

Abstract
A variety of techniques, including high-pressure unfolding monitored by Fourier transform infrared spectroscopy, fluorescence, circular dichroism, and surface plasmon resonance spectroscopy, have been used to investigate the equilibrium folding properties of six single-domain antigen binders derived from camelid heavy-chain antibodies with specificities for lysozymes, β-lactamases, and a dye (RR6). Various denaturing conditions (guanidinium chloride, urea, temperature, and pressure) provided complementary and independent methods for characterizing the stability and unfolding properties of the antibody fragments. With all binders, complete recovery of the biological activity after renaturation demonstrates that chemical-induced unfolding is fully reversible. Furthermore, denaturation experiments followed by optical spectroscopic methods and affinity measurements indicate that the antibody fragments are unfolded cooperatively in a single transition. Thus, unfolding/refolding equilibrium proceeds via a simple two-state mechanism (N⇋U), where only the native and the denatured states are significantly populated. Thermally-induced denaturation, however, is not completely reversible, and the partial loss of binding capacity might be due, at least in part, to incorrect refolding of the long loops (CDRs), which are responsible for antigen recognition. Most interestingly, all the fragments are rather resistant to heat-induced denaturation (apparent Tm = 60–80°C), and display high conformational stabilities (ΔG(H2O) = 30–60 kJ mole−1). Such high thermodynamic stability has never been reported for any functional conventional antibody fragment, even when engineered antigen binders are considered. Hence, the reduced size, improved solubility, and higher stability of the camelid heavy-chain antibody fragments are of special interest for biotechnological and medical applications.
Keywords: Camel heavy-chain antibodies, protein stability, protein folding, circular dichroism, fluorescence, Fourier transform infrared spectroscopy, surface plasmon resonance, high pressure
Antibodies and their derivative fragments have long been used as tools in a variety of applications, in fundamental research work, biotechnology, diagnosis, and even human therapy. Not surprisingly, immunoglobulins constitute at least 25% of the proteins in clinical trials (Hudson 1998; Glennie and Johnson 2000). Utilization of antibodies as drug delivery vehicles, or as triggers for human immune response in cancer therapy, are clearly successful applications (Green et al. 2000). Antibodies might also become useful in the treatment of amyloidosis associated with a range of debilitating conditions such as Alzheimer's and Creutzfeldt-Jakob diseases. Monoclonal antibodies can prevent the in vitro aggregation of the Alzheimer β-amyloid peptide, and also induce the solubilization of its aggregated pathological component (Solomon et al. 1996, 1997). For most applications, high-yield production, solubility, stability, and small size (when efficient biodistribution or reduced immunogenicity is required) are critical factors. Thus, many attempts to reduce the size of the conventional heterotetrameric IgG molecule (Mr ∼160 kD), while retaining its antigen-binding properties, have been reported. This resulted in a series of antibody fragment constructs, such as Fabs (Better et al. 1988), Fvs (Skerra and Plückthun 1988), scFvs (Bird et al. 1988), dsFvs (Reiter et al. 1996), and even single-domain VHs (Ward et al. 1989; Cai and Garen 1996), which can be expressed in E. coli, yeast (Horwitz et al. 1988) or myeloma cells (Riechmann et al. 1988).
Camels, dromedaries, and llamas (camelids) generate antibodies formed by two heavy chains, but no light chains (Hamers-Casterman et al. 1993). These immunoglobulins (Mr ∼95 kD), referred to as heavy-chain antibodies, constitute a major fraction of the functional antibodies in the serum of camelids (up to ∼50% in dromedaries). Refined structural changes (Muyldermans et al. 1994; Muyldermans and Lauwereys 1999; Muyldermans et al. 2001) in the variable domain of the naturally occurring heavy-chain antibodies (referred to as VHH) compensate for the absence of association with the light chain variable domain.
Following the immunization of dromedaries (Ghahroudi et al. 1997) and llamas (Frenken et al. 2000), recombinant antibody fragments (VHHs) can be isolated, which consist of a single domain only (118–136 residues). The X-ray structures of several of these minimum-sized antigen binders directed against various haptens or proteins are now available (Desmyter et al. 1996; Spinelli et al. 1996, 2000; Decanniere et al. 1999; Muyldermans et al. 2001). The VHH scaffold adopts the common immunoglobulin fold of conventional variable domains (VH), but the antigen-binding loops (CDRs) often deviate from the predicted canonical structures (Decannierre et al. 2000). The modifications in the VHH domain that compensate for the absence of a VL domain can be seen. In particular, three hydrophobic residues at positions 44, 45, and 47 (the Kabat numbering [Kabat et al. 1991] is used throughout the text) of the VHH surface, which interacts with the VL in conventional antibodies, are substituted by more hydrophilic amino acids. The single-domain VHH antibody fragments display unique properties (Muyldermans and Lauwereys 1999; Muyldermans et al. 2001), including their reduced size, good solubility and stability. They display a high level of specificity and affinity for their antigens (Lauwereys et al. 1998), with values of the dissociation constant (KD) in the nanomolar range, which is very similar to the affinity of most conventional antibodies. Remarkably, it appears that a significant fraction of heavy-chain antibodies raised against enzymes interact directly with the active site (Lauwereys et al. 1998; Conrath et al. 2001), indicating that the catalytic cleft of an enzyme is immunodominant for this class of immunoglobulins (Muyldermans and Lauwereys 1999). Thus, camelid antibodies recognize novel epitopes, such as enzyme active sites, that are not accessible to classical antibodies because of the size of the VH-VL binding site (Lauwereys et al. 1998; Transue et al. 1998). The enzyme inhibitory properties of VHHs offer high expectations for biotechnological and medical applications (Riechmann and Muyldermans 1999; Muyldermans 2001).
VHH fragments have been reported to be more stable than most conventional antibody fragments (Ghahroudi et al. 1997), even at temperatures as high as 90°C (van der Linden et al. 1999), and thermal unfolding was shown to be reversible (Perez et al. 2001), which also contrasts with conventional antibody fragments. No detailed information on the conformational stability of these fragments have, however, been reported to date. In the present work, the denaturant-induced unfolding transitions of six VHHs, elicited from dromedaries or llamas, were studied by using a combination of spectroscopic techniques and affinity measurements. A variety of denaturants were used, including chemicals (GdmCl and urea), temperature, and pressure. This survey gives insights into the mechanism of equilibrium unfolding, and provides independent estimates of the thermodynamic parameters.
Results
Characterization of cAb-HuL6 and cAb-NmcA2
Two new VHHs, cAb-HuL6 and cAb-NmcA2 (Fig. 1 ▶), with specificity for human lysozyme and the NmcA β-lactamase, respectively, have been selected from libraries of VHH genes cloned from immunized dromedaries. Following expression in E. coli, these antibody fragments were purified to homogeneity, together with four VHHs previously characterized, that is, cAb-Lys3, cAb-TEM2, cAb-BcII10, and cAb-R2, respectively, with hen lysozyme, TEM-1, and BcII β-lactamases, and azo dye RR6 specificity. Thus, five out of six VHHs have specificities for proteins, whereas cAb-R2 is a hapten binder. The kinetic rate constants for antigen–antibody association (ka) and dissociation (kd) were determined using an SPR detection system (BIAcore). The ka values are 8.6 × 105 M−1s−1 and 65 × 105 M−1s−1 for cAb-HuL6 and cAb-NmcA2, respectively, whereas the corresponding kd values are 5.9 × 10−4 s−1 and 21 × 10−4 s−1. Similar measurements were also performed with cAb-BcII10, cAb-Lys3, cAb-R2, and cAb-TEM2, yielding the values listed in Table 1. These are in reasonable agreement with those obtained using different equipment (IAsys), or by ELISA (Desmyter et al. 1996). The two newly isolated antigen binders display high affinities (i.e., low dissociation constants; Table 1) towards their respective antigens, in the typical order of magnitude measured with VHHs (1–50 nM).
Fig. 1.

Amino acid sequence alignment of the six VHHs studied in this work. The fragments, the CDRs, and the amino acid numbering (bottom line) are as defined in Kabat et al. (1991). The cystein residues involved in either an intradomain (C22 and C92) or an interloop disulphide bond (C33 and C100e in cAb-lys3) are in bold type. Note that in cAb-TEM2, C33, and C100a also probably form an interloop link.
Table 1.
Kinetic rate constants for association ( ka) and dissociation (kd), and equilibrium dissociation constant (KD) of VHH-antigen complexes as determined by SPR spectroscopy with a BIAcore X
| VHH fragments | Antigen | Trpa | ka (M−1 s−1) | kd (s−1) | KD (nM) |
| cAb-Lys3 | Hen lysozyme | 2 | 1.8 105 | 2.0 10−3 | 11 |
| cAb-HuL6 | Human lysozyme | 3 (Trp-100) | 8.6 105 | 5.9 10−4 | 0.7 |
| cAb-NmcA2 | NmcA (β-lactamase) | 3 (Trp-47) | 6.5 106 | 2.1 10−3 | 0.3 |
| cAb-BcII10 | BcII (β-lactamase) | 2 | 1.8 106 | 5.6 10−3 | 3 |
| cAb-TEM2 | TEM-1 (β-lactamase) | 2 | 1.1 106 | 5.0 10−4 | 0.45 |
| cAb-R2 | Azo dye RR6 | 4 (Trp-52a; Trp-58) | 1.6 105 | 4.0 10−4 | 2.5 |
The number of tryptophan residues is also indicated.
a Two tryptophan residues are strictly conserved, namely Trp-36 and Trp-103. Additional tryptophan residues are indicated.
Interestingly, it should be noted that, despite the high similarity between the hen and human lysozyme X-ray structures (Blake et al. 1965; Artymiuk and Blake 1981), no cross-reaction was observed, either between cAb-HuL6 and hen lysozyme, or between cAb-Lys3 and human lysozyme. Similarly, cAb-NmcA2 did not bind to the TEM-1 β-lactamase, although they display very similar overall folds (Swarén et al. 1998). These observations reflect the high antigen specificity of VHH fragments (Muyldermans and Lauwereys 1999). cAb-HuL6 and cAb-NmcA2 were also tested for their inhibitory potency (Lauwereys et al. 1998). cAb-NmcA2 did not inhibit the NmcA β-lactamase. With cAb-HuL6, a typical enzymatic test (Charlemagne and Jollés 1970) did not provide a clear answer as to the interaction between cAb-HuL6 and human lysozyme. Two- and three-dimensional NMR experiments (C. Redfield, M. Dumoulin, and C.M. Dobson, unpubl.), however, have indicated that the binding site of cAb-HuL6 to human lysozyme is opposite to the catalytic cleft of the enzyme.
Chemical-induced unfolding: Intrinsic fluorescence measurements
The fluorescence spectra of the six native VHHs show a single broad emission band with maxima at around 350 nm (λmax = 347–352 nm; Fig. 2a ▶). In 7 M GdmCl or 10 M urea, the maximum emission wavelength is shifted to 356–357 nm, indicating full solvent accessibility of the tryptophan indole groups (two to four; see Table 1). The high λmax values observed with the native VHHs indicate that the tryptophan residue(s) that significantly contribute(s) to the fluorescence emission of the native fragments are relatively exposed to the solvent. With all fragments but cAb-Lys3, excitation wavelengths of 280 and 295 nm give similar emission spectra, indicating that the tyrosine residues do not significantly contribute to the fluorescence spectra. With cAb-Lys3, however, excitation at 280 nm results in a second peak of relatively weak intensity in the emission spectrum at around 305 nm. This peak can be attributed to the contribution of the tyrosine residues (12 in cAb-Lys3 versus 4–9 in the other fragments) to the fluorescence emission spectrum.
