

Abstract
Small autonomously folding proteins are of interest as model systems to study protein folding, as the same molecule can be used for both experimental and computational approaches. The question remains as to how well these minimized peptide model systems represent larger native proteins. For example, is the core of a minimized protein tolerant to mutation like larger proteins are? Also, do minimized proteins use special strategies for specifying and stabilizing their folded structure? Here we examine these questions in the 35-residue autonomously folding villin headpiece subdomain (VHP subdomain). Specifically, we focus on a cluster of three conserved phenylalanine (F) residues F47, F51, and F58, that form most of the hydrophobic core. These three residues are oriented such that they may provide stabilizing aromatic–aromatic interactions that could be critical for specifying the fold. Circular dichroism and 1D-NMR spectroscopy show that point mutations that individually replace any of these three residues with leucine were destabilized, but retained the native VHP subdomain fold. In pair-wise replacements, the double mutant that retains F58 can adopt the native fold, while the two double mutants that lack F58 cannot. The folding of the double mutant that retains F58 demonstrates that aromatic–aromatic interactions within the aromatic cluster are not essential for specifying the VHP subdomain fold. The ability of the VHP subdomain to tolerate mutations within its hydrophobic core indicates that the information specifying the three dimensional structure is distributed throughout the sequence, as observed in larger proteins. Thus, the VHP subdomain is a legitimate model for larger, native proteins.
Keywords: Villin headpiece subdomain, protein folding, aromatic—aromatic interactions, hydrophobic core mutation
Understanding how the amino acid sequence codes for the final folded protein product is critical, especially given the tremendous amount of sequence information being generated by the human (and other) genome projects. Small, autonomously folding proteins are attractive as simplified model systems for testing our knowledge of the folding of larger, more complicated proteins (DeGrado and Sosnick 1996; Plaxco et al. 1998). These simplified and or minimized peptides are monomeric and fold autonomously, that is, they adopt a well-defined tertiary structure in the absence of disulfide bonds and without binding ligands or metals. Thus, all the information required for specifying and stabilizing the final three-dimensional structure is contained within their amino acid sequence. Several such autonomously folding peptides have been recently reported, including peptides designed de novo, from zinc fingers, and also isolated from the lipoamide dehydrogenase complex (Kalia et al. 1993; Struthers et al. 1996; Dahiyat and Mayo 1997; Fezoui et al. 1997; Bryson et al. 1998; Spector et al. 1998). We have previously reported the NMR structure of a 35-residue, autonomously folded peptide (VHP subdomain) derived from a villin headpiece, that forms a thermostable subdomain (McKnight et al. 1997).
These models systems have great potential to assist in deconvoluting the "second half of the genetic code," that is, how the linear sequence of amino acids encodes the information for the three-dimensional structure of the final protein. The reduced number of amino acids in these model systems makes them amenable to computational methods as well as direct experimental approaches. The VHP subdomain, for example, has been used in the longest dynamics simulation of folding to date (1 μs) (Duan and Kollman 1998) as well as other computational studies (Duan et al. 1998; Lee et al. 2000,2001; Gibbs et al. 2001; Sullivan and Kuntz 2001), in addition to experimental approaches (McKnight et al. 1997; Kristensen and Winter 1998; Hansen et al. 2000). Thus, the VHP subdomain is a useful system to bridge the gap between experimental and computational approaches to protein folding.
For model systems like the VHP subdomain to be most useful, it must be demonstrated that they are not simply curiosities, but are true protein folds in their own right. That is, they are not somehow "special sequences" that fold, but instead have the properties of larger proteins. For example, is the VHP subdomain, like larger proteins, resistant to amino acid mutations? Or conversely, does the short length of the VHP subdomain result in a higher constraint on each individual residue? Is the VHP subdomain structure stabilized by the same combination of forces as larger proteins (e.g., hydrophobic interactions, hydrogen bonds, charge–charge interactions, etc.), or is there a "special" interaction that allows the sequence to fold?
In examining the VHP subdomain structure, a feature that seems likely to be a "special interaction" is the cluster of three phenylalanine residues in the hydrophobic core. The NMR structure of the VHP subdomain reveals a unique 3-helix motif surrounding a hydrophobic core. Three phenylalanine residues F47, F51, and F58 make up the bulk of the hydrophobic core along with the hydrophobic residues L42, V50, and L69 (Fig. 1 ▶). These F residues are highly conserved: in a comparison of 23 different headpiece domains (Vardar et al. 1999), F47 is completely conserved, F51 is L in two sequences, and F58 is replaced W, Y, or L in three of the sequences. The three F residues are highly buried in the structure (F47, 0.7%; F51, 9.0%; F58, 4.1% solvent-accessible surface area). In addition, the geometry of the residues with respect to each other, while not optimal, suggest the possibility of a significant aromatic–aromatic interaction (Burley and Petsko 1986,1988; Hunter et al. 1991). For phenylalanine residues the maximal interaction energy occurs when the edge of one ring is perpendicular to the face of another. The energy of this optimal arrangement is estimated to contribute as much as 2 kcal mole−1 for each interaction. Even in the case of the VHP subdomain, where the geometry of the three interactions between F47, F51, and F58 are not optimal, the aromatic–aromatic interaction could contribute significantly to the overall stability of ∼3.3 kcal/mole (McKnight et al. 1996).
