

Abstract
It is well known that ultraviolet (UV) radiation may reduce or even abolish the biological activity of proteins and enzymes. UV light, as a component of sunlight, is illuminating all light-exposed parts of living organisms, partly composed of proteins and enzymes. Although a considerable amount of empirical evidence for UV damage has been compiled, no deeper understanding of this important phenomenon has yet emerged. The present paper presents a detailed analysis of a classical example of UV-induced changes in three-dimensional structure and activity of a model enzyme, cutinase from Fusarium solani pisi. The effect of illumination duration and power has been investigated. A photon-induced mechanism responsible for structural and functional changes is proposed. Tryptophan excitation energy disrupts a neighboring disulphide bridge, which in turn leads to altered biological activity and stability. The loss of the disulphide bridge has a pronounced effect on the fluorescence quantum yield, which has been monitored as a function of illumination power. A general theoretical model for slow two-state chemical exchange is formulated, which allows for calculation of both the mean number of photons involved in the process and the ratio between the quantum yields of the two states. It is clear from the present data that the likelihood for UV damage of proteins is directly proportional to the intensity of the UV radiation. Consistent with the loss of the disulphide bridge, a complex pH-dependent change in the fluorescence lifetimes is observed. Earlier studies in this laboratory indicate that proteins are prone to such UV-induced radiation damage because tryptophan residues typically are located as next spatial neighbors to disulphide bridges. We believe that these observations may have far-reaching implications for protein stability and for assessing the true risks involved in increasing UV radiation loads on living organisms.
Keywords: tryptophan fluorescence lifetime, fluorescence quenching, disulphide (disulfide) bridges, photochemical reaction, protein structure damaged by UV light, SS bond disruption, indole, theoretical model
Two divergent theories of the mechanisms involved in ultraviolet (UV) inactivation of enzymes have been developed over a period of years. One theory proposes that the random destruction of any amino acid residue causes inactivation (Augenstein and Riley 1964). The second emphasizes the importance of the disruption of a cluster of specific cystine residues (cysteines involved in disulphide bridges; Augenstein and Riley 1964). It was also found that the effective destruction of cystine, tryptophan, tyrosine, and phenylalanine occurs on UV irradiation of proteins (Kazutomo 1955; Augenstine and Ghiron 1961; McLaren and Luse 1961; Hideko and Kazutomo 1963; Dose 1964; Vladimirov and Roshchupkin 1964; Cooper and Davidson 1965; Fujimoro 1966; Piras and Vallee 1966; Vladimirov et al. 1970). The photosensitivity of protein-bound cystine residues has been shown to depend on their microenvironment (Augenstein and Ghiron 1961; Dose and Rajewsky 1962; Augenstein and Riley 1964; Dose 1964, 1967; Fiore and Dose 1965; Grist et al. 1965; Risi et al. 1967; Koudelka and Augenstein 1968). This correlates with the observation by Petersen et al. (1999) that cystine residues (cysteines involved in disulphide bridges) have a clear preference for aromatic residues as spatial neighbors. There is evidence that quanta primarily absorbed in the side-chains of aromatic amino acid residues of a given protein molecule contribute to the destruction of the cystine residues (Dose 1967; Risi et al. 1967; Koudelka and Augenstein 1968). It has been reported that the UV illumination of Trp and Tyr residues gives rise to electron or H-atom ejection from these amino acids (Vladimirov et al. 1970; Feitelson 1971). The role of the hydrated electron in photo-reduction of cystine in the presence of indole led to the proposal of a possible mechanism behind this phenomenon (Grossweiner and Usui 1970).
It is of major interest to describe and to better understand what UV light can induce in the structure and function of proteins. A partial answer on the effect of UV light on the fungal lipase/esterase cutinase from Fusarium solani pisi has been provided by Weisenborn et al. (1996) and Prompers et al. (1999). Weisenborn et al. (1996) reported that the fluorescence quantum yield of cutinase increases with the illumination time of cutinase, and that the short lifetime component, ∼40 ps, observed for Trp before extended illumination is substituted by longer lifetimes on prolonged UV irradiation of cutinase. The UV illumination has been reported to not affect cutinase lipolytic activity, in contradiction with our findings. The disruption by UV light of the disulphide bridge nearby the only Trp residue in cutinase has been correlated by Prompers et al. (1999), with the UV excitation of this aromatic residue. Because disulphide bridges are excellent quenchers of the excited state of aromatic residues, once disrupted by UV light, the above-mentioned disulphide bridge could no longer quench the Trp residue. Consistently, we have detected free SH groups (absent in nonirradiated native cutinase) on UV irradiation of cutinase, in amounts proportional to the increase of the fluorescence intensity.
In the present paper and for the first time, we present a novel physical model describing the change in the fluorescence quantum yield of the single Trp residue of cutinase as a function of excitation light power and illumination time. This simplified model allows us to correlate disruption of the disulphide bridge with the number of absorbed photons by the tryptophan residue. In conclusion, we were able to calculate the probability of the disruption process per absorbed quanta. Study of light power (intensity) dependence shows that probability is power independent and that observed disruption process is a statistical phenomenon. The dependence of the melting temperature of cutinase on the illumination time has been quantified, as well as the effect of UV illumination on the amount of secondary structural elements and on its activity. A preliminary quantification of the effect of illumination time on the distribution of the fluorescence lifetimes of the single endogenous aromatic residue Trp in cutinase, at different pH values, is presented.
The physical reasons behind the breakage of the disulphide bridge on UV illumination with light at 295 nm on cutinase will be further discussed. We will discuss the peculiar observation that nature prefers to have aromatic residues near cystine residues involved in disulphide bridges (Ioerger et al. 1999; Petersen et al. 1999) and that at the same time it seems that the close proximity of Trp, and Tyr residues to disulphide bridges enhances the SS bond destruction on UV illumination.
Results
Steady-state fluorescence emission
The time-dependent fluorescence intensity of cutinase at 350 nm on illumination with 295-nm light is shown in Figure 1 ▶ (see Materials and Methods). We can observe in the first 600 sec a very rapid increase in the number of counts per second, followed by a plateau where the fluorescence yield is stable. After the plateau region displayed in Figure 1 ▶, the fluorescence quantum yield was observed to drop if we further illuminate the cutinase sample.
Fig. 1.

Fluorescence intensity of cutinase versus illumination time. Excitation occurred at 295 nm, and emission was monitored at 350 nm. Measurements were performed using a SLM 8000 spectrofluorometer with a specially designed excitation beam as described in Materials and Methods.
Before illumination, an excitation spectrum (emission set to 350 nm) and an emission spectrum (excitation at 295 nm) of fresh cutinase were recorded. Afterward, excitation and emission spectra were recorded after 5, 20, and 60 min of continuous illumination at 295 nm (see Fig. 2 ▶). Spectra are corrected for Raman contribution. It can be observed from Figure 2 ▶ that an increase in the fluorescence counts of the excitation spectra is correlated with an increase in the fluorescence signal observed in the emission spectra. From Figure 3 ▶, it can be observed that the excitation spectra become increasingly red-shifted the longer we illuminate the cutinase sample.
Fig. 2.