Fig. 2.

(a) Tryptophan fluorescence spectra of cAb-Lys3 at various GdmCl concentrations. The spectra were recorded at 25°C and the protein concentration was 25 μg mL−1 (1.7 μM) in 20 mM HEPES, pH 7. The excitation wavelength was 295 nm. The inset shows the changes in tryptophan fluorescence intensity observed at 340 nm (filled circles) and 360 nm (open triangles). These data were analyzed on the basis of a two-state model, and the lines represent the best fit to Equation 2, calculated using ΔG(H2O) = 20 ± 5 kJ mole−1 and 16 ± 5 kJ mole−1, and m = 9 ± 2 kJ mole−1 M−1 and 7 ± 2 kJ mole−1 M−1, for data at 340 and 360 nm, respectively; (b) GdmCl-induced denaturation of cAb-Lys3 as shown by the changes in the mass center of the tryptophan fluorescence spectrum (csm). (c) GdmCl-induced unfolding followed by the changes in the fluorescence intensity maximum (λmax). The data in (b) and (c) were analyzed on the basis of a two-state model, and the lines represent the best fit to Equation 2, calculated using ΔG(H2O) = 29 ± 2 kJ mole−1 and 33 ± 2 kJ mole−1, and m = 12.6 ± 0.8 kJ mole−1 M−1 and 14.3 ± 0.8 kJ mole−1 M−1, for (b) and (c), respectively.
Concentrated solutions of the protein fragments (0.45 mg mL−1, i.e., 30–34 μM) left to equilibrate for 2 h in 7 M GdmCl and in 10 M urea (under these conditions, the half-life of the unfolding process is less than 2 min), and subsequently diluted to yield 25 μg mL−1 VHH solutions in 0.35 M GdmCl and 0.5 M urea, respectively, yielded fluorescence spectra virtually indistinguishable from those of the native fragments. This result suggests that the VHHs are unfolded by GdmCl and urea with full reversibility. With the six fragments, this could be unambiguously established by measuring the affinities after unfolding/refolding cycles, carried out with unfolded protein concentrations ranging from 25 μg mL−1 to 200 μg mL−1, which is similar to intrinsic fluorescence and CD measurements (see below). Figure 3 ▶ shows the sensograms obtained at different concentrations of native and unfolded/refolded cAb-Lys3. In all cases, the KD values of the native and refolded fragments were identical within the error limit. Similar results were obtained with all the fragments.
Fig. 3.

Analysis by surface plasmon resonance spectroscopy of the binding of native and unfolded/refolded cAb-Lys3 to hen lysozyme. (Solid lines) Native fragment; (dotted line) unfolded fragments (25 μg mL−1) in 6.5 M GdmCl/refolded in 50 mM sodium phosphate, pH 7. Different concentrations (20, 100, and 200 nM) of native and unfolded/refolded cAb-Lys3 in HBS buffer were injected at a flow rate of 30 μL min−1.
Figure 2a ▶ displays the spectra of cAb-Lys3 obtained at different GdmCl concentrations, and the inset shows the changes in fluorescence intensity at two single wavelengths as the GdmCl concentration is increased. Although the largest difference in fluorescence intensity between the native (in 0 M GdmCl) and unfolded (in 7 M GdmCl) states occurs at 360 nm, the largest intensity change in the transition zone (1.7–3.1 M, i.e., the region of interest) is observed at 340 nm. At both wavelengths, the amplitudes for the intensity change are weak, and similar values, although of opposite signs, are observed both in the pre- and post-transition regions, and in the transition zone itself. The GdmCl-induced unfolding data (Fig. 2a ▶, inset) seem to indicate, however, that cAb-Lys3 unfolds in a single transition, indicative of a cooperative two-state process. Satisfactory fits of the data could be obtained from Equation 2, which provide a ΔG(H2O) value of 18 ± 5 kJ mole−1. The small changes in fluorescence intensity, especially in the transition region, and the relatively large amplitudes of the pre- and post-transition baselines in comparison with that of the transition zone itself, might, however, lead to a wrong estimation of the thermodynamic parameters. To increase the confidence in the determination of these parameter values, both the maximum in fluorescence intensity (λmax), and the center of the spectral mass of the fluorescence spectrum (csm; Eq. 1) were calculated at varying GdmCl concentrations. Following this analysis, it can be seen in Figure 2b and c ▶ that better data could be obtained, where the relative amplitudes of the pre- and post-transition regions are significantly reduced. According to the two-state model hypothesis and Equation 2, analysis of the data in Figure 2b and c ▶ provided a ΔG(H2O) value of 31 ± 5 kJ mole−1. Noticeably, this value is significantly larger than that obtained from the analysis of fluorescence intensity changes at single emission wavelengths. The unfolding data obtained in a protein concentration range from 25 to 90 μg mL−1 (i.e., 1.7–6.1 μM) yielded identical values of the thermodynamic parameters, within the error limit, indicating that no significant aggregation of the protein fragment takes place in this concentration range. These findings are consistent with the SPR analysis (see above), which demonstrates that the biological activity of the VHHs fragments is fully recovered after a complete unfolding/refolding cycle.
With four out of the six VHH fragments, the change in fluorescence intensity within the transition zone proved to be more important than with cAb-Lys3. In those instances, the analysis of the data obtained by fluorescence measurements at a single emission wavelength yielded values of the thermodynamic parameters similar to those derived from λmax and csm measurements. Finally, each experimental unfolding curve was analyzed using at least two biophysical parameters; that is (a) the intensity at a given wavelength (not reliable with cAb-Lys3 and cAb-BcII10); (b) the wavelength at the maximum in fluorescence intensity (λmax; done with all fragments); and (c) the center of the spectral mass of the fluorescence spectrum (csm; done with all fragments). With the six fragments, the analysis of the same experiment based on two or three different fluorescence parameters resulted in similar values of the thermodynamic parameters, within the error limit, which were averaged to yield the data in Table 2.
Table 2.
Thermodynamic parameters of unfolding of VHH antibody fragments at pH 7, 25°C, as obtained from the analysis of the equilibrium transitions
| VHH fragments | ΔG(H2O) (kJ mol−1) | m (kJ mol−1 M−1) | Cm (M) |
| cAb-Lys3 | |||
| GdmCla | 31 ± 5 | 13.5 ± 2 | 2.3 ± 0.5 |
| Urea | 36 ± 4 | 5.3 ± 0.6 | 6.7 ± 1 |
| cAb-HuL6 | |||
| GdmCl | |||
| Fluorescence | 40 ± 3 | 12.7 ± 1 | 3.1 ± 0.3 |
| CD (209 nm) | 43 ± 6 | 14.5 ± 2 | 3 ± 0.5 |
| CD (229 nm) | 37 ± 4 | 12 ± 1.5 | 3 ± 0.6 |
| Ureaa | (—) | (—) | >9 |
| cAb-NmcA2 | |||
| GdmCla | 34 ± 5 | 11 ± 1.5 | 3.0 ± 0.6 |
| Ureaa | (—) | (—) | ∼8 |
| cAb-BCII 10 | |||
| GdmCla | 47 ± 2 | 14.2 ± 0.7 | 3.3 ± 0.2 |
| Ureaa | 49 ± 6 | 6.6 ± 0.8 | 7.5 ± 1 |
| cAb-TEM2 | |||
| GdmCla | 55 ± 3 | 17.6 ± 0.7 | 3.1 ± 0.2 |
| Ureaa | 61 ± 7 | 8.3 ± 1 | 7.5 ± 1 |
| cAb-R2 | |||
| GdmCl | |||
| Fluorescence | 36 ± 5 | 16 ± 2.5 | 2.3 ± 0.5 |
| CD (212 nm) | 30 ± 5 | 13 ± 2.5 | 2.3 ± 0.6 |
| CD (222 nm) | 33 ± 10 | 14 ± 5 | 2.3 ± 1 |
| CD (268 nm) | 42 ± 10 | 18 ± 4 | 2.3 ± 0.7 |
| Ureaa | 38 ± 3 | 6.5 ± 0.5 | 5.8 ± 0.4 |
a Fluorescence measurements. Errors are calculated at the 95% confidence limit.
— = not accessible.
The unfolding curves in Figures 4 and 5 ▶ ▶ indicate that with the six VHH fragments, single transitions between the initial and final states are observed in both GdmCl and urea. When GdmCl (Fig. 4 ▶) was used as denaturant, the characteristic thermodynamic parameters of all VHHs (Table 2) could be computed with the help of Equation 2, assuming a two-state model for the unfolding transitions. With urea (Fig. 5 ▶), the very high Cm values of cAb-NmcA2 (∼8 M) and cAb-HuL6 (>9 M) preclude any quantitative analysis of the data. With cAb-Lys3, cAb-BcII10, cAbTEM2, and cAb-R2, however, a two-state model analysis was performed and the fitting parameters are given in Table 2. Consistent data were obtained with the two denaturants, and the ΔG(H2O) values of the six fragments are comprised between 30 and 60 kJ mol−1, whereas rather high Cm values are calculated (≥2.3 M in GdmCl and ≥5.8 M in urea).
Fig. 4.

(a) GdmCl-induced unfolding transition of cAb-HuL6 (open squares), cAb-Lys3 (filled circles), cAb-NmcA2 (filled squares), cAb-BcII10 (filled triangles), cAb-TEM2 (open triangles), and cAb-R2 (open circles) at pH 7.0, 25°C, monitored by the change in the center of spectral mass (csm) of the fluorescence spectra recorded between 310 and 440 nm. Protein concentrations were 25 μg mL−1 in 20 mM HEPES. Excitation wavelengths were 280 nm (all fragments but cAb-Lys3) and 295 nm (cAb-Lys3). Data were analyzed according to a two-state reaction, and the lines represent the best fits to Equation 2, calculated using the thermodynamic parameters in Table 2; (b) Fraction of VHH unfolded, fU, as a function of GdmCl concentration. The values of fU were calculated from the data in (a), as described in Pace (1986, 1990a).
Fig. 5.

(a) Urea-induced unfolding transition of cAb-HuL6 (open squares), cAb-Lys3 (filled circles), cAb-NmcA2 (filled squares), cAb-BcII10 (filled triangles), cAb-TEM2 (open triangles), and cAb-R2 (open circles) at pH 7.0, 25°C, monitored by the change in the center of the spectral mass (csm) of the fluorescence spectra recorded between 310 and 440 nm. Protein concentrations were 25 μg mL−1 in 50 mM phosphate sodium. Excitation wavelengths were 280 nm (all fragments but cAb-Lys3) and 295 nm (cAb-Lys3). Data were analyzed according to a two-state reaction, and the lines represent the best fits to Equation 2, calculated using the thermodynamic parameters in Table 2; (b) Fraction of VHH unfolded, fU, as a function of urea concentration. The values of fU were calculated from the data in (a), as described in Pace (1986, 1990a).
Note that with cAb-Lys3, comparison of the parameter values obtained with the (His)6 tag-containing cAb-Lys3, and the same fragment devoid of poly-histidine tag indicated that the short C-terminal extension has no significant effect on the conformational stability of the fragment. Similar conclusions have been reached with various histidine tagged proteins (see, e.g. Milla et al. 1993; Reid et al. 1998), and this is assumed to be valid for all six VHHs as well.