Fig. 1.
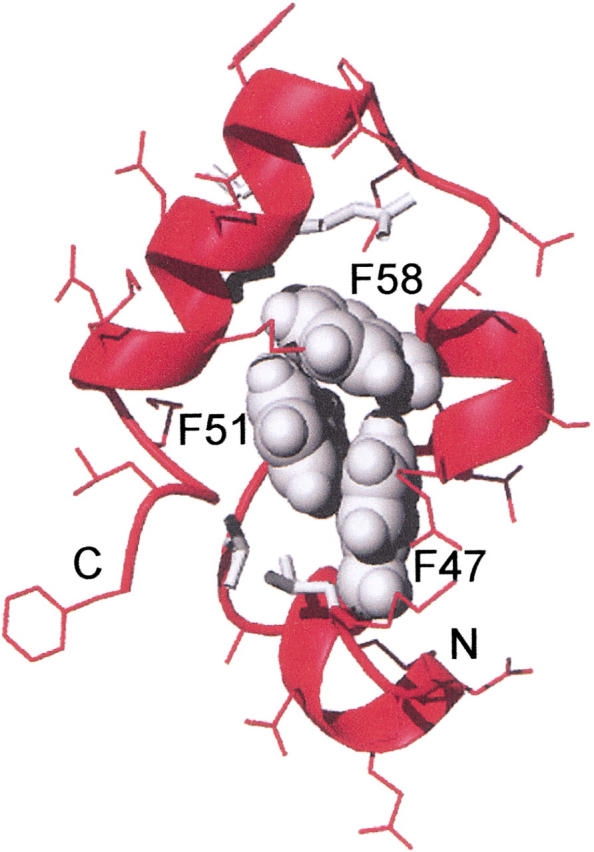
The aromatic cluster in the VHP subdomain. The side chains of phenylalanines 47, 51, and 58 are shown in space filling representation in gray. Other side chains are shown as sticks. Other side chains in the hydrophobic core are in gray. The backbone is shown as a ribbon. This figure was produced using MOLMOL (Koradi et al. 1996), from the Protein Data Bank file 1VII.PDB.
To test the role of these phenylalanine residues in stabilizing and specifying the folded structure of the VHP subdomain, we have replaced them with leucine, individually and in pairwise combination. Using circular dichroism and 1D-NMR spectroscopy, we show that each of the three possible F to L single mutants is destabilized, but still adopts the VHP subdomain fold. The F58L point mutant is the most destabilized, followed by F47L and F51L, which are less destabilized.
Because the point mutants still retain two phenylalanine residues in close proximity that could form aromatic–aromatic interactions, we also characterized the three possible double mutants that replace two of the three F residues with L. Only the double mutant F47,51L that retains F58 can adopt the VHP subdomain fold, although this mutant is highly destabilized. However, the ability of F47,51L to adopt even low levels of the native structure demonstrates that its sequence contains the information necessary for specifying the VHP subdomain fold, even in the absence of any aromatic–aromatic interactions.
Our results indicate that the VHP subdomain sequence, like larger "normal" proteins, is tolerant to mutations in the amino acids of its hydrophobic core. This demonstrates that its amino acid sequence contains more than the minimal amount of information for the native state. Like larger proteins, its folding code is redundant. Thus, the VHP subdomain is a legitimate model for larger native proteins.
Results
Expression of VHP subdomain mutants
VHP subdomain mutant proteins were expressed in Escherichia coli as fusion proteins with the TrpLE leader sequence (Schumacher et al. 1996). The TrpLE sequence targets the expressed fusion protein to inclusion bodies that can preserve even unstable peptides from cellular degradation. After isolating the inclusion bodies, the desired peptides were cleaved from the TrpLE leader by treatment with cyanogen bromide, yielding free amino termini on the peptides. The peptides expressed well with a final yield of HPLC purified proteins of 5 to 15 mg/L of culture.
To take advantage of this expression system, the amino acid sequence of the VHP subdomain was mutated to change the single methionine residue at position 53 to leucine (M53L) to prevent internal CNBr cleavage. Thus, the M53L mutant is the reference subdomain for all of the mutants described in this study (Table 1). All of the phenylalanine to leucine single and double mutants contain the M53L mutation as well.
Table 1.