Effect of illumination time on excitation and emission spectra. The excitation spectra were acquired setting emission at 350 nm, and emission spectra were acquired setting the excitation at 295 nm.
Fig. 3.

Normalized excitation spectra shown in Fig. 2 ▶, showing that the excitation spectra become increasingly red-shifted the longer the illumination time.
Interestingly, when observing the ratio between the normalized excitation spectra (see Fig. 4 ▶), it can be seen that illumination of the cutinase sample at 295 nm induces spectral changes between 290 and 306 nm, at which Trp absorbs. No spectral changes are observed in the wavelength region where Tyr and Phe are known to absorb (see Fig. 4 ▶, spectral ratio before 290 nm). The normalized emission spectra show no wavelength shift (data not shown).
Fig. 4.

Ratio between the normalized spectra displayed in Fig. 3 ▶. It can be seen that illumination induces spectral changes between 290 nm and 30 6nm, where Trp absorbs. No spectral changes are observed in the wavelength region where Tyr and Phe are known to absorb.
Frequency domain intensity decay measurements
The decay times (τi) and pre-exponential factors (αi) recovered from the global analysis of the frequency domain intensity decay measurements for the 14 μM cutinase solution on different irradiation times at different pH values are given in Table 1. Figure 5 ▶ shows the experimental data at pH 8.5 and the fit assuming four lifetime decays. The value of χ2 dropped significantly when progressing from a one lifetime component fit to two components, to three components, and even to four lifetime components. The fluorescence mean lifetime of cutinase on UV illumination at different pH values is also shown in Table 1. Assuming four lifetime components instead of three lifetime components decreases the value of the square root variance 50% to 100%. Assuming five lifetimes did not significantly lower the value of the square root variance. Therefore, four lifetimes were assumed to be consistent with our experimental data, in contrast to Weisenborn et al. (1996), who reported three lifetimes.
Table 1.
Lifetimes (τ1) and pre-exponential values (α1) of Trp from Fusarium solani pisi cutinase at pH 10.0 obtained by a nonlinear fit using the CFS software (Johnson 2000)
| Pre-exponential factors at illumination time with 295 nm light | ||||||
| 0 min | 5 min | 10 min | 15 min | 25 min | 60 min | |
| pH 4.0 tau (ns) | ||||||
| tau (ns) | ||||||
| τ1 = 5.96 | 0.01 | 0.01 | 0.02 | 0.06 | 0.10 | 0.12 |
| τ2 = 1.54 | 0.03 | 0.03 | 0.04 | 0.05 | 0.10 | 0.07 |
| τ3 = 0.33 | 0.03 | 0.02 | 0.04 | 0.06 | 0.03 | 0.07 |
| τ4 = 0.043 | 0.93 | 0.94 | 0.90 | 0.83 | 0.76 | 0.74 |
| Mean lifetime | 2.59 | 3.31 | 3.35 | 4.71 | 4.81 | 5.03 |
| pH 8.5 | ||||||
| tau (ns) | ||||||
| t1 = 4.76 | 0.01 | 0.01 | 0.02 | 0.05 | 0.10 | 0.11 |
| τ2 = 1.16 | 0.01 | 0.02 | 0.02 | 0.04 | 0.07 | 0.06 |
| τ3 = 0.12 | 0.16 | 0.10 | 0.12 | 0.11 | 0.08 | 0.12 |
| τ4 = 0.034 | 0.82 | 0.86 | 0.84 | 0.79 | 0.75 | 0.70 |
| Mean lifetime | 2.48 | 2.71 | 2.94 | 3.70 | 4.02 | 4.06 |
| pH 10.0 | ||||||
| tau (ns) | ||||||
| τ1 = 5.67 | 0.01 | 0.02 | 0.02 | 0.04 | 0.08 | 0.11 |
| τ2 = 1.49 | 0.03 | 0.03 | 0.03 | 0.05 | 0.06 | 0.06 |
| τ3 = 0.34 | 0.12 | 0.08 | 0.06 | 0.05 | 0.03 | 0.04 |
| τ4 = 0.041 | 0.85 | 0.87 | 0.89 | 0.86 | 0.83 | 0.79 |
| Mean lifetime | 2.79 | 3.01 | 3.24 | 4.096 | 4.64 | 4.88 |
The value of χ2R 2.4.
Fig. 5.

Frequency dependence of the phase shift and demodulation factors of the single endogenous Trp fluorescence of Fusarium solani pisi cutinase after illuminating cutinase with ultraviolet light. The solid lines represent the best four-exponential decays fit. Cutinase was excited by a frequency-doubled rhodamine 6G dye laser at 295 nm. Emission was monitored at 350 nm (see Materials and Methods). The protein concentration was 14 μM. Symbols refer to different illumination time before the experiment: 0 min (solid circles), 5 min (hollow triangles), 10 min (hollow circles), 15 min (solid triangles), 25 min (diamonds), and 60 min (crosses).
It can be observed in Table 1 that fresh cutinase (best approximation to 0-min illumination) displays a dominant, extremely short lifetime ranging from 34 to 43 ps, depending on pH. After illuminating the cutinase sample for 60 min with light at 295 nm, the contribution of the 43-ps species (pH 4.0) decreased from 93% to 74% (19% reduction). This effect seems to be pH dependent, because at pH 8.5 the contribution of the short-lifetime species (34 ps) decreases from 82% to 70% (12% reduction) after 60-min illumination. At pH 10, the contribution of the 41-ps lifetime species decreases from 85% to 79% (6% reduction). Figure 6 ▶ displays the drop of the short-lifetime component with the illumination time.
Fig. 6.

Dependence of the preexponential factors αi associated with the shortest lifetime component on the illumination time.
The contribution of the long components of the fluorescence decay is observed to increase on UV illumination, as observed in Table 1. The result is that the mean lifetime of Trp increases on UV illumination of cutinase (see Fig. 7 ▶). At pH 4.0, the mean lifetime of Trp increases from 2.6 to 5.0 ns; at pH 8.5, from 2.5 to 4.1 ns; and at pH 10.0, from 2.8 to 4.9 ns.
Fig. 7.

Dependence of the mean lifetime of the single Trp of cutinase on the illumination time (295 nm).
Steady-state fluorescence emission intensity measurements coupled with CD measurements
As mentioned in Materials and Methods, a 2 μM cutinase sample was irradiated with light at 295 nm for different periods of time. The fluorescence intensity of cutinase, monitored at 350 nm, after 0-, 1-, 2-, 3-, 4-, and 5-h illumination is shown in Figure 8 ▶. The relative changes in thermal stability of a 2 μM native cutinase sample exposed to light at 295 nm, using the steady-state fluorescence setup for different periods of time (see Materials and Methods), was subsequently investigated by circular dichroism (CD) spectroscopy at pH 8.5 at a scan rate of 90°C/h. Data reveals that the longer the illumination period, the lower the melting temperature of cutinase at pH 8.5. The calculated values of Tm and the spectrum half-width are shown in Table 2. The relative changes in the CD wavelength scans of cutinase at pH 8.5 and 25°C after illuminating a 4 μM native cutinase sample cutinase with light at 295 nm for different periods of time are displayed in Figure 9 ▶. Data reveals that cutinase looses secondary structural features the longer it is illuminated with 295-nm UV light.
Fig. 8.