Chemical-induced unfolding: ANS-bound fluorescence measurements
The enhancement of ANS fluorescence upon binding to exposed hydrophobic regions of partially unfolded protein molecules has been extensively used to detect intermediate species in protein folding (Kuwajima 1989; Ptitsyn et al. 1990; Semisotnov et al. 1991; Itzhaki et al. 1994). With all native fragments but cAb-R2, no binding of ANS could be observed. Urea- and GdmCl-induced denaturation in the presence of ANS, monitored by fluorescence, did not show any significant enhancement in intensity around 475 nm, that is, no ANS binding was detected throughout the unfolding transition. The binding of ANS to native cAb-R2 suggests that a significant hydrophobic surface area is accessible to the solvent, presumably at the surface region of the VHH, which normally interacts with the VL domain in conventional antibodies. Careful examination of the X-ray structure (Spinelli et al. 2000), however, does not provide a clear explanation for ANS binding at any site of the cAb-R2 surface. Nevertheless, because this fragment is a hapten binder involving hydrophobic binding forces (Spinelli et al. 2000), the antigen binding site might well be responsible for non-specific interaction with ANS. With this fragment, the ANS fluorescence decreases dramatically as GdmCl or urea is added, in a concentration range largely corresponding to the pre-transition zone observed in intrinsic fluorescence intensity measurements (∼90% of the fluorescence intensity is lost in the presence of 0.1 M GdmCl). At higher concentrations, no ANS-bound fluorescence is observed. Thus, with the six VHH fragments, unfolding experiments carried out in the presence of ANS confirm that no partially structured species are significantly populated, in good agreement with the intrinsic fluorescence data.
Chemical-induced unfolding: CD measurements
The GdmCl-induced unfolding transitions of cAb-R2 and cAb-HuL6 have been followed by far UV CD measurements that monitor the backbone secondary structures (212 and 209 nm, respectively) and, presumably the side-chain tertiary structures (222 and 229 nm, respectively). The CD spectra of cAb-R2 and cAb-HuL6 in the far UV region are shown in Figure 6a ▶. Both native fragments show negative maxima at 215 and 229 nm. The CD signal near 222 nm (positive with cAb-R2) and above (negative) is probably caused by the aromatic residues of the fragments. Indeed, Phe, Tyr, and Trp are known to contribute to the CD signal in this spectral region, especially when disulphide bonds are present (Venyaminov and Yang 1996). Interestingly, the conserved tryptophan residue at position 36 in the VHHs is in close proximity to the conserved disulphide bridge (C22–C92), and thus might well be responsible (at least in part) for the far UV CD positive band observed with some fragments around 220–230 nm. Note that in helical proteins, these non-peptidic contributions are masked by the strong negative ellipticity of the α-helices (Woody 1994). By contrast, the positive CD band near 203 nm and the negative band at around 215 nm are most likely due to the peptide backbone, that is, to the antiparallel β-sheet structure (Schindler et al. 1995; Venyaminov and Yang 1996).
Fig. 6.

(a) CD spectra in the far UV region of cAb-R2 and cAb-HuL6 (inset), at pH 7.0, 25°C. (Solid line) Native cAb-R2 and cAb-HuL6 in 10 mM MOPS and in 10 mM HEPES, respectively; (broken line) in 4 M GdmCl, same buffers. The protein concentrations were 0.2 mg mL−1 (14 μM) in a 0.1 cm cell; (b) GdmCl-induced unfolding transition of cAb-HuL6 followed by far UV CD measurements at 209 and 229 nm (inset). The protein concentrations were 1 mg mL−1 (72 μM) in a 0.01-cm cell. Data were analyzed on the basis of a two-state model, and the lines represent the best fit to Equation 2, calculated using ΔG(H2O) = 43 ± 6 kJ mole−1 and 37 ± 4 kJ mole−1, and m = 14.5 ± 2 kJ mole−1 M−1 and 12 ± 1.5 kJ mole−1 M−1, at 209 and 229 nm, respectively; (c) CD spectra in the near UV region of cAb-R2, at pH 7.0, 25°C. Solid line, in 10 mM HEPES; broken line, in 10 mM HEPES and 5.5 M GdmCl. The protein concentration was 0.2 mg mL−1 in a 1-cm cell. The equilibrium unfolding transition of cAb-R2 followed by CD measurements at 268 nm is shown as an inset. The lines represent the best fit to Equation 2, calculated using ΔG(H2O) = 42 ± 10 kJ mole−1 and m = 18 ± 4 kJ mole−1 M−1; (d) Fraction of cAb-HuL6 unfolded, fU, as a function of GdmCl concentration, at pH 7.0, 25°C. The fU values were calculated from data obtained by fluorescence (filled circles), far UV CD at 209 nm (open triangles), and far UV CD at 229 nm (filled triangles). The inset shows the unfolded fraction of cAb-R2 as monitored by intrinsic fluorescence (filled circles), far UV CD at 212 nm (open triangles), far UV CD at 222 nm (filled triangles), and near UV CD at 268 nm (open diamonds). The continuous lines were drawn using the parameters obtained in CD (at 229 nm; cAb-HuL6) and intrinsic fluorescence (cAb-R2) experiments.
In the presence of 4 M GdmCl, both fragments are unfolded, the positive band near 222 nm and the negative band near 229 nm in cAb-R2 and cAb-HuL6, respectively, are absent (Fig. 6a ▶), and the signal below 220 nm is characteristic of random coil structures. The denaturation of the two VHH fragments was followed at both 209 nm (cAb-HuL6) or 212 nm (cAb-R2), and 222 nm (cAb-R2) or 229 nm (cAb-HuL6), thus monitoring secondary and, presumably tertiary structure melting, respectively. In both cases, single transition curves were obtained (Fig. 6b ▶) and analyzed according to a two-state model. After calculation of the unfolded fraction at all GdmCl concentrations, the data obtained at 212 (or 209) and 222 (or 229) nm are superimposable. Furthermore, the values of the thermodynamic parameters (Table 2) derived from far UV CD and intrinsic fluorescence measurements are in good agreement. Finally, the CD spectrum of cAb-R2 in the aromatic (i.e., near UV) region (Fig. 6c ▶) shows several bands, the disappearance of which can easily be followed upon unfolding. Thus, as shown in Figure 6c ▶ (inset), the cooperative unfolding of the tertiary structure of cAb-R2 could unambiguously be followed by CD measurements at 268 nm, and yielded the thermodynamic parameters in Table 2. The transition curves of cAb-HuL6 and cAb-R2, obtained by intrinsic fluorescence and CD measurements at two and three different wavelengths, respectively, are shown in Figure 6d ▶. With both fragments, the various data superimpose, clearly indicating that secondary and tertiary structures melt simultaneously, that is that unfolding in the transition zone occurs between the native (N) and the unfolded (U) states, without population of any intermediate structural state (Kuwajima 1996; Fersht 1999).
Similar CD measurements could not be performed with the other four fragments, because either the spectral properties were not compatible, or not enough material was available.
Chemical-induced unfolding: SPR measurements
Using a BIAcore equipment, the GdmCl-induced unfolding of cAb-HuL6 could also be analyzed by measuring the affinity of the antibody fragment towards its antigen (human lysozyme; Fig. 7 ▶). At low GdmCl concentrations (0–2 M), the dissociation constant (KD) gradually increases with the denaturant concentration. This limited increase is most likely due to the effect of GdmCl on the antigen–antibody interaction, rather than to any significant conformational change of either of the proteins (Cm = 3 M and 3.5 M for cAb-HuL6 and human lysozyme, respectively). Then, at GdmCl concentrations above 2.5 M, a dramatic increase in KD occurs as the denaturant concentration is raised, clearly indicating that the protein fragment is destabilized in a cooperative transition reminiscent of the GdmCl-induced unfolding curves obtained by intrinsic fluorescence and CD measurements. This result suggests that unfolding of the loops (CDRs) responsible for antigen binding occurs concurrently with the destabilization of the whole protein core, and it brings further experimental evidence for two-state unfolding of cAb-HuL6.
Fig. 7.

GdmCl-induced unfolding of cAb-HuL6 followed by SPR spectroscopy, using a BIAcore X instrument. Protein fragments (0.07 mg mL−1, i.e., 5 μM in HBS buffer) were incubated overnight at 25°C in the presence of varying concentrations of GdmCl. VHHs were then diluted to yield concentrations in the range from 0.6 to 4800 nM, in varying GdmCl concentrations. Ninety microliters of each solution were injected at a flow rate allowing the equilibrium to be reached (i.e., 2–30 μL min−1), using GdmCl-containing HBS buffer. For each GdmCl concentration, at least eight protein concentrations were injected. Under these equilibrium conditions, the KD value corresponds to the concentration of VHH leading to half-saturation.
Pressure-induced unfolding
Fourier transform infrared (FTIR) spectroscopy, in combination with temperature- and pressure-induced unfolding, is a powerful technique for determining conformational changes in proteins under equilibrium conditions (Wong and Heremans 1988; Jackson and Mantsch 1995; Panick et al. 1998; Torrent et al. 2001). In particular, bandfitting of the Fourier-deconvoluted amide I` spectrum (Byler and Susi 1986; Smeller et al. 1995a) allows the changes of the secondary structure elements to be followed on protein unfolding. The deconvoluted IR spectra of the amide I` region of native cAb-R2 and cAb-HuL6 are shown in Figure 8a ▶. Spectral features are emphasized under Fourier self-deconvolution. The assignments of the component bands at different wavenumbers (Byler and Susi 1986; Jackson and Mantsch 1995; Haris and Chapman 1995) are shown in Table 3. In the case of cAb-R2, the band at 1678 cm−1 is likely due to an overlap of the turn and β-sheet structures, whereas for cAb-HuL6 the contribution of these bands could be separated (Table 3). The occurrence of a β-sheet absorption band at high wavenumber values is indicative of antiparallel β-sheet (Haris and Chapman 1995; Jackson and Mantsch 1995). The lack of α-helix, which is normally expected to absorb around 1650 cm−1 (Byler and Susi 1986), and the high β-sheet content are consistent with the X-ray structures of VHHs (Desmyter et al. 1996; Spinelli et al. 1996, 2000; Decanniere et al. 1999).
Fig. 8.