Amino acid sequence, thermal unfolding midpoints (Tm), and apparent molecular weight of VHP subdomains
| Protein | Sequence | Tm | Apparent MW | |||||||||||||||||||||||||||||||||||
| 42 | 47 | 51 | 53 | 58 | 76 | |||||||||||||||||||||||||||||||||
| HP36 | M | L | S | D | E | D | F | K | A | V | F | G | M | T | R | S | A | F | A | N | L | P | L | W | K | Q | Q | N | L | K | K | E | K | G | L | F | 70 | 3100 |
| M53L | | | | | L | | | 62 | 3100 | ||||||||||||||||||||||||||||||||
| F47L | L | | | L | | | 45 | 3400 | ||||||||||||||||||||||||||||||||
| F51L | | | L | L | | | 50 | 3400 | ||||||||||||||||||||||||||||||||
| F58L | | | | | L | L | 35 | 3400 | ||||||||||||||||||||||||||||||||
| F47, 51L | L | L | L | | | 45 | 4100 | ||||||||||||||||||||||||||||||||
| F47, 58L | L | | | L | L | (39) | 7400 | ||||||||||||||||||||||||||||||||
| F51, 58L | | | L | L | L | (37) | 7700 | ||||||||||||||||||||||||||||||||
| HP35 | L | S | D | E | D | F | K | A | V | F | G | M | T | R | S | A | F | A | N | L | P | L | W | K | Q | Q | N | L | K | K | E | K | G | L | F | |||
| 1 | 6 | 10 | 12 | 17 | 35 | |||||||||||||||||||||||||||||||||
Only the changed residues are shown for the mutant sequences. All of the leucine mutants also contain the M53L mutation. HP35 corresponds to last 35 residues (791 to 825) of intact chicken villin (Bazari et al. 1988). HP36 corresponds to the expressed VHP subdomain that retains an N-terminal methionine residue. The numbering at the top is relative to the 76-residue chicken villin headpiece, and corresponds to the numbering of the HP36 NMR structure 1VII.pdb (McKnight et al. 1997). The numbers at the bottom corresponds to the length of HP35. The Tm values were determined from the thermal unfolding experiments in Figure 2 ▶. The Tm values from the shallow unfolding curves of the double mutants that do not adopt the VHP subdomain native structure are in parentheses. The apparent molecular weights were calculated from the standards in the gel filtration experiments in Figure 5 ▶. The theoretical molecular weights are: HP36 = 4189.9; M53L = 4040.7; F47L, F51L, and F58L = 4006.6; F47, 51L, F47, 58L, and F51, 58L = 3972.6.
Comparison of the properties of M53L and HP36
The intensity of the near- and far-UV CD spectra of M53L is slightly reduced from that of the bacterially expressed VHP subdomain (HP36), which retains an N-terminal methionine (Figs. 2a,3a ▶ ▶). In addition, the thermal unfolding midpoint of M53L is reduced by ∼8°C from that of HP36 (Fig. 4a ▶), indicating that the side chain of M53, which is conserved among headpiece sequences (Vardar et al. 1999), makes a significant contribution to the thermostability of the VHP subdomain fold. The NMR chemical shifts of the backbone protons of M53L and HP36 are similar, as are the crosspeak patterns in NOESY spectra, indicating that the M53L mutation does not significantly alter the native structure of the VHP subdomain (not shown). A comparison of the downfield region of the 1D-NMR spectra of HP36 and M53L is shown in Figure 6 ▶.
Fig. 2.

Far-UV CD spectra of VHP subdomain mutants at 4°C. (a) Single leucine point mutants. (b) Double leucine mutants. Samples were 40 to 60 μM protein, in 50 mM phosphate buffer, pH 7.0. The M53L spectrum is shown in both (a) and (b) for comparison. HP36 (+); M53L (filled diamonds); F47L (open circles); F51L (open squares); F58L (open triangles); F47,51L (filled triangles); F47,58L (filled squares); F51,58L (filled circles).
Fig. 3.

Near-UV CD spectra of the VHP subdomain mutants at 4°C. (a) Single leucine point mutants. (b) Double leucine mutants. Samples were 100 to 225 μM protein, in 50 mM phosphate buffer, pH 7.0. The M53L spectrum is shown in both (a) and (b) for comparison. HP36 (+); M53L (filled diamonds); F47L (open circles); F51L (open squares); F58L (open triangles); F47,51L (filled triangles); F47,58L (filled squares); F51,58L (filled circles).
Fig. 4.

Thermal unfolding of the VHP subdomain mutants monitored by CD at 222 nm. (a) Single mutants. (b) Double mutants. Samples were 40 to 60 μM protein in 50 mM phosphate buffer, pH 7.0. For clarity, only every fourth data point is shown. For reference, the thermal unfolding of M53L is repeated in plot (b). HP36 (+); M53L (filled diamonds); F47L (open circles); F51L (open squares); F58L (open triangles); F47,51L (filled triangles); F47,58L (filled squares); F51,58L (filled circles).
Fig. 6.

Downfield region of the 1D-NMR spectra of VHP subdomains at 4°C. NMR samples contained approximately 1 mM protein, in 10 mM phosphate buffer pH 7.0.
CD spectra of VHP subdomain mutants
CD spectroscopy was used to assess the effect of the mutations on the native state of M53L. Far-UV CD spectroscopy is especially sensitive to the α-helical structure, which comprises a large fraction of the VHP subdomain fold. The far-UV CD spectra of the F47L and F51L mutants at 4°C are similar to that of M53L in shape and intensity, and so the secondary structure contents are also expected to be very similar (Fig. 2a ▶). The intensity of the F58L spectrum is slightly reduced relative to that of M53L, indicating either a lower secondary structure content for F58L, or an equilibrium of folded and unfolded molecules.