Steady-state measurement of the fluorescence intensity of cutinase using a 75-W Xenon arc lamp (RTC 2000 spectrometer from PTI; see Materials and Methods).
Table 2.
Melting temperature (Tm) and peak half-width of a 2 μM cutinase sample (pH 8.5) determined by CD spectroscopy at a rate of 90°C/h
| Illum. time (295 nm) | Tm (°C) | Decrease Tm (°C) | % Fluorescence maximuma | Peak half-width |
| 0 h | 56.6 | — | 18 | 8 |
| 1 h | 55.6 | 1.0 | 64 | 11.6 |
| 2 h | 54.0 | 2.6 | 86 | |
| 3 h | 53.6 | 3.0 | 97 | 14.4 |
| 4 h | 52.9 | 3.7 | 100 | 16.8 |
| 5 h | 52.4 | 4.2 | 97 | 14.8 |
a Fluorescence intensity reported as the percentage of maximum intensity observed during the 5-h continuous illumination.
Fig. 9.

Circular dichroism (CD) wavelength scans of cutinase at pH 8.5 and 25°C after illuminating cutinase solution with light at 295 nm for 0, 1, and 3 h. See Table 4 for details. *The CD wavelength scans for the 2- and 4-h illuminated cutinase were also obtained. Although there are some fluctuations, the overall trend is a loss of secondary structure the longer we illuminate cutinase with 295-nm ultraviolet light.
Detection of free thiol groups with DTNB on UV illumination of cutinase
Nonilluminated native cutinase does not display free thiol groups, because all four cysteine residues are involved in disulphide bridges. As expected, no free thiol groups were detected with 5,5`-dithiobis-(2-nitrobenzoic acid) (DTNB; see Materials and Methods) in nonilluminated cutinase samples. On UV illumination of cutinase with 295-nm light, free thiol groups were detected in cutinase in a concentration proportional to the increase in fluorescence intensity, as seen in Figure 10 ▶. These results support our theoretical model, which is based on the assumptions that the highly quenched Trp initial fluorescence intensity is caused by the presence of the nearby intact disulphide bridge and that the fluorescence intensity increase of the single endogenous Trp in cutinase is caused by the cleavage of the nearby disulphide bridge on illumination of cutinase with 295-nm light.
Fig. 10.

Positive correlation between the time-dependent Trp fluorescence emission intensity increase at 350 nm on excitation with 295-nm ultraviolet light and the detected concentration of free thiol groups formed before and after ultraviolet illumination of cutinase at different ratios of fluorescence emission increase (F/Fo). Free thiols were detected on reaction with 5,5`-dithiobis-(2-nitrobenzoic acid), and free thiol concentration is proportional to the absorbance value at 412 nm (Abs 412 nm), as described in Materials and Methods.
Reduction of native cutinase with dithiothreitol and Trp fluorescence emission intensity measurement
The control confirming the correlation between the increase in Trp fluorescence emission intensity (on excitation at 295 nm) and reduction of the disulphide bridges in cutinase is displayed in Figure 11 ▶. Four emission spectra were acquired: (A) cutinase Trp emission at 25°C without dithiothreitol (DTT), (B) cutinase Trp emission at 70°C without DTT, and (C) cutinase Trp emission at 25°C without DTT after cooling down from 70°C to 25°C. After adding DTT to the 1 μM cutinase solution at 70°C, the sample was allowed to cool to 25°C, and a fourth (D) emission spectra of cutinase Trp in the presence of DTT was obtained, showing the increase in Trp emission fluorescence intensity on reducing the disulphide bridges of cutinase with DTT compared with the emission spectrum of cutinase at 25°C in the absence of DTT (spectrum A). Spectra are corrected for Raman contribution. Data shows that the emission spectrum of cutinase at 25°C is the same as the emission spectrum of cutinase after cooling it down from 70°C to 25°C; that is, the thermal unfolding at pH 8.5 was reversible. When acquiring spectra A and C, an increase of the fluorescence emission on excitation of the Trp residue of cutinase with 295 nm was observed. This phenomenon was not observed when acquiring spectra B and D (see Discussion for data interpretation).
Fig. 11.