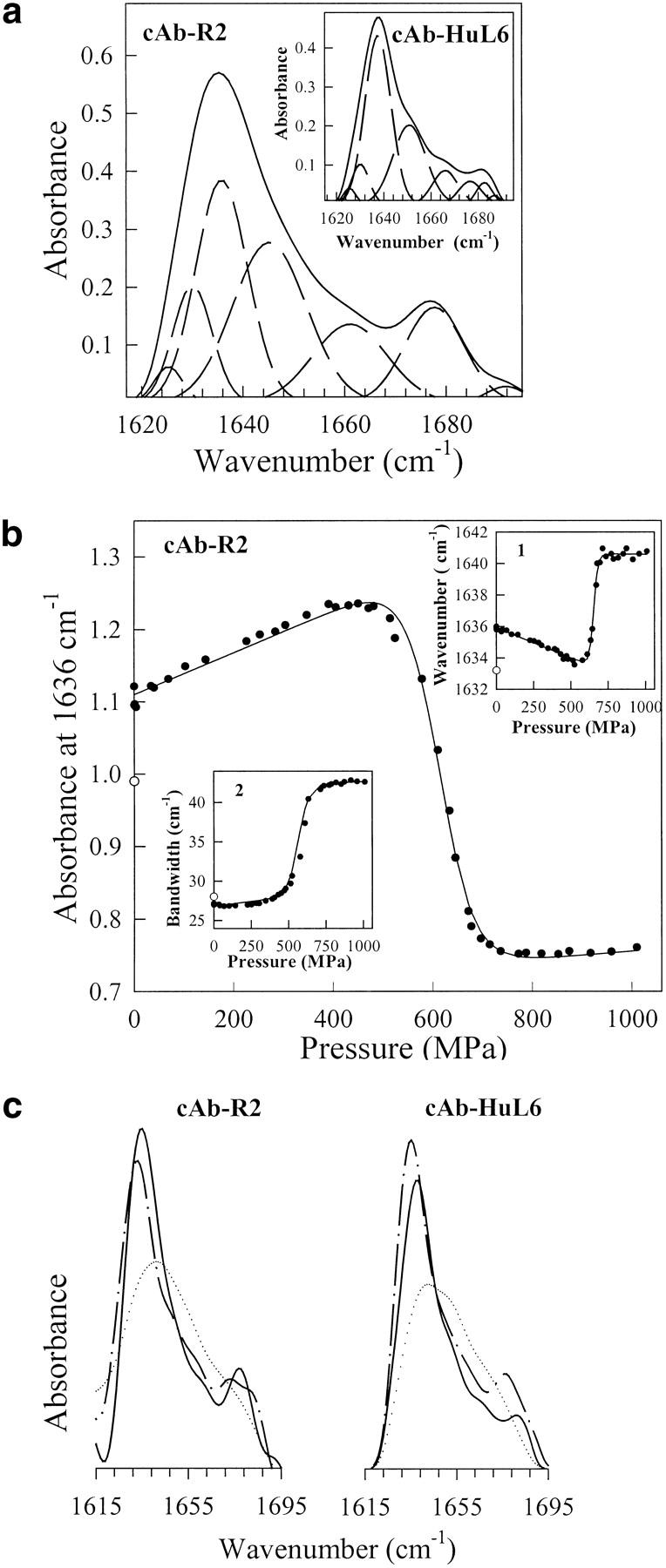
(a) Fourier-deconvoluted and fitted infrared spectra in the amide I` region (solid lines) with individual Gaussian components (broken lines) of cAb-R2 and cAb-HuL6 (inset). IR spectra were recorded at atmospheric pressure (0.1 MPa) and 25°C, using protein concentrations of about 50 mg mL−1 (∼3.6 mM) in 1 M 13C-urea and 10 mM TrisDCl, pD 7.6; (b) Pressure-induced unfolding curves of cAb-R2 followed by IR measurements. The absorbance at 1636 cm−1, the wavenumber corresponding to the maximum in absorbance (inset 1), and the amide I` bandwidth (inset 2) were used to monitor VHH unfolding. The solid lines represent the best fits of the data to Equation 3, as calculated from the parameters in Table 4. In each case, the value of the refolded state is indicated (open circles); (c) FTIR spectra of cAb-R2 and cAb-HuL6 under native (0.1 MPa; continuous line), unfolding (1060 MPa, dotted line) and refolding (0.1 MPa; broken line) conditions.
Table 3.
Band positions and secondary structure assignments for the amide I` band of cAb-R2 and cAb-HuL6 at 25°C and 0.1 MPa, after partial H/D exchange
| Position (cm−1) | Assignment | Band area (%) |
| cAb-R2 | ||
| 1625 | β-sheet | 2.5 |
| 1630 | β-sheet | 11 |
| 1636 | β-sheet | 28.5 |
| 1645 | Unordered | 29 |
| 1661 | turn | 14 |
| 1678 | turn/β-sheet | 13.5 |
| 1692 | turn | 1.5 |
| cAb-HuL6 | ||
| 1626 | β-sheet | 2 |
| 1630 | β-sheet | 7 |
| 1638 | β-sheet | 45 |
| 1651 | unordered | 27 |
| 1666 | turn | 9 |
| 1677 | turn | 5.5 |
| 1683 | β-sheet | 3.5 |
| 1687 | turn | 1 |
The band area is a measure of the relative amount of secondary structure present.
A possible limitation of the method arises from the very high protein concentration (∼50 mg mL−1, i.e., ∼3.6 mM) required. With both cAb-HuL6 and cAb-R2, this could be achieved without significant aggregation of the fragments, providing that 1 M 13C-labeled urea was added to the buffer. Considering the high Cm values (>9 and 5.8 M for cAb-HuL6 and cAb-R2, respectively) determined with urea, no significant effect of the denaturant is expected.
Pressure-induced changes in the deconvoluted amide I` region of the IR spectra were followed to obtain information about structural changes in the protein fragments. Three parameters were considered (Fig. 8b ▶), that is, the absorbance at a fixed wavenumber, the wavenumber corresponding to the absorbance maximum of the amide I` band, and the width of the band. With both VHHs, no significant modification in the IR spectrum is observed as the pressure is raised up to 400 MPa (Fig. 8b ▶). In this pressure range, only a limited decrease in the wavenumber of the band maximum occurs (inset 1), which results from the effect of compression (and hence strengthening) of the hydrogen bonds (Sandroff et al. 1984), and also from H/D exchange (Haris and Chapman 1995) due to forced water penetration inside the protein structure. The latter is clear from the decompression data for the amide I` band maximum at pressures below 400 MPa (not shown). Above 400 MPa, a cooperative displacement of the band maximum towards higher wavenumber values (Fig. 8b ▶, inset 1) is observed, together with a broadening of the band (Fig. 8b ▶, inset 2) and significant intensity changes. Thus, a cooperative decrease in band intensities is measured around 1636 cm−1 and 1678 cm−1, indicating the disappearance of the native structure, whereas the increase in band intensities observed between 1645 cm−1 and 1675 cm−1 are consistent with an increased amount of unordered structures. No indication of VHH aggregation could be observed, and the changes in the IR spectra described above proved to be largely reversible after pressure release (Fig. 8b,c ▶). Indeed, Figure 8b ▶ (inset 2) shows that the width of the amide I` band returns to its native value. In contrast, the absorbance at 1636 cm−1 (Fig. 8b ▶) and the wavenumber of the band maximum (Fig. 8b ▶, inset 1) characteristic of the native state are not completely restored. This phenomenon is due to the significant H/D exchange occurring on protein unfolding, which causes a slight shift of the amide I` band components towards lower wavenumbers (Haris and Chapman 1992). Thus, the pressure-induced unfolding transitions (Fig. 8b ▶) followed by IR intensity measurements at four different wavenumbers, and also by measuring bandwidth changes and band maximum displacements indicate that both fragments unfold reversibly, according to a cooperative two-state model. On this basis, the data in Figure 8b ▶ were analyzed by use of Equation 3, yielding the thermodynamic parameters in Table 4. The Pm values are ∼600 MPa and ∼750 MPa for cAb-R2 and cAb-HuL6, respectively, and the ΔG values measured in 1 M urea are ∼47 kJ mole−1 and ∼42 kJ mole−1 for cAb-R2 and cAb-HuL6, respectively. These values are in reasonable agreement with those found for chemical-induced unfolding (Table 2).
Table 4.
Thermodynamic parameters of cAb-HuL6 and cAb-R2 at pD 7.6, 25°C, as obtained from the analysis of pressure-induced unfolding transitions
| VHH fragments | ΔG* (kJ mol−1) | ΔV* (mL mol−1) | Pm* (MPa) |
| cAb-HuL6 | 42 ± 5 | −58 ± 8 | 740 ± 50 |
| cAb-R2 | 47 ± 9 | −77 ± 12 | 610 ± 30 |
a Values obtained in 1 M 13C-urea. Errors are calculated at the 95% confidence limit.
Heat-induced unfolding
In the absence of denaturant, the strong positive CD band at about 200 nm (Fig. 6a ▶) can be used as a sensitive probe for β-sheet unfolding. Thus, thermal unfolding of the six protein fragments was followed by CD measurements at 201–203 nm, using concentrations of 0.17 mg mL−1 (11–13 μM). With all fragments, apparently two-state unfolding curves were observed, with rather high melting temperature values (Tm ≥ 60°C; Table 5). With cAb-BcII10, heat-induced unfolding was followed at 202 and 223 nm for secondary and tertiary structure unfolding, respectively (see above). The unfolding curves at both wavelengths are largely superimposable, with high midpoint values (Tm = 68°C). These data suggest that thermal unfolding occurs with a simple two-state transition. In all cases, however, aggregation was observed at temperatures above the Tm values. Despite this phenomenon, a large proportion of the native far UV CD signal (80 to 90%) was recovered with four out of the six antibody fragments (Table 5) after cooling down the samples. This high regain in native ellipticity contrasts, however, with the twofold decrease in the apparent antigen-binding affinity measured with the corresponding samples (Table 5). The KD(heated) values in Table 5 were calculated, however, by assuming an homogeneous population of refolded molecules. Hence, this experiment does not discriminate between the situation in which the structure of all the VHH molecules is slightly modified (yielding an increased KD), and a second in which ∼50% of the fragments have fully refolded to the native state (with an unchanged KD), while the other ∼50% are incorrectly folded and no longer bind the antigen. However, the dissociation rate constants (kd) appear to be unchanged, and hence, the decreased KD values are merely due to decreased apparent association rate constants (ka). This result suggests that the second possibility might prevail. Whatever the explanation, this observation emphasizes that measurements of optical properties as a probe for protein structure can be largely misleading as far as the biological activity is concerned. With the cAb-Lys3 fragment, the antigen-binding properties could be fully recovered after thermal unfolding in the presence of ≥1 M GdmCl, but at the cost of a significant reduction in the apparent Tm values (ΔTm = 17°C in 1 M GdmCl).
Table 5.
Overview of the heat-induced unfolding experiments
| Reversibility | ||||
| VHHs | Temp. range (°C) | Tm (°C) | CD201–203 nm (%) | KD (native)/ KD (heated) |
| cAb-Lys3 | 25–80 | 62 | 90 | 0.5 |
| cAb-HuL6 | 25–92 | 78 | 91 | 0.63 |
| cAb-NmcA2 | 25–80 | 62 | 20 | ND |
| cAb-BcII10 | 25–80 | 68 | 30 | ND |
| cAb-TEM2 | 25–84 | 76 | 77 | 0.48 |
| cAb-R2 | 25–70 | 60 | 80 | 0.54 |
Tm is the temperature at the midpoint of the denaturation curve, i.e., the melting temperature. Apparent Tm values are given with standard deviations below 10%. KD values were measured using SPR spectroscopy, as described in the text, assuming a homogeneous population of refolded VHHs.
ND: not determined.
Discussion
The conformational properties of the VHH fragments were probed under three radically different unfolding conditions, using chemicals, temperature, and pressure as denaturants. In the presence of high GdmCl and urea concentrations, full reversibility of the unfolding process was established with all the fragments, by intrinsic tryptophan fluorescence and affinity measurements. Under these genuine equilibrium conditions, we demonstrated that cAb-HuL6 and cAb-R2 unfold cooperatively in a single two-state transition, where only the native and unfolded states are significantly populated. Thus, following the normalization of the fluorescence and CD (both in the far and near UV regions in the case of cAb-R2) data to give the fraction of unfolded protein at various denaturant concentrations (Fig. 6d ▶), it can be seen that coincident unfolding curves are obtained. This is usually taken as a stringent test for the absence of populated intermediate species under equilibrium conditions (Kuwajima 1989). Furthermore, the affinity of cAb-HuL6 for human lysozyme, measured by SPR spectroscopy at varying GdmCl concentrations suggests that the antigen-binding loops (CDRs) are disorganized coincidentally with the protein core (Fig. 7 ▶). Although no CD and SPR experiments were performed with the other four VHHs, the two-state model is most probably valid for all the fragments studied in this work. This assumption is supported by several lines of evidence. Thus, all fluorescence-detected unfolding curves are adequately described by Equation 2. With each VHH, identical free energy values (within the error limit; Table 2) were obtained for urea- and GdmCl-induced unfolding, and all these values are quite high (ΔG(H2O) = 30–60 kJ mole−1). In addition, the m values (Table 2), which account for the dependence of ΔG on the denaturant concentration (m = δΔG/δ[denaturant]), that is, for the slope of the transition, are found to be very similar for the six fragments, either in GdmCl or in urea. Interestingly, with both denaturants, the m values (11–18 kJ mole−1 M−1 and 5.3–8.3 kJ mole−1 M−1, respectively) are as high, or even higher than predicted (12–14 kJ mole−1 M−1 and 5.8–6.7 kJ mole−1 M−1, respectively) from their change in solvent-accessible surface area upon unfolding (estimated from their sizes; Myers et al. 1995). These findings are consistent with a two-state unfolding mechanism, from which any deviation should lower the m values (Pace 1986; Myers et al. 1995). Finally, the absence of ANS binding in the presence of denaturant is also a good indication of the lack of intermediate species at equilibrium.
