule and partially unfolded molecules detected under native conditions [published erratum appears in Nat. Struct. Biol. 1997 Jun;4(6):505]. Nat. Struct. Biol. 4 298–304. [DOI] [PubMed] [Google Scholar]
- Santoro, M.M. and Bolen, D.W. 1988. Unfolding free energy changes determined by the linear extrapolation method. 1. Unfolding of phenylmethanesulfonyl alpha-chymotrypsin using different denaturants. Biochemistry 27 8063–8068. [DOI] [PubMed] [Google Scholar]
- Wooll, J.O., Wrabl, J.O., and Hilser, V.J. 2000. Ensemble modulation as an origin of denaturant-independent hydrogen exchange in proteins. J. Mol. Biol. 301 247–256. [DOI] [PubMed] [Google Scholar]
- Xu, Y., Mayne, L., and Englander, S.W. 1998. Evidence for an unfolding and refolding pathway in cytochrome c. Nat. Struct. Biol. 5 774–778. [DOI] [PubMed] [Google Scholar]