Control confirming the correlation between the increase in Trp fluorescence emission intensity (on excitation at 295 nm) and reduction of the disulphide bridges in cutinase. Four emission spectra were obtained on excitation of a 1 μM cutinase sample with 295-nm light (RTC 2000 PTI spectrometer): (A) cutinase Trp emission at 25°C without dithiothreitol (DTT) (solid circles), (B) cutinase Trp emission at 70°C without DTT (solid black line), (C) cutinase Trp emission at 25°C without DTT after cooling down from 70°C to 25°C (hollow circles). After adding an excess of DTT to the 1 μM cutinase solution at 70°C, the sample was allowed to cool to 25°C, and a fourth (D) emission spectra of cutinase Trp in the presence of DTT (solid gray line) was obtained. All slits were set to 2-nm bandwidth.
Lipase activity assay
The specific lipolytic activity of cutinase before UV irradiation was 3056 μmole/min per mg, against tributyrin (pH 8.5) as described by Neves-Petersen et al. (2001). After exposing cutinase to light of 295 nm (second harmonic of R6G laser light) until the fluorescence intensity reached a plateau, its lipolytic activity against tributyrin (pH 8.5) dropped to 1545 μmole/min per mg (50% of the activity of fresh cutinase that has never been illuminated with UV light).
Theoretical model for two-state chemical exchange
As a result of UV light illumination, the fluorescence intensity of the protein increased >10 times. Our work reports a much faster process than that already reported by Weisenborn et al. (1996). In general, rate of fluorescence signal increase depends on excitation light intensity, protein volume, and diffusion rate (mixing). In standard experimental conditions (1-cm cuvette), the excitation volume is much smaller then total sample volume. Under such conditions, protein mixing and diffusion are main factors responsible for observed intensity increase. To overcome this problem for kinetic experiments, we have designed a special experimental setup in which the entire protein sample is illuminated with monochromatic light beam of uniform intensity. The fluorescence signal was also collected from all protein volume.
We will now describe the necessary theoretical foundation for analyzing the fluorescent yield, F(t), in a system consisting of two chemical species, A and B, that slowly interconvert.
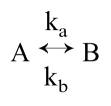 |
We will assume that ka ≫ kb and that 1/k(a or b) is long compared with the fluorescence lifetimes. The observed fluorescence is then the weighted average of the fluorescence of the two species.
Let us assume that the laser beam, which has an average power P, has been formed (expanded) to uniformly illuminate the full protein volume. One may calculate the number of photons per second, nf, which goes through the sample :
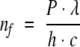 |
(1) |
where λ is the wavelength of the laser beam, h is the Planck constant, and c is the speed of light. The total number of protein molecules, nT, within the illumination volume, V, is as follows:
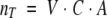 |
(2) |
where C is the molarity of the protein solution, and A is the Avogadro number.
The optical density, OD, of the protein sample at the excitation wavelength λ, is OD = ɛCl, where l is the path length, and ɛ is the molar extinction coefficient at the wavelength λ. Using the Beer-Lambert law, one may calculate the laser beam intensity change, ΔI:
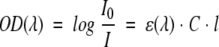 |
(3) |
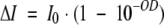 |
(4) |
where I0 is the initial intensity of the excitation beam. We also assume that the beam intensity I0 is relatively low and that ground-state depletion is negligible. Because P = I0S, where S is the illumination surface, for uniform beam and low optical density of the samples, one may calculate the number of absorbed photons per second by the sample:
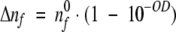 |
(5) |
The number of absorbed photons per second, Δnf, is equal to the number of excited molecules per second,
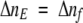 |
(6) |
At any given moment of time, t, an ensemble of molecules contains molecules with low fluorescence quantum yield, nL(t), and molecules with high fluorescence quantum yield, nH(t). Therefore, 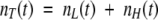 . For convenience, one may define the fraction of low, fL (t), and high, fH (t), fluorescence quantum yield:
. For convenience, one may define the fraction of low, fL (t), and high, fH (t), fluorescence quantum yield:
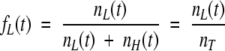 |
(7) |
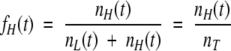 |
(8) |
and
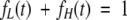 |
(9) |
Assuming that the excitation coefficient of the protein with low and high quantum yield is identical at a particular excitation wavelength (absorption spectrum is independent from the excited state quenching), one may calculate the number of molecules of low, ΔnL(t), and high, ΔnH(t), quantum yield excited at a given moment in time:
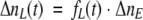 |
(10) |
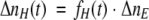 |
(11) |
In a simplified model, each molecule, after being excited, may relax to a ground state in two possible ways: nonradiative and radiative, in which the latter is emitting the photon detected as fluorescence. The observed fluorescence signal at time t will be proportional to the number of excited molecules and will be described by the following:
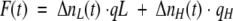 |
(12) |
or
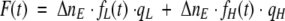 |
(13) |
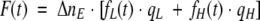 |
(14) |
where qL is the quantum yield of the molecules with low quantum yield, and qH is the quantum yield of the molecules with high quantum yield.
It is implicit that the low quantum yield species is native cutinase, and high quantum yield species is cutinase with the broken disulphide bridge.
Because
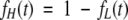 |
(15) |
then
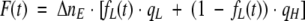 |
(16) |
We assume that before the illumination, the sample contains only molecules with low quantum yield (intact cutinase), so that at t = 0, fL0 = 1, and F(0) = ΔnE · qL.
For infinite illumination time, we assume that we have only molecules with high quantum yield, so that at t = ∞, f0H = 1 and F(∞) = ΔnE · qH.
Due to internal processes after tryptophan excitation, some of the excited low quantum yield tryptophans will convert to high quantum yield species. Let us assume that the probability of such an event is p. We can therefore describe the number of converting molecules in time by:
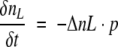 |
(17) |
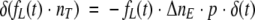 |
(18) |
or
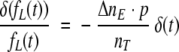 |
(19) |
Solving the above differential equation for fL(t) one will get:
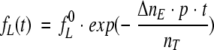 |
(20) |
Equation 20 describes the time-dependent fraction of the low quantum yield species, which according to equations 1 to 5 depend on the power of the excitation beam, P, and on the sample absorption, as shown below:
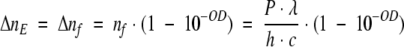 |
Substituting equation 20 in equation 16 one obtains the time dependent relative change of fluorescence intensity:
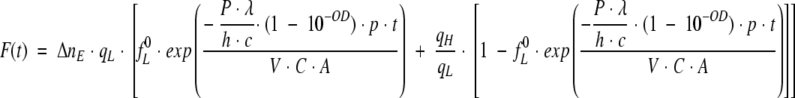 |
(21) |
Since 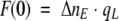 , where F(0) is the fluorescence signal for the system with only protein fraction with low quantum yield, equation 21 can be written as
, where F(0) is the fluorescence signal for the system with only protein fraction with low quantum yield, equation 21 can be written as
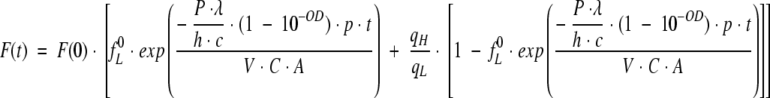 |
(22) |
Equation 22 describes the observed intensity of the fluorescence signal as a function of time, t, for a given beam power, P, and a probability of transition from low to high quantum yield species, p. We want to stress that the probability, p, represents the average number of transitions per average number of absorbed photons in a unit of time. Figure 12 ▶ shows examples of time-dependent fluorescence change F(t) for different power of the excitation light. When performing a Lavenberg-Marquardt fitting routine, the parameters we need to estimate are F(0), fL0, p, and qH/qL. Because the ratio qH/qL should correspond to the ratio between the long and the short lifetime components (∼4.5 ns versus 40 ps), we assumed qH/qL to be 110. Three experiments were performed, with three different powers of the illumination beam (see experimental setup described in Materials and Methods). The three different powers were 0.014, 0.1, and 0.14 mW. The initial fraction of the molecules with low quantum yield, fL 0, was expected to be close to 1, because the illumination time was minimal when F(t) = F(0). Lambda was set to 2.95 × 10−7 m, h is equal to 6.62 × 10−34 J.s, the speed of light is c = 3 × 108 m/s, the power in watt units, and the OD at 295 nm was set to 0.0022.
Fig. 12.