Considering the analysis carried out with the six antibody fragments, the values of their thermodynamic parameters (Table 2) can be considered with good confidence. Interestingly, simple two-state unfolding is observed, and all fragments exhibit high conformational stability. By contrast, the chemical-induced unfolding of conventional single-chain antibody fragments (scFv) is usually more complex, with several unfolding transitions (Wörn et al. 2000). Whether two or more transitions occur, the functionality of the scFv fragments is lost with the first transition, the Cm value of which is often in the range of 1–2 M and 2–3 M in GdmCl and urea, respectively (Proba et al. 1997; Wörn and Plückthun 1998, 1999; Wörn et al. 2000), that is, at denaturant concentrations significantly lower than with single-domain VHHs (2.3–3.3 M and ≥6 M in GdmCl and urea, respectively; Table 2). In comparison with antibody fragments derived from conventional immunoglobulin, VHH fragments from camelids combine excellent antigen-binding properties, with remarkably high conformational stabilities. In particular, such a high value (ΔG(H2O) ∼60 kJ mole−1, with Cm = 7.5 M urea) as measured with cAb-TEM2 has never been reported to date, even when tailored conventional antibody fragments (Jung and Plückthun 1997) are considered.
The exceptional stability of VHHs is also clearly evident from pressure- and heat-induced unfolding experiments. FTIR measurements reveal that high pressures (>400 MPa) are needed to unfold cAb-HuL6 and cAb-R2, and reversible unfolding of the two fragments allows high ΔG values (∼40 kJ mole−1 in 1 M urea) to be calculated, very close to those obtained from denaturant-induced unfolding. In contrast to the high values necessary to unfold VHHs, relatively low pressure values (∼50–250 MPa) are required to dissociate noncovalent protein complexes (Silva et al. 1986, 1992; Erijman et al. 1993). Therefore, in this pressure range, it should be feasible to disrupt antigen–VHH complexes without unfolding either molecule. This strategy can be advantageously used for immunoaffinity separation, where a specifically bound antigen is eluted from the immunoadsorbent after a controlled pressure increase (Olson et al. 1989; Sudaram et al. 1998). Compared with the harsh conditions usually used, such a "hyperbaric elution" extends the immunoadsorbent lifetime (Olson et al. 1989). The use of VHHs, which are made up of a single domain, and consequently are easier to express, purify, and handle, will render this technique even more valuable.
Although in our hands thermal unfolding of the six protein fragments did not appear to be fully reversible (≤60% of activity recovered; Table 5), high apparent Tm values could be estimated (60–80°C). These are similar to the values measured with a llama heavy-chain antibody fragment (VHH-H14, Tm = 60°C; Perez et al. 2001), but also with various Fab, Fv, and scFv fragments (Yasui et al. 1994; Shimba et al. 1995; Young et al. 1995; Welfle et al. 1999). These results confirm that scFv and VHH fragments may display comparable thermostabilities, but that the latter refold more efficiently following heat-induced denaturation, although not always completely, to the native conformation (van der Linden et al. 1999; Perez et al. 2001). Incubation of conventional antibody fragments at T > Tm results in dissociation of the native structures, with subsequent exposure of the hydrophobic interfaces of both the heavy and the light chains. These exposed "sticky" areas induce aggregation and precipitation, ultimately resulting in nonfunctional molecules. By contrast, the high solubility and thermostability of the VHH fragments are most probably largely due to specific amino acid substitutions at the VH–VL interface, which confer much more hydrophilicity to this normally very hydrophobic area (Muyldermans et al. 2001). This hypothesis is strongly supported by the results (i.e., enhanced solubility and thermostability) obtained by Davies and Riechmann (1994, 1996), with camelized human antibody VH domains.
Another distinct feature of VHHs is the occurrence of enlarged CDR1 and CDR3 loops, which is thought to compensate for both the lack of the antigen-binding surface contributed by the three hypervariable loops (CDRs) of the VL domain, and the absence of the VH-VL combinatorial diversity (Muyldermans and Lauwereys 1999; Muyldermans et al. 2001). Thus, the average length of the CDR3 sequence is 17, 12, and 9 amino acids in camelid VHHs, human VHs, and mouse VHs, respectively (Wu et al. 1993; Muyldermans et al. 1994; Vu et al. 1997), and remarkably, in the cAb-Lys3 fragment with specificity for hen lysozyme it is made up of 24 amino acids (Desmyter et al. 1996). This very long antigen-binding loop is constrained by an interloop disulphide bond, which is expected to impose conformational restraints on the loop flexibility in the absence of antigen (Desmyter et al. 1996; Muyldermans et al. 2001). Beside the strictly conserved intradomain disulphide bridge (C22–C92), which is characteristic for the immunoglobulin fold, the presence of a second interloop disulphide bond is of common occurrence in VHHs of the dromedary. Although this bond has been shown to stabilize camelized human antibody VH domains with long CDR3 (Davies and Riechmann 1996), it is clear that the high stability of camelid VHHs cannot be merely attributed to this additional linkage. Indeed, all the VHH antibody fragments studied by others (van der Linden et al. 1999; Perez et al. 2001), or in the present work display very high stabilities, whether they contain one or two disulphide bridges. A comparison of the available VHH sequences indicates that the sequences with the longest CDR3 have most frequently two additional cysteine residues, one within the CDR1 (or at position 45 in the framework 2 region) and one in the CDR3, which most probably form a disulphide bridge. Interestingly, knocking out this interloop bond yields nonfunctional VHHs, at least in case of cAb-Lys3 (K.B. Vu and S. Muyldermans, unpubl. obs.). Thus, it seems that the role of this tightening disulphide link is to compensate for the possible drawback of long CDR3 loops, which might affect the affinity, the specificity, and the stability of VHH antigen binders. In consequence, the net stability of VHHs with long CDR3 loops and one extra-disulphide bond is neither reduced nor enhanced when compared with other VHHs. The interloop disulphide bridge is especially common in dromedary VHHs, which show longer CDR3 in comparison with llama sequences (Vu et al. 1997). It is noteworthy that with some fragments (Table 5), only ∼50% of the antigen affinity could be restored after heat-induced unfolding, whereas a large proportion of the native far UV CD signal (80 to 90%) was recovered. This phenomenon might be due to efficient refolding of the core of the protein fragments, which would explain the large recovery of native far UV CD signal, and incorrect refolding of the long CDR loops, which are responsible for antigen binding.
The present results highlight the remarkable stability of VHHs, and confirm their superiority to conventional antigen-binders in a number of biotechnological and clinical applications because of their smaller size, higher solubility, broad and unique antigen binding capacity, and increased stability.
Materials and methods
Enzymes and chemicals
Guanidinium chloride (GdmCl) (>99%), bovine serum albumin (BSA), and 8-anilino-1-naphtalene-sulfonic acid (ANS) were purchased from Sigma Chemical Co. Urea (>99%) and 13C-urea (>99%) were from Merck and Isotec IMC, respectively. Ni2+ NTA-agarose was obtained from Affiland. Azo-dye reactive red-6 (RR6, Procion Rubine MX-B) was from ICI. All solutions were prepared with milli-Q water, and filtered through 0.22-μm filters before use.
Hen egg white lysozyme was purchased from Sigma Chemical Co., and the human lysozyme preparation was kindly given by professor C.M. Dobson (O.C.M.S., New Chemistry Laboratory, University of Oxford, UK). The TEM-1, NmcA and Bacillus cereus 569H (BcII) β-lactamases were purified as described in Raquet et al. (1994), Swarén et al. (1998), and Carfi et al. (1995), respectively. The human lysozyme and NmcA preparations were used for dromedary immunization, as described in Conrath et al. (2001).
Selected VHH fragments
The various single-domain antibody fragments studied in the present work are listed in Table 1. Five VHHs (cAb-Lys3, cAb-HuL6, cAb-TEM2, cAb-NmcA2, and cAb-BcII10) were derived from dromedary heavy-chain antibodies, and one (cAb-R2) originated from llama heavy-chain antibodies. In the case of dromedaries, specific VHHs were isolated by biopanning from phage libraries containing the genes coding for the variable domains of the heavy-chain antibodies. The phage libraries were generated using the mRNA extracted from the lymphocytes of immunized dromedaries (Ghahroudi et al. 1997). According to this procedure, VHHs that bind specifically to hen (cAb-Lys3; Desmyter et al. 1996) and human (cAb-HuL6) lysozyme, the TEM-1 (cAb-TEM2; Conrath et al. 2001) and NmcA (cAb-NmcA2) β-lactamases, and the BcII metallo-β-lactamase (cAb-BcII10; Conrath et al. 2001) were isolated. The cAb-R2 fragment, with specificity for the azo dye reactive red-6 (RR6) was constructed and selected as described in Spinelli et al. (2000).
Expression and purification of VHH fragments
cAb-TEM2 was produced and purified as previously described (Conrath et al. 2001). cAb-R2 was produced in Saccharomyces cerevisiae as a fusion protein (Spinelli et al. 2000), and purified from the crude yeast culture broth by combining ion exchange (Q-Sepharose, Amersham Pharmacia Biotech) and gel filtration (Sephadex S75, Amersham Pharmacia Biotech) chromatography techniques. The VHH genes of the other four selected binders were recloned into an expression vector pHEN6 (Conrath et al. 2001), and were transformed into the WK6 nonsuppressor strain of E. coli. The recombinant VHHs were expressed and purified as follows: a 15 L fermentor (BioFlo 4500, New Brunswick), containing 12 L of SB-ampicillin medium (32 g L−1 tryptone, 20 g L−1 yeast extract, 5 g L−1 NaCl, 1 g L−1 glucose, and 100 μg L−1 of ampicillin) was inoculated with 750 mL of an approximately 5 h preculture. The culture was grown at 37°C, pH 7.2, and cell development was followed by monitoring both the oxygen consumption and the turbidimetry. At the end of the exponential phase, expression was induced by the addition of 1 mM IPTG, and cell growth was allowed for an additional 3–6 h at 25°C. After harvesting the cells by centrifugation, the periplasmic proteins were extracted according to the procedure described in Skerra and Plückthun (1988). The fusion fragments containing a C-terminal (His)6 tag were then purified in a single step, by metal chelate affinity chromatography on a Ni2+ NTA–agarose matrix. The VHH fragments were eluted with a linear imidazole gradient (0–240 mM) in 50 mM potassium phosphate, pH 7. The fractions in the major peak were pooled, and imidazole was removed by dialysis. With all six fragment preparations, no trace of contaminating protein was found, either by SDS-PAGE or by mass spectrometry analysis, indicating a purity higher than 98%. The VHH concentrations were determined spectrophotometrically using their computed extinction coefficients and Mr (PC Gene, IntelliGenetics), and a yield in the range of 1 to 5 mg of purified protein per litre of bacterial culture was calculated. The final cAb-HuL6 and cAb-R2 preparations were stored lyophilized at −80 °C, whereas the other fragments were conserved at −80 °C in 50 mM sodium phosphate, pH 7. Under these conditions, the six VHH fragments were shown to be fully stable, at least for the duration of this work.