Three double mutants that replace the F residues with L in pairs were constructed to test whether interactions between aromatic residues are critical for specifying the VHP subdomain fold. In the far-UV CD spectra of the F-to-L double mutants, the two mutants that lack F58 (F47,58L and F51,58L) have greatly reduced CD intensity compared to M53L (Fig. 2b ▶). The reduced intensities of these CD signals indicate that these two mutants have a significantly lower secondary structure content than M53L, or are largely unfolded. The far-UV spectrum of the F47,51L mutant that retains F58 displays significantly more intensity than the two double mutants that lack F58, but less than that of M53L. Indeed, the far-UV spectrum of F47,51L is very similar to that of the F58L single mutant.
Near-UV CD spectra are sensitive to tertiary interactions involving aromatic side chains. The near-UV CD spectra of the VHP subdomains are dominated by the signal from tryptophan 64 as the only other aromatic residues are phenylalanines that contribute little to the near- (or far)-UV CD signal (Woody 1995). The near-UV CD spectra of F47L and F51L are quite similar to that of M53L, while the intensity of the F58L mutation is significantly reduced (Fig. 3a ▶). For the double mutants, only F47,51L has an appreciable near-UV CD spectrum. Indeed, the intensity of the F47,51L mutant is greater than the F58L single mutant. The lack of any near-UV CD spectrum indicates that the two double mutants that lack F58 (F47,58L and F51,58L) lack significant tertiary structure, are substantially unstructured, and do not adopt the VHP subdomain native state.
The reduced intensity of the near- and far-UV CD spectra of the F58L and F47,51L mutants may arise from loss of secondary structure (i.e., an altered native state) or from incomplete folding at 4°C. A slight additional loss of signal in the near- and far-UV CD spectra may arise from the loss of the aromatic phenylalanine rings in the mutants. NMR evidence described below suggests that F58L and F47,51L fold to the native VHP subdomain structure. Thus, the loss of intensity in the far-UV CD spectra of F58 and F47,51L are most likely the result of an equilibrium population of folded and unfolded molecules.
Thermal unfolding of VHP subdomain mutants
To assess the effect of the point mutations on the stability of M53L, thermal unfolding experiments monitored by CD were performed (Fig. 4a ▶). The thermal unfolding midpoints (Tm) were from the derivative of the thermal unfolding curves, as this method is relatively independent from the choice of folded and unfolded baseline values. All of the mutants were destabilized with respect to M53L, which has a Tm of 58°C (Table 1). The order of the thermostability of the point mutants decreased from F51L (Tm = 46°C) to F47L (Tm = 43°C) to F58L (Tm = 30°C). The slopes in the transition region of the unfolding curves were similar, suggesting no major changes in the enthalpy of unfolding for these phenylalanine to leucine mutants.
For the double mutant F47,51L the Tm is 41°C (Fig. 4b ▶). The two double mutants lacking F58 (F47,58L and F51,58L) both appear to undergo a cooperative thermal unfolding transition, but the folded state differs from that of the VHP subdomain. The apparent Tm for F47,58L is 37°C and 39°C for F51,58L. However, it is not clear that the "folded" baseline region is truly folded because it contains a significantly reduced secondary structure, and the near-UV CD indicates that there is essentially no tertiary structure.
Gel filtration of VHP subdomain mutants
To ensure that the mutants studied remained monomeric, each mutant was run on an analytical gel filtration FPLC column (Fig. 5 ▶). The aromatic point mutations elute at a slightly earlier volume (i.e., higher molecular weight) than does M53L, but the molecular weights calculated from the retention times of globular protein standards are still consistent with monomers (Table 1). The increase in the apparent molecular weights of the aromatic point mutations may reflect an increase in the hydrodynamic radius of these mutants, or some contribution from the unfolded state whose extended structure should elute much earlier than the globular native state.
Fig. 5.

Gel filtration chromatography of the VHP subdomain single (a) and double (b) mutants. The column was run at 4°C at 0.5 mL/min in 10 mM phosphate buffer pH 7.0 with 150 mM NaCl. Traces corresponding to each peptide are labeled on the figure. The gray traces are molecular weight standards that were spiked with HP36 in (a) only. Note that the chromatograms in (a) and (b) were run on different, but identical HiPrep 16/60 Sephacryl S-100 columns (Pharmacia Biotech) so the elution volumes of both the peptides and molecular weight markers vary slightly between the two figures.
All three double mutants show a significant increase in their apparent molecular weights on the gel filtration column. This effect is most pronounced for the double mutants lacking F58 (F47,58L and F51,58L), which elute at over twice the apparent molecular weight of M53L (Table 1). These mutants are either predominately extended or, potentially, dimeric. In contrast, the apparent molecular weight of F47,51L (4100 D) is low enough that it can be safely assumed to be monomeric like M53L. As with the point mutants, the increase in the apparent molecular weight of F47,51L may arise from an increase in hydrodynamic radius or contributions from the unfolded state.