Fitted experimental kinetic traces with equation 22 obtained for three different powers of the incoming exciting beam (295 nm). Fluorescence was monitored at 350 nm. Measurements were obtained using a SLM 8000 spectrofluorometer with a specially designed excitation beam (see Materials and Methods).
The fitted parameters F(0), fL0, p, and qH/qL are displayed in Table 3.
Table 3.
Estimated parameters as a function of light power
| Power (mW) | F(0) | fL0 | P | qH/qL |
| 0.014 ± 0.002 | 90 ± 0.06 | 0.94 ± 0.001 | 0.40 ± 0.002 | 110 ± 0.01 |
| 0.1 ± 0.012 | 98 ± 0.08 | 0.92 ± 0.002 | 0.42 ± 0.001 | 110 ± 0.01 |
| 0.14 ± 0.02 | 125 ± 0.07 | 0.93 ± 0.002 | 0.38 ± 0.002 | 110 ± 0.01 |
Discussion
The fluorescence signal of the single tryptophan residue of F. solani pisi cutinase, in its native conformation, is highly quenched because of the presence of an adjacent disulphide bridge. Disulphide bridges are known to be excellent quenchers of excited-state aromatic residues. However, selective prolonged irradiation of the Trp residue at 295 nm increases its fluorescence quantum yield, in agreement with previous results by Weisenborn et al. (1996) and Prompers et al. (1999). It was also observed that on irradiation of cutinase with light at 295 nm, free thiol groups are present in cutinase (see Fig. 10 ▶), which did not exist in the native nonirradiated cutinase (F. solani pisi cutinase has four cysteine residues involved in two disulphide bridges). The destruction of the disulphide bond adjacent to the Trp residue is believed to cause the increased quantum yield of the Trp residue, because it no longer can quench the excited-state Trp residue.
Our data also shows that direct reduction of the disulphide bridge next to the Trp residue with the reducing agent DTT leads to an increase of Trp quantum yield, further supporting our initial hypothesis that the fluorescence signal of the single tryptophan residue of F. solani pisi cutinase, in its native conformation, is highly quenched because of the presence of an adjacent disulphide bridge. Data displayed in Figure 11 ▶ shows that the emission spectrum of cutinase at 25°C is the same as the emission spectrum of cutinase after cooling it down from 70°C to 25°C; that is, the thermal unfolding at pH 8.5 was reversible. When acquiring spectra A (cutinase Trp emission at 25°C without DTT) and C (cutinase Trp emission at 25°C without DTT after cooling down from 70°C to 25°C), an increase of the fluorescence emission on excitation of the Trp residue of cutinase with 295 nm was observed, confirming the close proximity between the Trp and the disulphide bridge observed in the three-dimensional structure of native cutinase on refolding. This phenomenon was not observed when acquiring spectra B (cutinase Trp emission at 70°C without DTT) and D (emission spectra of cutinase Trp in the presence of DTT after being cooled down from 70°C ro 25°C), revealing that both on unfolding and on reduction of the nearby disulphide bridge, the photon-induced mechanism is no longer observed. The unfolded cutinase at 70°C no longer has the disulphide bridge in close contact to the Trp residue, proximity necessary for the photo-induced mechanism. This is most likely the reason why the Trp fluorescence quantum yield of cutinase is higher at 70°C than at 25°C, despite the fact that we would expect a lower quantum yield because of the higher temperature (because of collisional quenching between the Trp and solvent molecules).
The present paper also quantifies the change in fluorescence response as a function of time and power of the excitation light. Linear power dependence of steady-state experiments shows statistical character of the disruption process. We have presented a theoretical model that provides a quantitative interpretation of the effect of UV light on the fluorescence quantum yield of cutinase. The longer the illumination time, the lower the number of molecules with their SS bridge adjacent to the Trp residue intact, leading to a proportional increase of the number of molecules with a broken SS bridge. The model led to the deduction of an equation that describes the change of the fluorescence intensity of cutinase versus illumination time, and such equation is dependent on the probability of the photons at 295 nm to break the bridge. When the experimental plot of the cutinase fluorescence intensity at 350 nm versus illumination time (excitation at 295 nm) is fitted with the derived equation, the probability of single photon disulphide bond breakage under our conditions is close to 40%, independent of the beam power. The probability p for this event is a general physical parameter that is independent of light intensity, beam geometry, and protein concentration. Because of significantly different experimental conditions, our results unfortunately can not be quantitatively compared with results reported by Weisenborn et al. (1996) and Prompers et al. (1999). The latter paper reports results obtained under severe inner filter effects, the former paper reports results obtained under partial illumination of the sample (i.e., diffusion related effects may distort the observations).
Because Prompers et al. (1999) reported none of the above parameters on which p depends, we can not compare their findings with ours. Also, the concentration of protein used by Prompers et al. (500 μM cutinase solution) is drastically higher then ours (2, 4, 14, and 17 μM), making comparison impossible. We believe that the data presented by Prompers et al. (1999) is distorted by serious inner filter effects, which for cutinase start at 20 μM at 280 nm and 22°C. However, all results are qualitatively consistent, indicating the same process responsible for fluorescence change.
Time-resolved experiments under continuous illumination additionally support the suggested mechanism of fluorescence increase. Four lifetime components were necessary to fit experimental data. The dominant ultrafast fluorescence component is consistent with the earlier observation of Weisenborn et al. (1996). They report only three lifetime components, which we believe is caused by lower instrumental resolution available at that time. It is observed that at pH 4, on average 93% of the population of Trp residues is characterized by a 43-ps lifetime. This extremely short lifetime, as well as the low quantum yield of cutinase, is caused by the efficient quenching of the excited-state Trp by the adjacent disulphide bridge. It is important to stress that under illumination the contribution of the 43-ps species quickly decreases. After 60-min exposure, the 43-ps fraction dropped by ∼19%. Our results also show that the fractional change seems to be pH dependent, because at pH 8.5, a 12% reduction of the contribution of the short lifetime species (34 ps) is observed after 60-min illumination, and at pH 10, the contribution of the 41-ps lifetime species decreases only 6%. We believe that the observed pH dependence has important functional meaning and requires more careful steady-state and time-resolved investigations.
It is important to note that illumination conditions for time-resolved measurements are different and cannot be compared with the steady-state experiments. In particular, the excitation beam diameter for lifetime measurement is small (∼1 mm), and the sample volume is big (∼2.5 mL). Much bigger sample volumes than beam-exposed volume and efficient stirring were necessary to maintain constant signal and fractions during time-resolved measurements.
The loss of secondary structure, thermal stability, and lipolytic activity by cutinase on UV illumination
The loss of secondary structural elements on UV illumination reveals that the overall structure of cutinase is being damaged by local phenomena between the Trp and the adjacent disulphide bridge. The lower melting temperature, Tm, of cutinase on irradiation is therefore likely to be caused by not only the disruption of one disulphide bridge but also the overall damage of structure of cutinase. The 50% loss of lipolytic activity after illumination of cutinase with 295-nm light is correlated with the overall damage of the structure of the enzyme, because the active site region and the Trp residue are located at opposite poles of the molecule. The observed drop in activity contradicts prior results by Weisenborn et al. (1996). This clearly indicates that UV illumination is also affecting the conformation of the active site, located far away from the Trp residue.
Concluding remarks
The fluorescence signal of the single tryptophan residue of F. solani pisi cutinase, in its native conformation, is highly quenched, most likely because of the presence of an adjacent disulphide bridge. Besides disulphide bridges, other interactions, for example, aromatic interactions, can also contribute to the low quantum yield of a tryptophan (Chen et al. 1996; Chen and Barkley 1998; Nanda and Brand 2000). For the first time, a novel physical model describing the change in the fluorescence quantum yield of the single Trp residue of cutinase on UV excitation as a function of excitation light power and illumination time is presented. This simplified model allows us to correlate disruption of the disulphide bridge with the number of absorbed photons by the tryptophan residue. Our model clearly shows that under our conditions, we have 40% probability of disrupting the disulphide bridge per absorbed quanta, independent of the beam power. It is also clear from the present data that the likelihood for UV damage of proteins is directly proportional to the intensity of the UV radiation.
In general, we can only conclude that the observed increase in the mean lifetime of Trp on UV illumination is most likely because of the light-induced breakage of the disulphide bridge adjacent to the Trp residue. Once broken, it can no longer quench the excited state Trp, and a long-lifetime component is observed. Gradual disappearance of τ4, the shortest lifetime component, with illumination time is caused by the conversion of the disulphide bond into SH groups.
Possibly, excited-state dipole moment of Trp on UV illumination induces a dipole moment in the adjacent SS bond (highly polarizable, a property correlated with good fluorescence quenchers), and energy can be transferred from the Trp to the SS bond. This could induce vibrational modes in the SS bond that lead to its disruption. Another possible mechanism responsible for the induced disruption of the disulphide bridge involves electron transfer from the excited-state tryptophan to the disulphide bridge (Mathews and Durley 1996; Chen and Barkley 1998). Further insight into the likely mechanism responsible for the disulphide bridge disruption is being investigated in our laboratories.
We do not rule out that the disulphide bridge adjacent to the Trp residue in cutinase itself can absorb light at 295 nm. The reason for this belief is that cyclic disulphide compounds, such as the one shown in Figure 13 ▶, have a maximum absorption at 295 nm. However, its extinction coefficient at that wavelength is 300 M−1cm−1, whereas the extinction coefficient of Trp at 295nm is ∼18 times larger (5600 M−1cm−1 at neutral pH).
Fig. 13.

