

Abstract
Esterases form a diverse class of enzymes of largely unknown physiological role. Because many drugs and pesticides carry ester functions, the hydrolysis of such compounds forms at least one potential biological function. Carboxylesterases catalyze the hydrolysis of short chain aliphatic and aromatic carboxylic ester compounds. Esterases, d-alanyl-d-alanine-peptidases (DD-peptidases) and β-lactamases can be grouped into two distinct classes of hydrolases with different folds and topologically unrelated catalytic residues, the one class comprising of esterases, the other one of β-lactamases and DD-peptidases. The chemical reactivities of esters and β-lactams towards hydrolysis are quite similar, which raises the question of which factors prevent esterases from displaying β-lactamase activity and vice versa. Here we describe the crystal structure of EstB, an esterase isolated from Burkholderia gladioli. It shows the protein to belong to a novel class of esterases with homology to Penicillin binding proteins, notably DD-peptidase and class C β-lactamases. Site-directed mutagenesis and the crystal structure of the complex with diisopropyl-fluorophosphate suggest Ser75 within the "β-lactamase" Ser-x-x-Lys motif to act as catalytic nucleophile. Despite its structural homology to β-lactamases, EstB shows no β-lactamase activity. Although the nature and arrangement of active-site residues is very similar between EstB and homologous β-lactamases, there are considerable differences in the shape of the active site tunnel. Modeling studies suggest steric factors to account for the enzyme's selectivity for ester hydrolysis versus β-lactam cleavage.
Keywords: α/β-Hydrolase, β-lactamase, enzyme mechanism, esterase
Esterases are widely distributed in animals, plants, and micro-organisms (Okuda 1991). With the notable exception of lipases, the physiological role of most members of this large class of hydrolytic enzymes is still unknown. In recent years, esterases and lipases have attracted interest from the side of organic chemistry and biotechnology, where they are exploited to catalyze stereo- and enantioselective chemical reactions thanks to their broad substrate specifity and high stereoselectivity (Toone et al. 1990; Faber 1997; Schulze and Wubbolts 1999; Faber 2000). Lipases already now account for about 20% of all biotransformations (Faber 2000).
Most esterases belong to the α/β hydrolase superfamily, which share the short consensus sequence Gly-x-Ser-x-Gly. Its central serine is the terminal residue of the catalytic triad consisting of residues Ser-His-Asp(Glu) (Ollis et al. 1992), whose function consists in orienting and activating the Serine-Oγ (Wharton 1998) to act as nucleophile in this class of hydrolases.
EstB is an esterase whose gene was identified by screening a genomic library of Burkholderia gladioli (previously Pseudomonas marginata) on tributyrin plates (Stubenrauch 1991). It was cloned and expressed in E. coli. The water-soluble enzyme (392 amino acid residues, MW = 40,000 Daltons) shows very specific activity on short-chain (C4–C6) fatty acid esters and triglycerides (Petersen 1995; Petersen et al. 2001). Notably, it is able to hydrolize esters of tertiary alcohols, such as linalyl acetate (Schlacher et al. 1998). Its biological function is as yet unknown, but may be related to the enzyme's high activity for the esterolysis of 7-aminocephalosporinic acid (Petersen et al. 2001). The primary structure exhibits homology to esterases from family VIII (Arpigny and Jaeger 1999) and modest homologies to β-lactamases, DD-peptidases, and penicillin-binding proteins. Both the serine hydrolase motif Gly-x-Ser-x-Gly and the motif Ser-x-x-Lys, characteristic for β-lactamases, are present in its sequence (Petersen 1995; Petersen et al. 2001). However, although β-lactamases of classes A and C also contain a conserved triad of Lys-Ser/Thr-Gly (KTG-box) between the Ser-x-x-Lys motif and the C-terminus, no such signature exists in the sequence of EstB.
Site-directed mutagenesis yielded only modest reduction of the activity upon replacement of the serine within the G-x-S-x-G motif by alanine, but complete loss of activity as a result of the corresponding exchange within the Ser-x-x-Lys motif (Petersen et al. 2001). Thus, the catalytic residue appears to be located within the "β-lactamase" motif and not within the "esterase" motif.