1D-NMR spectra of VHP subdomain mutants
The downfield region of the 1D-NMR spectra of all three point mutants are qualitatively similar to that of M53L (Fig. 6 ▶). There is a decrease in chemical shift dispersion, which may arise in part from the loss of the aromatic ring in the mutants. The resonances are also slightly broadened relative to that of M53L, suggesting that the point mutants are approaching the intermediate exchange time regime with the unfolded state. Thus, the folded and unfolded states of the VHP subdomain and mutants are interconverting on the submillisecond-to-millisecond time scale, which is consistent with their compact size and helical structure. These effects are seen throughout the spectrum, but are most pronounced at the high field and low field extremes. In particular, the highly downfield shifted amide resonances of serine 43 and aspartate 44 (9.85 and 9.40 ppm in M53L, respectively) are broadened but still clearly present in F58L. The S43 and D44 resonances are also evident in the spectrum of the F47,51L mutant. In contrast, these characteristic resonances are shifted or absent from the spectra of the double mutants that lack F58 (F47,58L and F51,58L). The changes in the NMR spectra of the double mutants lacking F58 indicate that they do not adopt the VHP subdomain fold.
Additional support for the ability of the single point mutants and F47,51L to adopt the near-native VHP subdomain fold comes from the chemical shift of the indole ring of tryptophan 64 located on the opposite side of the VHP subdomain structure from S43 and D44. The Nɛ1-H resonance for all the mutants have similar chemical shifts (10.55 to 10.65 ppm) except for the double mutants lacking F58 where this resonance is closer to random coil chemical shift values. Thus, it appears that the all three point mutants (F47L, F51L, F58L) and the double mutant that retains F58 can all adopt a near-native VHP subdomain fold, even though they are destabilized. The double mutants that lack F58 cannot adopt the VHP subdomain fold.
Discussion
To test whether the hydrophobic core of the VHP subdomain is resistant to mutation, like larger proteins, and to test the importance of the aromatic cluster of the VHP subdomain, we have replaced these three phenylalanine residues with leucine and examined the resulting mutants for folded structure. The replacement of phenylalanine with leucine is a disruptive mutation as the branching of the leucine side chain may cause steric clashes (Richards and Lim 1994). In the absence of any rearrangement in the VHP subdomain structure, the F-to-L mutations would also be expected to leave a void of 35Å3 (Chothia 1975).
Replacement of any individual F with L results in destabilization of the VHP subdomain fold, as evidenced by a decrease in the thermal unfolding midpoint monitored by CD. However, all three F-to-L point mutants retain the wild-type VHP subdomain fold as their native state, as evidenced by CD and 1D-NMR spectroscopy at lower temperature. Thus, replacement of any individual buried phenylalanine residue is tolerated by the VHP subdomain fold. This tolerance to hydrophobic substitution is typical of what is routinely observed in larger proteins, and indicates the VHP subdomain behaves like a native protein with respect to the toleration of mutation.
Replacement of a second phenylalanine residue with leucine results in much more serious structural consequences. Only one of the three possible double mutants, F47,51L, can adopt the VHP subdomain fold, albeit with much lower stability, and only at low temperatures. The F47,51L mutant has secondary and tertiary structure like the native VHP subdomain in the far- and near-UV CD spectra. The F47,51L 1D-NMR spectrum also shows the characteristic downfield-shifted NMR resonances of the VHP subdomain, although they are broadened. It is likely that even at low temperature there is a significant population of the unfolded state of F47,51L in exchange with the native conformation.
The other two double mutants, F47,58L and F51,58L, cannot adopt the native VHP subdomain structure even at 0°C, suggesting that F58 is the most critical of the three phenylalanine residues for specifying the VHP subdomain fold. This is consistent with the order of the thermostability of the three single leucine mutants, where replacement of F58 is the most destabilizing mutation. The F47,58L and F51,58L mutants appear to be predominately unfolded, as evidenced by their lack of near-UV CD signal, low intensity far-UV CD, and lack of dispersion in 1D-NMR spectra. However, there may be residual local structure, or potential molten globule formation, because there is still some intensity in the helical region of the far-UV CD spectrum, and the resonances in the 1D NMR spectrum are broader than those expected for a fully unfolded polypeptide. The elution volume of the F47,58L and F51,58L mutants in the gel filtration experiments are also consistent with a significant amount of unstructured residues resulting in a larger hydrodynamic radius. Although this early elution could also be explained by the formation of fully folded and tightly packed dimers by these mutants, we feel this is unlikely because their CD spectra and 1D-NMR spectra suggest significant unfolding.
The presence of native VHP subdomain structure in the F47,51L double mutant implies that aromatic–aromatic interactions are not essential for specifying the VHP subdomain fold because they cannot be formed by the single remaining aromatic residue in the core. We cannot, however, rule out aromatic–aromatic interactions as important for stabilizing the VHP subdomain. It is important to note that the effect of the individual F47L and F51L mutations on the Tm of F47,51L are not additive. Indeed, the Tm of F47,51L is the same as F47L, which may indicate that F47 and F51 interact strongly, and that loss of F47 in the F47L mutant results in the loss of stabilization by F51 as well. There may also be rearrangements in the packing of other residues in the core that partially compensate for these mutations. The precise contribution of aromatic–aromatic interactions to the VHP subdomain stability awaits studies with nonaromatic isosteres replacing the phenylalanine residues.