Cyclic disulphide compound.
The close proximity between aromatic residues and disulphide bridges
Trp has been reported to be the preferred amino acid in the spatial vicinity of disulphide bridges, together with Tyr and Phe (Petersen et al. 1999). Why should nature place aromatic residues and disulphide bridges in such close spatial contact if, according to our knowledge in cutinase, the Trp excitation triggers the disruption of the adjacent disulphide bridge? UV light has been shown to trigger electron or H ejection from both Trp residues and Tyr residues. Such photo-reactions are likely to give rise to radical formation, which usually leads to other reactions involving those radicals and usually damages the protein structure. On the other hand, thiols are known to retard photo-degradation by interfering with the propagation steps of the photo-reactions (radical scavengers; Wayne and Wayne 1996). Disruption of the disulphide bridges is indeed providing the molecule with free thiol groups, possibly preventing further protein damage.
Materials and methods
Experimental setup used to obtained fluorescence intensity versus illumination time traces displayed in Figures 1 and 12 ▶ ▶
Steady-state measurements were performed using a SLM 8000 spectrofluorometer with the specially designed excitation beam. The excitation was provided by the frequency-doubled output of a cavity-dumped rhodamine 6G dye laser tuned at 295 nm synchronously pumped by a mode-locked argon ion laser. The laser beam was expanded by a set of mirrors and lenses, and the central uniform part of the beam was used for sample excitation. The beam powers used within this spot were measured and were ∼0.014, 0.1, and 0.14 mW (10% to 15% error). The size of the excitation beam (10 mm × 5 mm) closely matched the side surface of the sample, so that the entire protein volume was uniformly illuminated (Fig. 14 ▶). It is necessary to uniformly illuminate the full protein volume to avoid the secondary processes like diffusion or mixing (stirring), which lead to sample exchange within the excitation volume. Steady-state fluorescence measurements were performed under the magic angle conditions.
Fig. 14.