Chemical-induced unfolding transitions
Samples of VHH fragments were incubated overnight at 25°C in the presence of various concentrations of urea or guanidinium chloride (GdmCl). Unfolding curves were determined by monitoring the intrinsic fluorescence emission or circular dichroism (CD) at 25°C. The pH was checked to ensure a constant value throughout the whole transition, and the denaturant concentration was determined from refractive index measurements (Pace 1986), using a R5000 hand refractometer from Atago.
Fluorescence measurements
Both intrinsic fluorescence and ANS-bound fluorescence emission spectra were recorded on a Perkin-Elmer LS50B spectrofluorimeter. Excitation and emission slit widths were 3 and 5 nm, respectively, and the scan speed was 350 nm min−1. Cuvettes with 1-cm pathlength were used.
Intrinsic fluorescence measurements were performed using a protein concentration of 25 μg mL−1 (1.7–1.9 μM), with excitation wavelength at either 280 or 295 nm, and emission spectra recorded from 310 to 440 nm. The buffers used were 50 mM phosphate sodium, pH 7, and 20 mM HEPES, pH 7, in the presence of urea and GdmCl, respectively. With all samples, fluorescence spectra were corrected for the background fluorescence of the solution (buffer + denaturant). Three fluorescence parameters have been considered in this work: the fluorescence intensity at single excitation and emission wavelengths, the wavelength corresponding to the maximum in fluorescence intensity (λmax), and the center of the spectral mass of the fluorescence spectrum (csm). A five-parameter weibull function (provided with the software SigmaPlot 5.0) was fitted to the fluorescence intensity spectra to obtain the associated λmax values. The csm values were computed according to the following equation (Royer 1995):
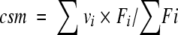 |
(1) |
where vi is the wavenumber (i.e., inverse wavelength) and Fi the fluorescence intensity at vi. The csm values were calculated between 310 and 440 nm.
ANS-bound fluorescence measurements were performed with the samples used for intrinsic fluorescence measurements, with excitation at 350 nm, and emission spectra recorded from 420 to 600 nm. The fluorescence spectra were corrected for the background fluorescence of ANS. The ANS concentration (determined from the molar extinction coefficient of 4950 M−1 cm−1 at 350 nm; Merck Index, Merck & Co.) was 255–360 μM and hence, [ANS]/[VHH] ≈ 140–230.
Circular dichroism measurements
CD measurements were performed with a Jobin-Yvon CD6 spectropolarimeter, either in the far UV (205–250 nm) or in the near UV (250–350 nm) regions, using a protein fragment concentration of 0.2 mg mL−1 (13–15 μM), and 0.1 cm or 1 cm cell pathlengths, respectively. With cAb-HuL6, experiments in the far UV were also performed at a 1 mg mL−1 (70 μM) concentration, in a 0.01-cm cell pathlength. The buffers used were 10 mM MOPS, pH 7, and 10 mM HEPES, pH 7, with cAb-R2 and cAb-HuL6, respectively. The instrument was calibrated with d-10-camphorsulfonic acid (Schmid 1997). Spectra were acquired at a scan speed of 12 nm min−1, with a 2-nm bandwidth and a 1-s integration time. The spectra were measured five times, averaged, and corrected by subtraction of the solvent spectrum obtained under identical conditions.
GdmCl and urea unfolding curves were recorded at fixed wavelengths of 212, 222, and 268 nm with cAb-R2, and 209 and 229 nm with cAb-HuL6, using a 2-nm bandwidth. At all denaturant concentrations, at least 30 data points were acquired with a reading frequency of 1/15 s−1 and a 2-s integration time, and averaged. The resulting values were corrected for the contribution of the solvent.
Heat-induced unfolding transitions were monitored at 201–203 and 223 nm, using a protein concentration of 0.17 mg mL−1 (11–13 μM) in 50 mM sodium phosphate, pH 7, and a 0.1-cm cell pathlength. The temperature was increased monotonically in the range from 25 to 65–84°C, at a rate of 0.55°C min−1. The reversibility of the phenomenon was assayed by cooling the sample down to 25°C, at a rate of 0.6°C min−1. Data were acquired with a reading frequency of 1/20 s−1, a 1-s integration time and a 2-nm bandwidth.
Affinity measurements
The kinetics of binding were analyzed by surface plasmon resonance (SPR) spectroscopy, using a BIAcore X instrument (Biacore AB). For immobilization, the azo dye RR6 was coupled to BSA, as described in van der Linden et al. (2000). The individual antigenic proteins (RR6-BSA, lysozymes, and β-lactamases) were immobilized on a carboxymethylated dextran-coated sensor chip (CM5, Biacore AB), using the amine coupling chemistry (EDC/NHS) according to the instructions of the manufacturer. In general, 250–350 resonance units (RU) were immobilized, except with RR6-BSA (5000 RU immobilized). Hen lysozyme was immobilized as the reference protein for measurements with human lysozyme, and vice versa, whereas NmcA was taken as the reference for BcII, and vice versa. BcII and BSA were used as references for TEM-1 and RR6-BSA, respectively. Hence, blank sensograms could be obtained for subtraction of bulk refractive index background. The binding/regeneration cycles were performed at 25°C in HBS (10 mM HEPES, pH 7.4, 150 mM NaCl, 3 mM EDTA, 0.005% surfactant P20), at a constant flow rate of 30 μL min−1. Under these conditions, the mass transfer effects proved to be negligible. Regeneration of the surfaces was achieved by injection of 30 μL of 10 mM NaOH or 5 M GdmCl. Binding traces were recorded in duplicate, with at least six different concentrations. The sensograms were analyzed by nonlinear least-squares fitting, with the help of the BIAevaluation 3.0 software (Amersham Pharmacia Biotech), on the basis of a homogeneous 1:1 association model, with simultaneous fitting of the dissociation (kd) and association (ka) rate constants. The equilibrium dissociation constant (KD) was then calculated from the ratio of the individual rate constants (kd/ka). Note that the concentration of antibody in the test solution must be introduced as a known parameter to perform the fitting.
Enzymatic assays
The inhibitory capacity of cAb-NmcA2 was assayed by measuring the rate of hydrolysis of nitrocefin, after preincubation of the NmcA β-lactamase with varying concentrations of antibody (Conrath et al. 2001).
Pressure-induced unfolding
Lyophilized cAb-HuL6 and cAb-R2 fragments were dissolved in deuterated 10 mM TrisDCl, pD 7.6, at a protein concentration of 50 mg mL−1 (3.6 mM), in the presence of 1 M 13C-labeled urea to minimize aggregation. The samples were stored overnight at 25°C, hence ensuring complete H/D exchange of all solvent-accessible protons, and then mounted into the well of a stainless steel gasket of a diamond anvil cell (Diacell Products). The cell was placed into a cell holder, and the pressure was built up by means of a screw mechanism. The pressure in the cell was measured with BaSO4 by following the shift of the 983 cm−1 sulphate peak in the deconvoluted spectrum (Wong and Moffat 1989). The infrared spectra were recorded on a Bruker IFS66 FTIR spectrometer, equipped with a liquid nitrogen cooled broad band mercury–cadmium–telluride solid-state detector. A total of 250 interferograms were co-added at a resolution of 2 cm−1.
The secondary structure of the protein fragments was determined by fitting the resolution enhanced amide I` band of the spectrum (Byler and Susi 1986; Smeller et al. 1995a). The overlapping components of the amide I` band were narrowed by the Fourier self-deconvolution developed by Kauppinen et al. (1981). The optimal parameters were determined from the observation of the power spectrum, as described in Smeller et al. (1995b). A resolution enhancement factor (Kauppinen et al. 1981) of 1.5 was reached using the Lorentzian band shape of 20-cm−1 bandwidth. A triangular square apodization function was used. The deconvoluted spectra were then fitted with Gaussian functions. The fitting of component peaks was performed by a program developed in the Leuven laboratory, using the Levenberg-Marquard algorithm (Press et al. 1986).
Three variable parameters were considered, that is, the amide I` bandwidth, the wavenumber of the band maximum, and the absorbance at given wavenumbers.
Data analysis
The thermodynamic parameters for chemical unfolding were computed on the assumption of a two-state model for the unfolding reaction (N⇋U). On this basis, the transition curves were analyzed according to the following equation (Santoro and Bolen 1988; Pace 1990a):
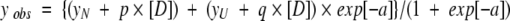 |
(2) |
where
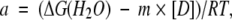 |
and where yobs is the measured variable parameter at a given denaturant concentration, and yN and yU represent the values of this parameter for the native and denatured states, respectively. ΔG(H2O) is the difference in free energy between the folded and unfolded conformations under physiological conditions (also defined as the conformational stability of a globular protein; Pace 1990b); m is a measure of the dependence of the free energy on the denaturant concentration, and [D] is the denaturant concentration. p and q are the slopes of the pre- and post-unfolding baselines, respectively, R is the gas constant, and T is the absolute temperature. The midpoint of the denaturation curve ([U]/[N] = 1) is given by Cm = ΔG(H2O)/m.
Similarly, the pressure-induced unfolding experiments were analyzed assuming an all-or-none transition between the unfolded and native states (N⇋U), and the corresponding thermodynamic parameters were determined using the following equation (Dumoulin et al. 1999):
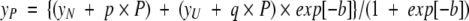 |
(3) |
where
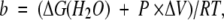 |
and where P is the pressure, ΔV is the volume change, and ΔG(H2O), yN, yU, p, q, R and T are the same parameters as defined in Equation (2). The melting pressure of half transition ([U]/[N] = 1) is given by Pm = −ΔG(H2O)/ΔV.
The programs GraFit 3.09 (Erithacus software Ltd.) and Sigmaplot 5.0 (SPSS Inc.) were used to carry out nonlinear least-squares fitting of the data. Errors are calculated at the 95% confidence limit.
Acknowledgments
We thank Klaas Decanniere, Aline Desmyter, Christopher M. Dobson, Jean-Marie Frére, Marc Lauwereys, Annabelle Lejeune, and Roger H. Pain for many helpful discussions. We acknowledge Bart Devreese for performing the mass spectrometry experiments, and Cees van Vliet for producing cAb-R2 in Saccharomyces cerevisiae. We are grateful to Jean-Marie Frére and Roger H. Pain for critical reading of the manuscript. A.M. and M.D. also wish to acknowledge C.M. Dobson for his encouragement and support. This work was supported by the RTD programme on Biotechnology of the 4th EC framework program, grant number BIO4-98-0048. The purchase of BIAcore X equipment was supported in part by a grant from the Fonds de la Recherche Fondamentale et Collective (contract number 3.4589.96). A.M. is a Research Associate of the National Fund for Scientific Research (F.N.R.S., Belgium).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
ANS, 8-anilino-1-naphtalene-sulfonic acid
BSA, bovine serum albumin
CD, circular dichroism
CDR, complementary determining region
csm, center of the spectral mass
Fab, Fv, scFv, and dsFv, antigen-binding fragment, variable fragment, single-chain variable fragment, and disulphide stabilized variable fragment of conventional antibodies, respectively
FTIR, Fourier transform infrared
GdmCl, guanidinium chloride
HEPES, N-(2-hydroxyethyl)piperazine-N`-2-ethanesulfonic acid
IPTG, isopropyl β-D-thiogalactopyranoside
IR, infrared
MOPS, 3-N-morpholinopropanosulfonic acid
RU, resonance units
SPR, surface plasmon resonance
VH, variable domain of immunoglobulin heavy chain
VL, variable domain of immunoglobulin light chain
VHH, variable domain of camelid heavy-chain antibody
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.34602.