Our results indicate that the hydrophobic core of the VHP subdomain is tolerant to mutations like larger "normal" proteins. Therefore, it has the properties of a native protein with respect to the mutability of its hydrophobic core. Our results indicate that the VHP subdomain is a truly minimal protein, and uses the informational code interspersed throughout its sequence to adopt and stabilize its three-dimensional fold.
Mutations have been shown to be tolerated on the surface and at interfacial positions of other minimized/simplified subdomains, for example, in a designed metal-independent 28-residue zinc-finger peptide (Sarisky and Mayo 2001). Here we show that hydrophobic residues buried in the core of the 35-residue VHP subdomain can be mutated without loss of native protein structure. Our findings demonstrate that the VHP subdomain is a useful and viable minimalist model system for both experimental and computational approaches to studying protein folding and stability.
Materials and methods
Construction of VHP subdomain mutants
Molecular biology procedures used standard protocols (Sambrook et al. 1989). The VHP subdomain coding sequence was first cloned into the pTMHa30–51 vector (Schumacher et al. 1996) that encodes a portion of the TrpLE protein with methionine residues replaced with alanine, to yield the VHP subdomain expression plasmid pFHP35. The TrpLE sequence targets the expressed fusion protein to inclusion bodies where even unstable proteins are protected from proteolysis. The VHP subdomain sequence was amplified by the polymerase chain reaction (PCR) from plasmid pVHP42–76b (McKnight et al. 1996) with complementary primers encoding an Nde I site at the 5` end and an EcoR I site at the 3` end. The pTMHa30–51 plasmid was double digested with HinD III and EcoR I, and the VHP subdomain PCR product was double digested with Nde I and EcoR I. The digested PCR product and vector were annealed and ligated in the presence of a HinD III/Nde I adaptor oligonucleotide containing a unique Pst I site. The adaptor oligonucleotide added five in frame amino acids (KLAAA) between the end of the TrpLE fusion protein and the methionine residue to be cleaved by CNBr. Kanamycin resistant colonies were screened for the presence of insert by Pst I digestion and then sequenced (Boston University School of Medicine Protein/DNA Core Facility). Mutations in the VHP subdomain sequence were constructed by PCR with primers containing the desired sequence changes using the QuikChange™ mutagenesis system (Stratagene). All mutations were confirmed by DNA sequencing.
Expression, CNBr cleavage, and purification of mutant VHP subdomains
Recombinant VHP subdomain, which retains an N-terminal methionine (HP36), was expressed and purified as previously described (McKnight et al. 1996). Mutants of the VHP subdomain were produced in BL21 pLysS E. coli cells (Novagen) using the T7 expression system (Studier et al. 1990). Typically, 1 μL cultures grown at 37°C to an O.D. of 0.6 at 600 nm were induced with isopropyl β-d-thiogalactopyranoside (120 μg/mL), and harvested 4–5 h postinduction by centrifugation. The pellets were resuspended in 20 mL of 20 mM tris-hydroxymethylaminomethane (TRIS), 5 mM MgCl2, and 0.01 mg/mL DNaseI at pH 7.5 and sonicated to lyse the cells, then spun at 4°C for 30 min at 35,000 × g. The inclusion body pellet was resuspended in 20 mL of 20 mM TRIS, 1 mM ethylenediamine tetraacetic acid, 1% Triton X-100, and 1% deoxycholic acid at pH 7.5 then sonicated to homogeneity and spun for 30 min at 20,000 × g at 4°C. The pellet was solubilized in 10 mL of 20 mM TRIS, 6 M guanidine hydrochloride, and 1 mM dithiothreitol at pH 7.5. The mixture was diluted 1:10 with water and spun for 30 min at 17,000 × g at 4°C to recover the purified inclusion body pellet.
The inclusion body pellet was resolubilized in 5 mL of 70% formic acid, and solid CNBr (0.5 to 1 g) was added and allowed to react for 2 h at room temperature in the dark with constant stirring. After cleavage, 5 mL of water was added to the reaction, and the CNBr was removed by rotary evaporation to near dryness. The cleavage products were then extensively dialyzed in 3500 MW cutoff dialysis tubing against 10 mM phosphate, 1 mM DTT at pH 7. Cleaved TrpLE leader sequence and uncleaved fusion protein precipitated during the dialysis leaving the desired point mutants in solution. The cleaved point mutants were then further purified by reverse-phase HPLC on a C18 preparative column (Vydac 2 × 25 cm) using an acetonitrile, 0.1% trifluoroacetic acid gradient. The molecular weights of the purified mutant subdomains were verified by mass spectral analysis (Boston University School of Medicine Mass Spectrometry Resource).
Circular dichroism of VHP subdomain mutants
CD spectra were acquired on an Aviv 62DS or 60DS spectrometer equipped with a solid-state temperature controlled cell holder. For far-UV wavelength scans and thermal unfolding experiments, samples were 40 to 60 μM protein in 50 mM phosphate, pH 7.0. For near-UV CD spectra, samples contained 100–225 μM peptide also in 50 mM phosphate buffer, pH 7.0. The samples were dialyzed against 50 mM phosphate pH 7.0 before their exact concentrations were determined by absorbance at 280 nm with the dialysis buffer as a reference and using an extinction coefficient of 5690 cm−1 M−1 (Edelhoch 1967).