Experimental setup used to obtain fluorescence intensity versus illumination time traces displayed in Figs. 1 and 12 ▶ ▶. The size of the excitation beam closely matched the side surface of the sample, so that the entire protein volume was uniformly illuminated.
Because of meniscus effect on the sample surface we have some inaccuracy for the absolute power determination (10% to 15% uncertainty). A coherent digital power meter was used to measure the power of the incoming beam. For measurements, 10-mm × 4-mm cuvettes were used, and the sample height was ∼3 mm. As shown in Figure 14 ▶, the sample was illuminated along the 4-mm pathway, and the fluorescence was observed along the 10-mm pathway. The sample temperature was stabilized at 18°C, and it was constant during the experiment, independent of the excitation power. Under such conditions, sample stirring made no difference, and the sample maintained its homogeneity during the experiment.
Time-resolved fluorescence measurements
Time-resolved fluorescence measurements were performed under the magic angle conditions. Time-resolved fluorescence measurements were made using a frequency-domain 10-GHz fluorometer equipped with Hammatsu 6-μm microchannel plate detector (MCP-PMT) as previously described (Laczko et al. 1990). The instrument covered a wide frequency range (4 to 5000 MHz), which allowed detection of lifetimes ranging from several nanoseconds to a few picoseconds. Samples were placed in a 10-mm pathlength cuvette. The excitation was provided by the frequency-doubled output of a cavity-dumped rhodamine 6G dye laser tuned at 295 nm synchronously pumped by a mode-locked argon ion laser. Sample emission was filtered through the Oriel interference filter centered at 340 nm of Corning 7 to 60 broadband filter. For the reference signal, we used the scatter of the sample filtered through an Oriel interference filter at 289 nm, together with neutral density filters. Because observed tryptophan fluorescence lifetimes of cutinase protein contain very short picosecond components, the filters used for emission and reference have to be calibrated to identical optical length at 295 and 340 nm. To measure short lifetime properly, it is important to have well-defined excitation spots to avoid geometrical artefacts (Gryczynski and Bucci 1993). For this reason and to slow down protein degradation, the laser beam was expanded to 1 to 2 mm square.
The governing equations for the time-resolved intensity decay data were assumed to be a sum of discrete exponentials as in the following:
 |
where I(t) is the intensity decay, αi is the amplitude (preexponential factor), τi the fluorescence lifetime of the i-th discrete component, and Σ αi = 1.0. The fractional intensity fi of each decay time is given by
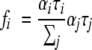 |
and the mean lifetime is
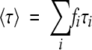 |
In the frequency domain, the measured quantities at each frequency ω are the phase shift (in degrees) and demodulation factor (m) of the emitted light versus the reference light. Amplitudes and lifetime parameters were calculated by a nonlinear least square fit using the CFS software (Johnson 2000). For calculations of the goodness-of-fit parameter χ2R, the uncertainties in the differential phase angles and modulation ratios were assumed to be 0.2° and 0.005, respectively, during the nonlinear least square analysis.
Experimental setup used to obtain the data presented in the Steady-State Fluorescence Emission Intensity Measurements Coupled with CD Measurements section
The far-UV region of CD spectrum provides a qualitative measure of the average secondary structure content. Therefore, CD was used to study the relative changes in thermal stability of cutinase at pH 8.5 after illumination of cutinase with UV light for different time periods. Three milliliters of a 2 μM protein stock solution was illuminated in a quartz macro cuvette (1-cm pathlength) for different time periods: 0, 1, 2, 3, 4, and 5 h. Continuous illumination at 295 nm was performed using a 75-W Xenon arc lamp from a RTC 2000 PTI spectrometer equipped with a thermostated cuvette holder. The 295-nm excitation wavelength was provided by a Xenon arc lamp coupled with a monochromator. The sample was continuously magnetically stirred at 700 rpm to maintain the homogeneity of the solution, and the temperature was kept constant at 25°C. Excitation and emission slits were set at 6 nm. The fluorescence intensity of cutinase at 350 nm was monitored throughout 5 h of illumination. Before illumination, the melting temperature of cutinase was determined using CD spectroscopy (see description below). After 1, 2, 3, 4, and 5 h of illumination at 295 nm, the melting temperature of cutinase was checked once more with CD.
The experimental parameters used for acquiring standard CD spectra (wavelength scan at 25°C and temperature scan at the fixed wavelength of 223 nm) are listed in Table 4. Seven hundred microliters of 2 μM irradiated sample was placed in a quartz microcuvette, with a pathlength of 0.1 cm to determine the melting temperature of cutinase as a function of illumination time. Two hundred fifty microliters of 4 μM irradiated sample was placed in a quartz microcuvette, with a pathlength of 0.1 cm to check the amount of secondary structural features (CD wavelength scan) of cutinase as a function of illumination time. Each measurement was controlled by the JASCO J-700 hardware manager. Analysis was performed using the software program JASCO J-700 for Windows standard analysis and JASCO J-700 for Windows temperature scan analysis.
Table 4.
Experimental parameters for the standard CD spectra of a wavelength scan at 25°C and temperature scan at 223 nm
| Parameters | Wave scan | Temperature scan |
| Mode | CD/HT | CD/HT |
| Range | 300–195 nm | 20–80°C (223 nm, 90°C/h) |
| Band width (nm) | 1.0 | 5.0 |
| Resolution | 0.1 | 0.2 |
| Accumulation | 6 | 1 |
| Speed (nm/min) | 50 | — |
| Sensitivity (mdeg) | 200 | 20 |
| Response (sec) | 1 | 2 |
| Buffer | 20 mM Tris (pH 8.5) | 20 mM Tris (pH 8.5) |
| Sample | 4 μM Cutinase | 2 μM cutinase |
| Test | 0-, 1-, 2-, 3-, 4-h irradiation | 0-, 1-, 2-, 3-, 4-, 5-h irradiation |
The obtained CD traces as a function of temperature were fitted to the equations described in Otzen et al. (1999) to extract Tm.
Detection of free thiol groups with DTNB on UV illumination of cutinase
Detection and quantification of the protein thiol groups was performed with the spectrophotometric assay based on the reaction of thiol groups with DTNB (or Ellman's reagent; Ellman 1959; Hu 1994; Riddles et al. 1983). For the purpose, 3 mL of a 17.3 μM protein solution in 20 mM Tris-HCl (pH 8.5) was illuminated with 295-nm light, in a quartz macrocuvette (1-cm pathlength) for different time periods using a RTC 2000 PTI spectrometer, as described in the previous section. The time-dependent fluorescence emission intensity at 350 nm was acquired (see Fig. 10 ▶). All slits were set to 2-nm bandwidth. An excess of DTNB (100 μL of a 8.5 mM stock solution of DTNB in absolute methanol) was added to 900 μL of cutinase solution, before and after illumination of cutinase with 295-nm light. The DTNB stock in methanol is stable up to 2 wk at 4°C (Hu 1994). The absorbance at 412 nm of the released nitrotehiobenzoate ion (ɛ412nm = 13,600 M−1cm−1) was measured with a UV/Visible Pharmacia spectrophotometer immediately after mixing the two components, and after 20-min and 24-min reaction times. The readings were stable between 20 and 24 min. Each data point is an average of three measurements after 24 min of mixing time at 25°C. The absorbance at 412 nm, proportional to the amount of thiol groups released by the photo-induced reaction, was measured as a function of the fluorescence intensity increase observed on different illumination times, as described above (see Fig. 10 ▶). As blank, 100 μL of an 8.5 mM stock solution of DTNB in absolute methanol was added to 900 μL of nonirradiated cutinase (17.3 μM cutinase solution in 20 mM Tris-HCl at pH 8.5). The concentration of illuminated cutinase sample had to be higher than that in the previously described experiments to secure that enough free thiol group was formed on illumination to be detected by the DTNB method.
Reduction of native cutinase with DTT and Trp fluorescence emission intensity measurement
The control confirming the correlation between the increase in Trp fluorescence emission intensity (on excitation at 295 nm) and reduction of the disulphide bridges in the protein was performed by measuring Trp fluorescence emission on reduction of the disulphide bridges of cutinase with the reducing agent DTT. Because cutinase, when folded, can not be directly reduced by DTT because the disulphide bridges are not solvent accessible, we had to facilitate the access of DTT to the disulphide bridges by warming up the solution of the protein. To avoid precipitation of the protein on heating, the control was performed with 650 μL of a dilute 1 μM cutinase solution in 20 mM Tris-HCl (pH 8.5). The solution was heated from 25°C to 70°C, and excess DTT was added (8.5 μL from a 3.7 M stock of DTT in 20 mM Tris-HCl at pH 8.5). The final concentration of DTT was therefore 50 mM.