References
- Artymiuk, P.J. and Blake, C.C.F. 1981. Refinement of human lysozyme at 1.5 Å resolution. J. Mol. Biol. 152: 737–762. [DOI] [PubMed] [Google Scholar]
- Better, M., Chang, C.P., Robinson, R.R., and Horwitz, A.H. 1988. Escherichia coli secretion of an active chimeric antibody fragment. Science 240: 1041–1043. [DOI] [PubMed] [Google Scholar]
- Bird, R.E., Hardman, K.D., Jacobson, J.W., Johnson, S., Kaufman, B.M., Lee, S.M., Lee, T., Pope, S.H., Riordan, G.S., and Whitlow, M. 1988. Single-chain antigen-binding proteins. Science 242: 423–426. [DOI] [PubMed] [Google Scholar]
- Blake, C.C.F., Koenig, D.F., Mair, G.A., and Sarma, R. 1965. Crystal structure of lysozyme by X-ray diffraction. Nature 206: 757–761. [DOI] [PubMed] [Google Scholar]
- Byler, D.M. and Susi., H. 1986. Examination of the secondary structure of proteins by deconvolved FTIR spectra. Biopolymers 25: 469–487. [DOI] [PubMed] [Google Scholar]
- Cai, X. and Garen, A. 1996. A melanoma-specific VH antibody cloned from a fusion phage library of a vaccinated melanoma patient. Proc. Natl. Acad. Sci. 93: 6280–6285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carfi, A., Pares, S., Duée, E., Galleni, M., Duez, C., Frére, J.M., and Dideberg, O. 1995. The 3-D structure of a zinc metallo-β-lactamase from Bacillus cereus reveals a new type of protein fold. EMBO J. 14: 4914–4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlemagne, D. and Jollés, P. 1970. Inhibition par des polyméres de la N-acetylglucosamine de l'activité lysante à pH 6,2 de lysozymes d'origines différentes vis-à-vis de Micrococcus lysodeikticus. C.R. Acad. Sci. Hebd. Seances Acad. Sci. D 270: 2721–2723. [PubMed] [Google Scholar]
- Conrath, K.E., Lauwereys, M., Galleni, M., Matagne, A., Frére, J.M., Kinne, J., Wyns, L., and Muyldermans, S. 2001. β-Lactamase inhibitors derived from single-domain antibody fragments elicited in Camelidae. Antimicrob. Agents Chemother. 45: 2807–2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies, J. and Riechmann, L. 1994. "Camelising" human antibody fragments: NMR studies on VH domains. FEBS Lett. 339: 285–290. [DOI] [PubMed] [Google Scholar]
- Davies, J. and Riechmann, L. 1996. Single antibody domains as small recognition units: Design and in vitro antigen selection of camelized, human VH domains with improved protein stability. Protein Eng. 6: 531–537. [DOI] [PubMed] [Google Scholar]
- Decanniere, K., Desmyter, A., Lauwereys, M., Ghahroudi, M.A., Muyldermans, S., and Wyns, L. 1999. A single-domain antibody fragment in complex with RNase A: Non-canonical loop structures and nanomolar affinity using two CDR loops. Struct. Fold. Des. 7: 361–370. [DOI] [PubMed] [Google Scholar]
- Decanniere, K., Muyldermans, S., and Wyns, L. 2000. Canonical antigen-binding loop structures in immunoglobulins: More structures, more canonical classes? J. Mol. Biol. 300: 83–91. [DOI] [PubMed] [Google Scholar]
- Desmyter, A., Transue, T.R., Ghahroudi, M.A., Thi, M.H., Poortmans, F., Hamers, R., Muyldermans, S., and Wyns, L. 1996. Crystal structure of a camel single-domain VH antibody fragment in complex with lysozyme. Nat. Struct. Biol. 3: 803–811. [DOI] [PubMed] [Google Scholar]
- Dumoulin, M., Ueno, H., Hayashi, R., and Balny, C. 1999. Contribution of the carbohydrate moiety to conformational stability of the carboxypeptidase Y. High pressure study. Eur. J. Biochem. 262: 475–483. [DOI] [PubMed] [Google Scholar]
- Erijman, L., Lorimer, G.H., and Weber, G. 1993. Reversible dissociation and conformational stability of dimeric ribulose bisphosphate carboxylase. Biochemistry 32: 5187–5195. [DOI] [PubMed] [Google Scholar]
- Fersht, A.R. 1999. Protein stability. In Structure and mechanism in protein science—A guide to enzyme catalysis and protein folding, pp. 508–539. W.H. Freeman and Co., New York.
- Frenken, L.G.J., van der Linden, R.H.J., Hermans, P.W., Bos, J.W., Ruuls, R.C., de Geus, B., and Verrips, C.T. 2000. Isolation of antigen specific llama VH antibody fragments and their high level secretion by Saccharomyces cerevisiae. J. Biotechnol. 7811–21. [DOI] [PubMed] [Google Scholar]
- Ghahroudi, M., Desmyter, A., Wyns, L., Hamers, R., and Muyldermans, S. 1997. Selection and identification of single domain antibody fragments from camel heavy-chain antibodies. FEBS Lett. 414: 521–526. [DOI] [PubMed] [Google Scholar]
- Glennie, M.J. and Johnson, P.W. 2000. Clinical trials of antibody therapy. Immunol. Today 21: 403–410. [DOI] [PubMed] [Google Scholar]
- Green, M.C., Murray, J.L., and Hortobagyi, G.N. 2000. Monoclonal antibody therapy for solid tumors. Cancer Treat. Rev. 26: 269–286. [DOI] [PubMed] [Google Scholar]
- Hamers-Casterman, C., Atarhouch, T., Muyldermans, S., Robinson, G., Hamers, C., Songa, E.B., Bendahman, N., and Hamers, R. 1993. Naturally occurring antibodies devoid of light chains. Nature 363: 446–448. [DOI] [PubMed] [Google Scholar]
- Haris, P.I. and Chapman, D. 1992. Does Fourier-transform infrared-spectroscopy provide useful information on protein structures. Trends Biochem. Sci. 17: 328–333. [DOI] [PubMed] [Google Scholar]
- ——. 1995. The conformational analysis of peptides using Fourier transform IR spectroscopy. Biopolymers 37: 251–263. [DOI] [PubMed] [Google Scholar]
- Horwitz, A.H., Chang, C.P., Better, M., Hellstrom, K.E., and Robinson, R.R. 1988. Secretion of functional antibody and Fab fragment from yeast cells. Proc. Natl. Acad. Sci. 85: 8678–8682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, P.J. 1998. Recombinant antibody fragments. Curr. Opin. Biotechnol. 9: 395–402. [DOI] [PubMed] [Google Scholar]
- Itzhaki, L.S., Evans, P.A., Dobson, C.M., and Radford, S.E. 1994. Tertiary interactions in the folding pathway of hen lysozyme: Kinetic studies using fluorescent probes. Biochemistry 33: 5212–5220. [DOI] [PubMed] [Google Scholar]
- Jackson, M. and Mantsch, H.H. 1995. The use and misuse of FTIR spectroscopy in the determination of protein structure. Crit. Rev. Biochem. Mol. Biol. 30: 95–120. [DOI] [PubMed] [Google Scholar]
- Jung, S. and Plückthun, A. 1997. Improving in vivo folding and stability of a single-chain Fv antibody fragment by loop grafting. Protein Eng. 10: 959–966. [DOI] [PubMed] [Google Scholar]
- Kabat, E., Wu, T.T., Perry, H.M., Gottesman, K.S. and Foeller, C. 1991. Sequence of proteins of immunological interest. U.S. Public Health Sevices, NIH Bethesda, MD, Publication No. 91–3242.
- Kauppinen, J.K., Moffat, D.J., Mantsch, H.H., and Cameron, D.G. 1981. Fourier self-deconvolution—A method for resolving intrinsically overlapped bands. Appl. Spectrosc. 35: 271–276. [Google Scholar]
- Kuwajima, K. 1989. The molten globule state as a clue for understanding the folding and cooperativity of globular–protein structure. Protein Struct. Funct. Genet. 6: 87–103. [DOI] [PubMed] [Google Scholar]
- Kuwajima, K. 1996. Stopped-flow circular dichroism. In Circular dichroism and the conformational analysis of biomolecules (ed. G.D. Fasman), pp. 159–182. Plenum, New York.
- Lauwereys, M., Ghahroudi, M.A., Desmyter, A., Kinne, J., Holzer, W., De Genst, E., Wyns, L., and Muyldermans, S. 1998. Potent enzyme inhibitors derived from dromedary heavy-chain antibodies. EMBO J. 17: 3512–3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milla, M.E., Brown, B.M., and Sauer, R.T. 1993. P22 Arc repressor: Enhanced expression of unstable mutants by addition of polar C-terminal sequences. Protein Sci. 2: 2198–2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muyldermans S. 2001. Single-domain camel antibodies: Current status. Rev. Mol. Biotechnol. 74: 277–302. [DOI] [PubMed] [Google Scholar]
- Muyldermans, S. and Lauwereys, M. 1999. Unique single-domain binding fragments derived from naturally occuring camel heavy-chain antibodies. J. Mol. Recognit. 12: 131–140. [DOI] [PubMed] [Google Scholar]
- Muyldermans, S., Atarhouch, T., Saldanha, J., Barbosa, J.A., and Hamers, R. 1994. Sequence and structure of VH domain from naturally occurring camel heavy chain immunoglobulins lacking light chains. Protein Eng. 7: 1129–1135. [DOI] [PubMed] [Google Scholar]
- Muyldermans, S., Cambillau, C., and Wyns, L. 2001. Recognition of antigens by single-domain antibody fragments: The superfluous luxury of paired domains. Trends Biochem. Sci. 26: 230–235. [DOI] [PubMed] [Google Scholar]
- Myers, J.K., Pace, C.N., and Scholtz, J.M. 1995. Denaturant m values and heat capacity changes: Relation to changes in accessible surface areas of protein unfolding. Protein Sci. 4: 2138–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson, W.C., Leung, S.K., and Yarmush, M.L. 1989. Recovery of antigens from immunoadsorbents using high pressure. Biotechnology 7: 369–373. [Google Scholar]
- Pace, C.N. 1986. Determination and analysis of urea and guanidine hydrochloride denaturation curves. Methods Enzymol. 131: 266–280. [DOI] [PubMed] [Google Scholar]
- —. 1990a. Measuring and increasing protein stability. Trends Biotechnol. 8: 93–98. [DOI] [PubMed] [Google Scholar]
- —. 1990b. Conformational stability of globular proteins. Trends Biochem. Sci. 15: 14–17. [DOI] [PubMed] [Google Scholar]
- Panick, G., Malessa, R., Winter, R., Rapp, G., Frye, K.J., and Royer, C.A. 1998. Structural characterization of the pressure-unfolded state of staphylococcal nuclease by synchrotron small-angle X-ray scattering and Fourier-transform infrared spectroscopy. J. Mol. Biol. 275: 389–402. [DOI] [PubMed] [Google Scholar]
- Perez, J.M., Renisio, J.G., Prompers, J.J., van Platerink, C.J., Cambillau, C., Darbon, H., and Frenken, L.G. 2001. Thermal unfolding of a llama antibody fragment: A two-state reversible process. Biochemistry 40: 74–83. [DOI] [PubMed] [Google Scholar]
- Press, W.H., Flannery, B.P., Teukolsky, S.A., and Vetterling, W.T. 1986. Optimal (Wiener) filtering with the FFT. In Numerical recepies in pascal: The art of scientific computing, pp. 459–462. Cambridge University Press, Cambridge.