Far-UV scans were collected at 1 nm intervals from 260 to 190 nm at 4°C as the average of three scans with a 5 s integration time in a 1 mm path length cell. Near-UV scans were collected at 4°C as the average of five scans with a 5 s integration time in a 10 mm path length cell, from 310 to 250 nm, using a 0.5 nm step size. For near- and far-UV CD scans, a spectrum of buffer alone was subtracted from each scan to correct for the baseline signal.
For thermal unfolding experiments, data points were collected at 222 nm, with 20 s of signal averaging at 1° intervals from 0 to 100°C after a 45 s temperature equilibration time in a 2 mm path length cell. Each thermal unfolding experiment was baseline corrected against an experiment with buffer alone. To determine the midpoint (Tm) of the thermal unfolding curves, the derivative of the signal with respect to temperature was used to pinpoint the inflection point (Cantor and Schimmel 1980).
Gel filtration chromatography
Peptides (10 to 80 nmole) in 200 μL of 150 mM NaCl, 10 mM phosphate buffer, pH 7.0 were injected on to a HiPrep 16/60 Sephacryl S-100 FPLC column (Pharmacia Biotech) equilibrated in the same buffer. The column was run at 0.5 mL/min at 4°C, and the eluate was monitored at 280 nm. Low molecular weight gel filtration standards (Pharmacia Biotech) spiked with HP36, which has been shown to be monomeric by analytical ultracentrifugation (McKnight et al. 1996), were used to calibrate the column.
NMR spectroscopy
NMR spectra were acquired on a Bruker DMX500 spectrometer with approximately 1 mM peptide. All 1D spectra were taken at 4°C, in 10 mM phosphate buffer, pH 7.0 and referenced against 1 mM 3-(trimethylsilyl) tetradeutero sodium propionate. Water suppression was achieved by presaturation of the water signal, or with gradients using the WATERGATE pulse sequence (Piotto et al. 1992).
Acknowledgments
This work was supported by the Department of Biophysics at BUSM, and NIH Grant HL07291 (B.S.F.). We thank M. Milhollen and Dr. P. Kim for the gift of the pTMHa30–51 plasmid, Dr. C. Costello for assistance with mass spectroscopy, and Drs. M. Cordes and R. Sauer for CD spectrometer time.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
CD, circular dichroism
DTT, dithiothreitol
FPLC, fast-performance liquid chromatography
HPLC, high-performance liquid chromatography
L, leucine
M, methionine
NMR, nuclear magnetic resonance
OD, optical density
F, phenylalanine
PCR, polymerase chain reaction
TRIS, tris-hydroxymethylaminomethane
V, valine
W, tryptophan
Y, tyrosine
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.22202.
References
- Bazari, W.L., Matsudaira, P.T., Wallek, M., Smeal, T., Jakes, R., and Ahmed, Y. 1988. Villin sequence and peptide map identify six homologous domains. Proc. Natl. Acad. Sci. 85 4986–4990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryson, J.W., Desjarlais, J.R., Handel, T.M., and DeGrado, W.F. 1998. From coiled coils to small globular proteins: Design of a native-like three-helix bundle. Protein Sci. 7 1404–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burley, S. and Petsko, G. 1986. Dimerization energetics of benzene and aromatic amino acid side chains. J. Am. Chem. Soc. 108 7995–8001. [Google Scholar]
- Burley, S.K. and Petsko, G.A. 1988. Weakly polar interactions in proteins. Adv. Protein Chem. 39 125–189. [DOI] [PubMed] [Google Scholar]
- Cantor, C.R. and Schimmel, P.R. 1980. Biophysical chemistry, vol. 3. W.H. Freeman and Co., New York.