Four emission spectra were obtained (RTC 2000 PTI spectrometer, slits set up at 2-nm bandwidth): (A) cutinase Trp emission at 25°C without DTT, (B) cutinase Trp emission at 70°C without DTT, and (C) cutinase Trp emission at 25°C without DTT after cooling down from 70°C to 25°C. After adding DTT to the 1 μM cutinase solution at 70°C, the sample was allowed to cool to 25°C, and a fourth (D) emission spectra of cutinase Trp in the presence of DTT was obtained (see Fig. 11 ▶).
Preparation of buffers
Tris-HCl (20 mM) was used at pH 8.5. This buffer displays a minimal pH drift in relation to temperature and volume changes when performing thermal denaturation experiments. Tris-HCl has minimal enthalpy of ionization. For the frequency domain intensity decay measurements, 20 mM sodium acetate buffer was used at pH 4.0; 20 mM glycine buffer was used at pH 10.0.
Preparation of protein samples
Protein samples of 2 μM and 4 μM were prepared in 20 mM Tris-HCl buffer (pH 8.5) to obtain the data presented in the Steady-State Fluorescence Emission Intensity Measurements Coupled With CD Measurements section. Cutinase samples (17.3 μM) were prepared in 20 mM Tris-HCl buffer (pH 8.5), and 14 μM protein samples were prepared in 20 mM buffer (pH 4.0, 8.5, and 10) before obtaining the kinetic traces displayed in Figures 1 and 12 ▶ ▶. OD was measured at 280 nm, and concentration was estimated using the extinction coefficient of cutinase at 280 nm (13,500 M−1cm−1).
Lipase activity assay and preparation of protein samples for the lipase activity assay and protein concentration determination
Lipase activity assay and sample preparation procedures are described in Neves-Petersen et al. (2001).
F. solani pisi cutinase expression and purification
The expression system and the purification procedure are described in Petersen et al. (1998).
Acknowledgments
M.T.N-P. acknowledges the support from the European Commission, TMR grant, Marie Curie Program, contract No. ERBFMBICT972574; the Danish Research Agency; Novo Nordisk A/S; and Novozymes A/S. S.B.P. thanks the Nordjyllands Energifond, the Obelsk Familiefond, and Mål-2 for their generous support. Z.G. and J.L. acknowledge the support from the National Institutes of Health (RR-08119).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.06002.
References
- Augenstein, L.G. and Ghiron, C.A. 1961. The inactivation of trypsin by ultraviolet light, I: The correlation of inactivation with the disruption of constituent cystine. Proc. Natl. Acad. Sci. 47 1530–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augenstein, L. and Riley, P. 1964. The inactivation of enzymes by ultraviolet light, IV: The nature and involvement of cystine disruption. Photochem. Photobiol. 3 353–367. [Google Scholar]
- Chen, Y. and Barkley, M.D. 1998. Toward understanding tryptophan fluorescence in proteins. Biochemistry 37 9976–9982. [DOI] [PubMed] [Google Scholar]
- Chen, Y., Liu, B., Yu, H.-T., and Barkley, M.D. 1996. The peptide bond quenches indole fluorescence. J. Am. Chem. Soc. 118 9271–9278. [Google Scholar]
- Cooper, D.R. and Davidson, R.J. 1965.The effect of ultraviolet irradiation on soluble collagen. Biochem. J. 97 139–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dose, K. 1964. Die Rolle der Disulfidbrücken bei der Inaktivierung von Porteinen durch Strahlung. Biophysik 1 316–324. [Google Scholar]
- ———. 1967. Theoretical aspects of the UV inactivation of proteins containing disulphide bonds. Photochem. Photobiol. 6 437–443. [DOI] [PubMed] [Google Scholar]
- Dose, K. and Rajewsky, B. 1962. Photochemistry of sulphur containing amino acids and peptides. Photochem. Photobiol. 2 181–189. [Google Scholar]
- Ellman, G.G. 1959. Tissue sulphydryl groups. Arch. Biochem. Biophys. 82 70–77. [DOI] [PubMed] [Google Scholar]
- Feitelson, J. 1971. The formation of hydrated electrons from the excited state of indole derivatives. Photochem. Photobiol. 13 87–96. [Google Scholar]
- Fiore, C. and Dose, K. 1965. Biological chemical effects of ultraviolet light on insulin. Biophysik 2 340–346. [PubMed] [Google Scholar]
- Fujimoro, E. 1966. Ultraviolet light irradiated collagen macromolecules. Biochemistry 5 1034–1040. [DOI] [PubMed] [Google Scholar]
- Grist, K., Taylor, T., and Augenstein, L. 1965. The inactivation of enzymes by ultraviolet light, V: The disruption of specific cystines in ribonuclease. Radiat. Res. 26 198–210. [PubMed] [Google Scholar]
- Grossweiner, L.I. and Usui, Y. 1970. Flash photolysis and inactivation of aqueous lysozyme. Photochem. Photobiol. 13 195–214. [DOI] [PubMed] [Google Scholar]
- Gryczynski, Z. and Bucci, E. 1993. Design and application of a new optical cell for measuring weak fluorescent emission with time resolution in the picosecond time scale. Biophys. Chem. 48 31–38. [DOI] [PubMed] [Google Scholar]
- Hideko, F. and Kazutomo, I. 1963. Studies on ultraviolet irradiation of carboxypeptidase A. J. Biochem. 53(5): 20341–347. [DOI] [PubMed]
- Hu, M.L. 1994. Measurement of protein thiol groups and glutathione in plasma. Meth. Enzymol. 233 380–385. [DOI] [PubMed] [Google Scholar]
- Ioerger, T.R., Du, C., and Linthicum, D.S. 1999. Conservation of cys-cys trp structural triads and their geometry in the protein domains of immunoglobulin superfamily members. Mol. Immunol. 36 373–386. [DOI] [PubMed] [Google Scholar]
- Johnson, M.L. 2000. Non-linear least-squares program, University of Virginia, Charlotesville, VA.
- Kazutomo, I. 1955. The ultraviolet inactivation of pepsin. Biochim. Biophys. Acta 18 216–220. [DOI] [PubMed] [Google Scholar]
- Koudelka, J. and Augenstein, L. 1968. The importance of the microenvironment surrounding a chromophore in determining its spectroscopic behaviour. Photochem. Photobiol. 7 613–617. [Google Scholar]
- Lackzo, G., Gryczyniski, I., Wiczk, W., Malak, H., and Lakowicz, J.R. 1990. A 10-GHz frequency-domain fluorometer. Rev. Sci. Instrum. 61 2331–2337. [Google Scholar]
- Mathews, F.S. and Durley, R.C.E. 1996. Structure of electron transfer proteins and their complexes. In Protein electron transfer (D.S. Bendall, ed.). Bios Scientific Publishers, Oxford, U.K., pp. 99–123.
- McLaren, A.D. and Luse, R.A. 1961. Mechanisms of inactivation of enzyme proteins by ultraviolet light. Science 134 836–837. [DOI] [PubMed] [Google Scholar]
- Nanda, V. and Brand, L. 2000. Aromatic interactions in homeodomains contribute to the low quantum yield of a conserved, buried tryptophan. Proteins 40 112–125. [DOI] [PubMed] [Google Scholar]
- Neves-Petersen, M.T., Petersen, E., Fojan, P., Noronha, M., Madsen, R.G., and Petersen, S.B. 2001. Engineering the pH-optimum of a triglyceride lipase: From predictions based on electrostatic computations to experimental results. J. Biotechnol. 87 225–254. [DOI] [PubMed] [Google Scholar]
- Otzen, D.E., Kristensen, O., Proctor, M., and Oliveberg, M. 1999. Structural changes in the transition state of protein folding: Alternative interpretations of curved chevron plots. Biochemistry 38 6499–6511. [DOI] [PubMed] [Google Scholar]
- Petersen, S.B., Jonson, P.H., Fojan, P., Petersen, E.I., Petersen, M.T.N., Hansen, S., Ishak, R.J., and Hough, E. 1998. Protein engineering the surface of enzymes. J. Biotechnol. 66 11–26. [DOI] [PubMed] [Google Scholar]
- Petersen, M.T.N., Jonson, P.H., and Petersen, S.B. 1999. Detailed analysis of the amino acid neighbours and conformation of cysteines in proteins. Protein Eng. 12 535–548. [DOI] [PubMed] [Google Scholar]
- Piras, R., and Vallee, B.L. 1966. Effect of ultraviolet irradiation on composition and function of carboxypeptidase A. Biochemistry 5 849–54. [DOI] [PubMed] [Google Scholar]
- Prompers, J.J., Hilbers, C.W., and Pepermans, H.A.M. 1999. Tryptophan mediated photoreduction of disulphide bond causes unusual fluorescence behavior of Fusarium solani pisi cutinase. FEBS Lett. 45:6 409–16. [DOI] [PubMed] [Google Scholar]
- Riddles, P.W., Blakeley, R.L., and Zerner, B. 1983. Reassessment of Ellman's reagent. Meth. Enzymol. 91 49–60. [DOI] [PubMed] [Google Scholar]
- Risi, S., Dose, K., Rathinasamy, T.K., and Augenstein, L. 1967. The effect of environment on cystine disruption by ultraviolet light. Photochem. Photobiol. 6 423–36. [DOI] [PubMed] [Google Scholar]
- Vladimirov, Y.A and Roshchupkin, D.I. 1964. Study of primary photochemical processes in proteins, 1: Two photochemical reactions in aromatic amino acids. Biofizika 9 282–292. [PubMed] [Google Scholar]
- Vladimirov, Y.A., Roshchupkin, D.I., and Fesenko, E.E. 1970. Photochemical reactions in amino acid residues and inactivation of enzymes during UV irradiation: A review. Photochem. Photobiol. 11 227–246. [DOI] [PubMed] [Google Scholar]
- Wayne, C.E. and Wayne, R.P. 1996. Photochemistry. Oxford University Press, New York. NY.
- Weisenborn, P.C.M., Meder, H., Egmond, M.R., Visser, T.J.W.G., and van Hoek, A. 1996. Photophysics of the single tryptophan residue in Fusarium solani Cutinase: Evidence for the occurrence of conformational substates with unusual fluorescence behaviour. Biophysic. Chem. 58 281–288. [DOI] [PubMed] [Google Scholar]