β-Lactamases and the phylogenetically related d-alanyl-d-alanine carboxypeptidase/transpeptidases (DD-peptidases; Kelly et al. 1986; Samraoui et al. 1986; Knox et al. 1996) have a very distinct fold compared to the above α/β-hydrolases. Although they are also α/β structures containing a β-sheet core flanked by α-helices, their β-sheet consists mainly of antiparallel strands, and the catalytic serine—which is not part of a triad—is at the beginning of an α-helix adjacent to the central sheet. Two other esterases were found to show sequence homology to class C penicillin-binding proteins (PBPs; McKay et al. 1992; Holm and Sander 1996), and cross-reactivities between "esterases" and β-lactamases have been noted (Pratt and Govardhan 1984; Govardhan and Pratt 1987; Jones and Page 1991).
Despite the sequence similarity to β-lactamases and DD-peptidases, EstB shows no β-lactamase or peptidase activity. In fact, when incubated with 7-aminocephalosporinic acid (7-ACA), which contains a β-lactam ring and an ester group, it cleaves the ester bond but leaves the β-lactam unaffected (Petersen et al. 2001). Following the crystallization of this enzyme (Wagner et al. 1997), we now report its crystal structure, which should yield insight into the structural factors discriminating between esterolytic and β-lactam-cleaving activity. To identify the active-site nucleophile, and to probe possible conformational changes during catalysis, we also determined the structure of the reaction product between EstB and diisopropyl-fluorophosphate (DFP). The complex can be considered as an analog of the tetrahedral intermediate formed as a result of the nucleophilic attack by the enzyme on the substrate's carbonyl group (Lobkovsky et al. 1994).
Results and Discussion
Solution and refinement of the crystal structure
The structure was solved by multiple isomorphous replacement using seven derivatives (Table 1), and refined against diffraction data extending to 2.0 Å resolution. A final map (Fig. 1 ▶) shows well-defined density throughout most of the structure, with the exception of the 14 N-terminal and the last C-terminal residues, which could not be observed in either of the two crystallographically unrelated molecules, and of a β-sheet covering the active site (residues 251 to 254), where diffuse density indicates disorder. The final model also includes 357 water and 4 isopropanol molecules. The side chains of several residues (Cys217 in both monomers and Asp 97 in monomer A) had to be refined in two alternate conformations. The stereochemical parameters of the model are satisfactory (Table 2); a Ramachandran plot shows no residues in "disallowed" regions of φ/Ψ space, with Ala74 and Asp294 (in both molecules) being the only residues in "disfavoured" regions (Laskowski et al. 1993). Ala74 is preceding the active Serine residue, and shows this apparently strained φ/Ψ combination also in other β-lactamase structures (Knox and Moews 1991; Lobkovsky et al. 1993).
Table 1.
Data collection and MIR analysis
| Data set | Maximum resolution | Measured reflections | Unique reflections | Rmergea,b (%) | Completeness (%) | Number of sites | Rderivc (%) | Rcullisd (%) | Phasing powere | Used resolution |
| PCMBSf | 2.80 | 36749 | 14245 | 4.5 (6.8) | 72 (48) | 2 | 22.4 | 0.83 | 1.09 | 25–3.0 |
| EMPh | 2.80 | 75904 | 18955 | 6.0 (8.1) | 96 (91) | 2 | 24.3 | 0.79 | 1.22 | 25–3.0 |
| UO3Ac2 • 2H2O | 3.00 | 55982 | 14633 | 6.3 (9.5) | 91 (86) | 2 | 22.9 | 0.72 | 1.77 | 25–3.0 |
| PIPg | 2.80 | 51718 | 15007 | 6.6 (13.6) | 76 (54) | 2 | 23.8 | 0.71 | 1.75 | 25–3.0 |
| K2PtCl4 | 2.80 | 19944 | 12951 | 5.1 (9.0) | 64 (40) | 2 | 17.7 | 0.88 | 0.85 | 25–3.0 |
| K[Au(II)(CN)2] | 2.80 | 31518 | 15553 | 4.5 (6.4) | 79 (60) | 2 | 25.8 | 0.92 | 0.68 | 25–3.0 |
| PdCl2 | 2.80 | 27849 | 16425 | 3.8 (5.5) | 83 (62) | 2 | 16.4 | 0.94 | 0.56 | 25–3.0 |
| DFP-derivative | 1.80 | 235197 | 69158 | 2.4 (10.8) | 94 (97) | — | — | — | — | 15–1.8 |
| Native | 1.80 | 188976 | 49177 | 5.3 (13.8) | 68 (27) | — | — | — | — | 20–2.0 |
a Numbers in parentheses in the columns for Rmerge and Completeness correspond to data from the highest resolution shell.
bRmerge = ∑|Ii − Im/∑|m where Ii and Im are the observed intensity and mean intensity of related reflections respectively.
cRderiv = ∑|FPH − FP|/∑|FP|, where FPH is the structure–factor amplitude of the derivative crystal and FP is that of the native crystal.
dRcullis = ∑||FPH ± FP| − |FH(calc)||/∑|FPH − FP|, where FPH and FP are defined as above and FH(calc) is the calculated heavy atom structure–factor amplitude.
e Phasing power = FH/E, the root mean square (rms) heavy-atom structure–factor amplitude divided by the lack of closure.
f PCMBS = p-chlormercuriphenylsulfonic acid monosodium salt.
g PIP = Di-μ-iodobis(ethylenediamine)-di-platinum (II) nitrate.
h EMP = ethylene mercury phsophate.