- Chothia, C. 1975. Structural invariants in protein folding. Nature 254 304–308. [DOI] [PubMed] [Google Scholar]
- Dahiyat, B.I. and Mayo, S.L. 1997. De novo protein design: Fully automated sequence selection. Science 278 82–87. [DOI] [PubMed] [Google Scholar]
- DeGrado, W. and Sosnick, T. 1996. Protein minimization: Downsizing through mutation. Proc. Natl. Acad. Sci. 93 5680–5681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan, Y. and Kollman, P. 1998. Pathways to a protein folding intermediate observed in a 1 microsecond simulation in aqueous solution. Science 282 740–744. [DOI] [PubMed] [Google Scholar]
- Duan, Y., Wang, L., and Kollman, P.A. 1998. The early stage of folding of villin headpiece subdomain observed in a 200-nanosecond fully solvated molecular dynamics simulation. Proc. Natl. Acad. Sci. 95 9897–9902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelhoch, H. 1967. Spectroscopic determination of tryptophan and tyrosine in proteins. Biochemistry 6 1948–1954. [DOI] [PubMed] [Google Scholar]
- Fezoui, Y., Connolly, P.J., and Osterhout, J.J. 1997. Solution structure of alpha t alpha, a helical hairpin peptide of de novo design. Protein Sci. 6 1869–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs, N., Clarke, A.R., and Sessions, R.B. 2001. Ab initio protein structure prediction using physicochemical potentials and a simplified off-lattice model. Proteins Struct. Funct. Genet. 43 186–202. [DOI] [PubMed] [Google Scholar]
- Hansen, K., Rock, R., Larsen, R., and Chan, S. 2000. A method for photoinitating protein folding in a nondenaturing environment. J. Am. Chem. Soc. 122 11567–11568. [Google Scholar]
- Hunter, C.A., Singh, J., and Thornton, J.M. 1991. Pi-pi interactions: The geometry and energetics of phenylalanine–phenylalanine interactions in proteins. J. Mol. Biol. 218 837–846. [DOI] [PubMed] [Google Scholar]
- Kalia, Y.N., Brocklehurst, S.M., Hipps, D.S., Appella, E., Sakaguchi, K., and Perham, R.N. 1993. The high-resolution structure of the peripheral subunit-binding domain of dihydrolipoamide acetyltransferase from the pyruvate dehydrogenase multienzyme complex of Bacillus stearothermophilus. J. Mol. Biol. 230 323–341. [DOI] [PubMed] [Google Scholar]
- Koradi, R., Billeter, M., and Wüthrich, K. 1996. MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graph. 14 51–55. [DOI] [PubMed] [Google Scholar]
- Kristensen, P. and Winter, G. 1998. Proteolytic selection for protein folding using filamentous bacteriophages. Fold. Des. 3 321–328. [DOI] [PubMed] [Google Scholar]
- Lee, M.R., Baker, D., and Kollman, P.A. 2001. 2.1 and 1.8 angstrom average C-alpha RMSD structure predictions on two small proteins, HP-36 and S15. J. Am. Chem. Soc. 123 1040–1046. [DOI] [PubMed] [Google Scholar]
- Lee, M.R., Duan, Y., and Kollman, P.A. 2000. Use of MM-PB/SA in estimating the free energies of proteins: Application to native, intermediates, and unfolded villin headpiece. Proteins Struct. Funct. Genet. 39 309–316. [PubMed] [Google Scholar]
- McKnight, C.J., Doering, D.S., Matsudaira, P.T., and Kim, P.S. 1996. A thermostable 35-residue subdomain within villin headpiece. J. Mol. Biol. 260 126–134. [DOI] [PubMed] [Google Scholar]
- McKnight, C.J., Matsudaira, P.T., and Kim, P.S. 1997. NMR structure of the 35-residue villin headpiece subdomain. Nat. Struct. Biol. 4 180–184. [DOI] [PubMed] [Google Scholar]
- Piotto, M., Saudek, V., and Sklenar, V. 1992. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR 2 661–665. [DOI] [PubMed] [Google Scholar]
- Plaxco, K.W., Riddle, D.S., Grantcharova, V., and Baker, D. 1998. Simplified proteins: Minimalist solutions to the "protein folding problem." Curr. Opin. Struct. Biol. 8 80–85. [DOI] [PubMed] [Google Scholar]
- Richards, F.M. and Lim, W. 1994. An analysis of packing in the protein folding problem. Q. Rev. Biophys. 26 423–498. [DOI] [PubMed] [Google Scholar]
- Sambrook, J., Fritsch, E.F., and Maniatis, T. 1989. Molecular cloning: A laboratory course, 2nd ed. Cold Spring Harbor Press, Cold Spring Harbor, NY.
- Sarisky, C.A. and Mayo, S.L. 2001. The beta-beta-alpha fold: Explorations in sequence space. J. Mol. Biol. 307 1411–1418. [DOI] [PubMed] [Google Scholar]
- Schumacher, T.N., Mayr, L.M., Minor, Jr., D.L. , Milhollen, M.A., Burgess, M.W., and Kim, P.S. 1996. Identification of D-peptide ligands through mirror-image phage display. Science 271 1854–1857. [DOI] [PubMed] [Google Scholar]
- Spector, S., Kuhlman, B., Fairman, R., Wong, E., Boice, J.A., and Raleigh, D.P. 1998. Cooperative folding of a protein mini domain: The peripheral subunit-binding domain of the pyruvate dehydrogenase multienzyme complex. J. Mol. Biol. 276 479–489. [DOI] [PubMed] [Google Scholar]
- Struthers, M.D., Cheng, R.P., and Imperiali, B. 1996. Design of a monomeric 23-residue polypeptide with defined tertiary structure. Science 271 342–345. [DOI] [PubMed] [Google Scholar]
- Studier, F.W., Rosenberg, A.H., Dunn, J.J., and Dubendorff, J.W. 1990. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 185 60–89. [DOI] [PubMed] [Google Scholar]
- Sullivan, D.C. and Kuntz, I.D. 2001. Conformation spaces of proteins. Proteins Struct. Funct. Genet. 42 495–511. [PubMed] [Google Scholar]
- Vardar, D., Buckley, D.A., Frank, B.S., and McKnight, C.J. 1999. NMR structure of an F-actin binding "headpiece" motif from villin. J. Mol. Biol. 249 1299–1310. [DOI] [PubMed] [Google Scholar]
- Woody, R.W. 1995. Circular dichroism. Methods Enzymol. 246 34–71. [DOI] [PubMed] [Google Scholar]