- Proba, K., Honegger, A., and Plückthun, A. 1997. Natural antibody missing a cysteine in VH: Consequences for thermodynamic stability and folding. J. Mol. Biol. 265: 161–172. [DOI] [PubMed] [Google Scholar]
- Ptitsyn, O.B., Pain, R.H., Semisotnov, G.V., Zerovnik, E., and Razgulyaev, O.I. 1990. Evidence for a molten globule state as a general intermediate in protein folding. FEBS Lett. 262: 20–24. [DOI] [PubMed] [Google Scholar]
- Raquet, X., Lamotte-Brasseur, J., Fonzé, E., Goussard, S., Courvalin, P., and Frére, J.M. 1994. TEM β-lactamase mutants hydrolysing third-generation cephalosporins. A kinetic and molecular modelling analysis. J. Mol. Biol. 244: 625–639. [DOI] [PubMed] [Google Scholar]
- Reid, K.L., Rodriguez, H.M., Hillier, B.J., and Gregoret, L.M. 1998. Stability and folding properties of a model β-sheet protein, Escherichia coli CspA. Protein Sci. 7: 470–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter, Y., Brinkmann, U., Lee, B., and Pastan, I. 1996. Engineering antibody Fv fragments for cancer detection and therapy: Disulfide-stabilized Fv fragments. Nat. Biotechnol. 14: 1239–1245. [DOI] [PubMed] [Google Scholar]
- Riechmann, L. and Muyldermans, S. 1999. Single domain antibodies: Comparison of camel VH and camelised human VH domains. J. Immunol. Methods 231: 25–38. [DOI] [PubMed] [Google Scholar]
- Riechmann, L., Foote, J., and Winter, G. 1988. Expression of an antibody Fv fragment in myeloma cells. J. Mol. Biol. 203: 825–828. [DOI] [PubMed] [Google Scholar]
- Royer, C. 1995. Fluorescence spectroscopy. In Methods in molecular biology, vol. 40, protein stability and folding (ed. B.A. Shirley), pp. 65–90. Humana Press Inc., Totowa, NJ. [DOI] [PubMed]
- Sandroff, C.J., King, H.E., and Herschbach, D.R. 1984. High pressure study of the liquid/solid interface: Surface enhanced raman scattering from adsorbed molecules. J. Phys. Chem. 88: 5647–5653. [Google Scholar]
- Santoro, M.M. and Bolen, D.W. 1988. Unfolding free energy changes determined by the linear extrapolation method. 1. Unfolding of phenylmethanesulfonyl alpha-chymotrypsin using different denaturants. Biochemistry 27: 8063–8068. [DOI] [PubMed] [Google Scholar]
- Schindler, T., Herrler, M., Marahiel, M.A., and Schmid, F.X. 1995. Extremely rapid protein folding in the absence of intermediates. Nat. Struct. Biol. 2: 663–673. [DOI] [PubMed] [Google Scholar]
- Schmid, F.X. 1997. Optical spectroscopy to characterize protein conformation and conformational changes. In Protein structure, a practical approach, 2nd ed. (ed. T.E. Creighton), pp. 299–321. Oxford University Press, Oxford.
- Semisotnov, G.V., Rodionova, N.A., Razgulyaev, O.I., Uversky, V.N., Gripas, A.F., and Gilmanshin, R.I. 1991. Study of the "molten globule" intermediate state in protein folding by a hydrophobic fluorescent probe. Biopolymers 31: 119–128. [DOI] [PubMed] [Google Scholar]
- Shimba, N., Torigoe, H., Takahashi, H., Masuda, K., Shimada, I., Arata, Y., and Sarai, A. 1995. Comparative thermodynamic of the Fv, Fab*, and Fab fragments of anti-dansyl mouse monoclonal antibody. FEBS Lett. 360: 247–250. [DOI] [PubMed] [Google Scholar]
- Silva, J.L., Miles, E.W., and Weber, G. 1986. Pressure dissociation and conformational drift of the beta dimer of tryptophan synthetase. Biochemistry 25: 5780–5786. [DOI] [PubMed] [Google Scholar]
- Silva, J.L., Silveira, C.F., Correia, J.A., and Pontes, L. 1992. Dissociation of a native dimer to a molten globule monomer. Effects of pressure and dilution on the association equilibrium of arc repressor. J. Mol. Biol. 223: 545–555. [DOI] [PubMed] [Google Scholar]
- Skerra, A. and Plückthun., A. 1988. Assembly of a functional immunoglobulin Fv fragment in Escherichia coli. Science 240: 1038–1041. [DOI] [PubMed] [Google Scholar]
- Smeller, L., Goossens, K., and Heremans, K. 1995a. Determination of the secondary structure of proteins at high pressure. Vib. Spectrosc. 8: 199–203. [Google Scholar]
- ———. 1995b. How to minimize certain artifacts in Fourier self-deconvolution. Appl. Spectrosc. 49: 1538–1542. [Google Scholar]
- Solomon, B., Koppel, R., Frankel, D., and Hanan-Aharon, E. 1997. Disaggregation of Alzheimer beta-amyloid by site-directed mAb. Proc. Natl. Acad. Sci. 94: 4109–4112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon, B., Koppel, R., Hanan, E., and Katzav, T. 1996. Monoclonal antibodies inhibit in vitro fibrillar aggregation of the Alzheimer beta-amyloid peptide. Proc. Natl. Acad. Sci. 93: 452–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinelli, S., Frenken, L.G., Bourgeois, D., de Ron, L., Bos, W., Verrips, T., Anguille, C., Cambillau, C., and Tegoni, M. 1996. The crystal structure of a llama heavy chain variable domain. Nat. Struct. Biol. 9: 752–757. [DOI] [PubMed] [Google Scholar]
- Spinelli, S., Frenken, L.G., Hermans, P., Verrips, T., Brown, K., Tegoni, M., and Cambillau, C. 2000. Camelid heavy-chain variable domains provide efficient combining sites to haptens. Biochemistry 39: 1217–1222. [DOI] [PubMed] [Google Scholar]
- Sudaram, S., Roth, C.M., and Yarmush, M.L. 1998. Pressure-induced dissociation of antigen-antibody complexes. Biotechnol. Prog. 14: 773–781. [DOI] [PubMed] [Google Scholar]
- Swarén, P., Maveyraud, L., Raquet, X., Cabantous, S., Duez, C., Pédelacq, J.D., Mariotte-Boyer, S., Labia, R., Nicolas-Chanoine, M.H., Mourey, L., Frére, J.M., and Samama, J.P. 1998. X-ray analysis of the NMC-A β-lactamase at 1.64-Å resolution, a class A carbapenemase with broad substrate specificity. J. Biol. Chem. 273: 26714–26721. [DOI] [PubMed] [Google Scholar]
- Torrent, J., Rubens, P., Ribo, M., Heremans, K., and Vilanova, M. 2001. Pressure versus temperature unfolding of ribonuclease A: An FTIR spectroscopic characterization of 10 variants at the carboxy-terminal site. Protein Sci. 10: 725–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Transue, T.R., De Genst, E., Ghahroudi, M.A., Wyns, L., and Muyldermans, S. 1998. Camel single-domain antibody inhibits enzyme by mimicking carbohydrate substrate. Protein Struct. Funct. Genet. 32: 515–522. [DOI] [PubMed] [Google Scholar]
- van der Linden, R.H., Frenken, L.G., de Geus, B., Harmsen, M.M., Ruuls, R.C., Stok, W., de Ron, L., Wilson, S., Davis, P., and Verrips, C.T. 1999. Comparison of physical chemical properties of llama VHH antibody fragments and mouse monoclonal antibodies. Biochim. Biophys. Acta 1431: 37–46. [DOI] [PubMed] [Google Scholar]
- van der Linden, R.H.J., de Geus, B., Stok, W., Bos, W., van Wassenaar, D., Verrips, T., and Frenken, L. 2000. Induction of immune responses and molecular cloning of the heavy chain antibody repertoire of Lama glama. J. Immunol. Methods 240: 185–195. [DOI] [PubMed] [Google Scholar]
- Venyaminov, S.Y. and Yang, J.T. 1996. Determination of protein secondary structure. In Circular dichroism and the conformational analysis of biomolecules (ed. G.D. Fasman), pp. 69–108. Plenum, New York.
- Vu, K.B., Ghahroudi, M.A., Wyns, L., and Muyldermans, S. 1997. Comparison of llama VH sequences from conventional and heavy chain antibodies. Mol. Immunol. 34: 1121–1131. [DOI] [PubMed] [Google Scholar]
- Ward, E.S., Gussow, D., Griffiths, A.D., Jones, P.T., and Winter, G. 1989. Binding activities of a repertoire of single immunoglobulin variable domains secreted from Escherichia coli. Nature 341: 544–546. [DOI] [PubMed] [Google Scholar]
- Welfle, K., Misselwitz, R., Hausdorf, G., Höhne, W., and Welfle, H. 1999. Conformation, pH-induced conformational changes and thermal unfolding of anti-p25 (HIV-1) monoclonal antibody CB4–1 and its Fab and Fc fragments. Biochim. Biophys. Acta 1431: 120–131. [DOI] [PubMed] [Google Scholar]
- Wong, P.T. and Heremans, K. 1988. Pressure effects on protein secondary structure and hydrogen deuterium exchange in chymotrypsinogen: A Fourier transform infrared spectroscopic study. Biochim. Biophys. Acta 956: 1–9. [DOI] [PubMed] [Google Scholar]
- Wong, P.T.T. and Moffat, D.J. 1989. A new internal pressure calibrant for high-pressure infrared spectroscopy of aqueous systems. Appl. Spectrosc. 43: 1279–1281. [Google Scholar]
- Woody, R.W. 1994. Contributions of tryptophan side chains to the far-ultraviolet circular dichroism of proteins. Eur. Biophys. J. 23: 253–262. [DOI] [PubMed] [Google Scholar]
- Wörn, A. and Plückthun, A. 1998. Mutual stabilisation of VL and VH in single-chain antibody fragments, investigated with mutants engineered for stability. Biochemistry 37: 13122–13127. [DOI] [PubMed] [Google Scholar]
- ——. 1999. Different equilibrium stability behavior of scFv fragments: Identification, classification, and improvement by protein engineering. Biochemistry 38: 8739–8750. [DOI] [PubMed] [Google Scholar]
- Wörn, A., Auf der Maur, A., Escher, D., Honegger, A., Barberis, A., and Plückthun, A. 2000. Correlation between in vitro stability and in vivo performance of anti-GCN4 intrabodies as cytoplasmic inhibitors. J. Biol. Chem. 275: 2795–2803. [DOI] [PubMed] [Google Scholar]
- Wu, T.T., Johnson, G., and Kabat, E.A. 1993. Length distribution of CDR H3 in antibodies, Protein Struct. Funct. Genet. 16: 1–7. [DOI] [PubMed] [Google Scholar]
- Yasui, H., Ito, W., and Kurosawa, Y. 1994. Effects of substitutions of amino acids on the thermal stability of the Fv fragments of antibodies. FEBS Lett. 353: 143–146. [DOI] [PubMed] [Google Scholar]
- Young, N.M., MacKenzie, C.R., Narang, S.A., Oomen, R.P., and Baenziger, J.E. 1995. Thermal stabilisation of a single-chain Fv antibody fragment by introduction of a disulfide bond. FEBS Lett. 377: 135–139. [DOI] [PubMed] [Google Scholar]