Fig. 1.
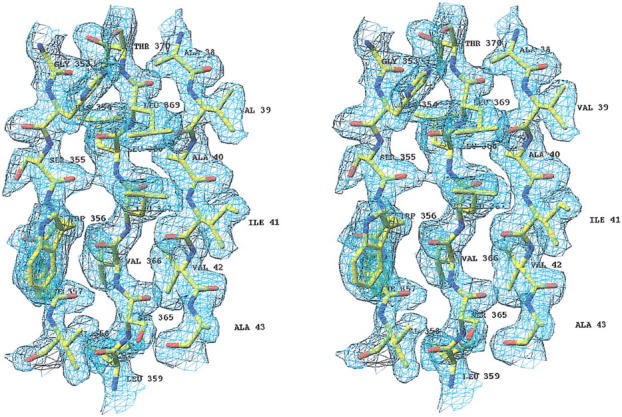
Difference density (3Fo–2Fc map, 1σ level) of a portion of the EstB structure to illustrate the quality of the final phases. Figure produced with RIBBONS (Carson 1997).
Table 2.
Structure statistics
| Structure refinement and model | Native | DFP-derivate |
| Residue range (molA/molB) | 15–391/15–391 | 15–391/15–391 |
| No. nonhydrogen atoms (molA/molB) | 2836/2836 | 2846/2846 |
| No. water molecules | 354 | 571 |
| Hetero atoms | 16 | |
| Resolution range (Å) (completeness in %) | 20.0–2.0 (83) | 15–1.8 (95) |
| R/Rfree | 18.4/24.7 | 17.6/23.5 |
| rms deviation from ideal geometry | ||
| Bond length (Å) | 0.012 | 0.027 |
| Bond angles (°) | 1.73 | 1.89 |
| Dihedral angles (°) | 24.64 | 25.5 |
| Improper torsions (°) | 1.81 | 1.37 |
| Ramachandran plot (with PROCHECK) | ||
| Core regions (%) | 89 | 90 |
| Allowed regions (%) | 10 | 10 |
| Generously allowed regions (%) | 0.7 | 0.2 |
| Not allowed regions (%) | 0.0 | 0.0 |
| Temperatur factor model | ||
| Average temperatur factor (all atoms ) (Å2) | 12 | 23 |
| Temperatur factor (mainchain) (Å2) | ||
| Average (molA/molB) | 10.3/11.2 | 19/21 |
| Maximum (molA/molB) | 41.5/34.2 | 93/75 |
| Temperatur factor (sidechain) (Å2) | ||
| Average (molA/molB) | 11.2/12.4 | 23/25 |
| Maximum (molA/molB) | 45.3/50.4 | 101/117 |
| Superposition of NCS-related residues 15–391 | ||
| Deviation Cα atoms rms | 0.27 | 0.27 |
| Deviation all atoms rms | 0.74 | 0.77 |
There are two crystallographically nonequivalent molecules (A and B) within the asymmetric unit, which are related by a pseudo-twofold rotation of 177.6° without a translation component. The two molecules superimpose with a root mean square (RMS) deviation of 0.27 Å for 377 main-chain atoms. Only the vicinities of residues 113 and 235 show deviations larger than 1.2 Å. Other regions with RMS deviations exceeding 0.6 Å are loop regions with poorly defined density.
Molecular architecture
Because molecules A and B are very similiar, the description of the structure will be confined to molecule A, which yielded a slightly clearer density. Figure 2a ▶ shows a ribbon-type representation of the molecule, drawn in the same orientation as the Cα-trace shown in Figure 2b ▶. The central part of the structure consists of a seven stranded, antiparallel β-sheet, with the N- and the C-terminal helices covering the "front" and two other helices covering the "back" of this sheet. Residue Ser75, which occurs within the Ser-x-x-Lys motif, is located at the beginning of the second helix.
Fig. 2.
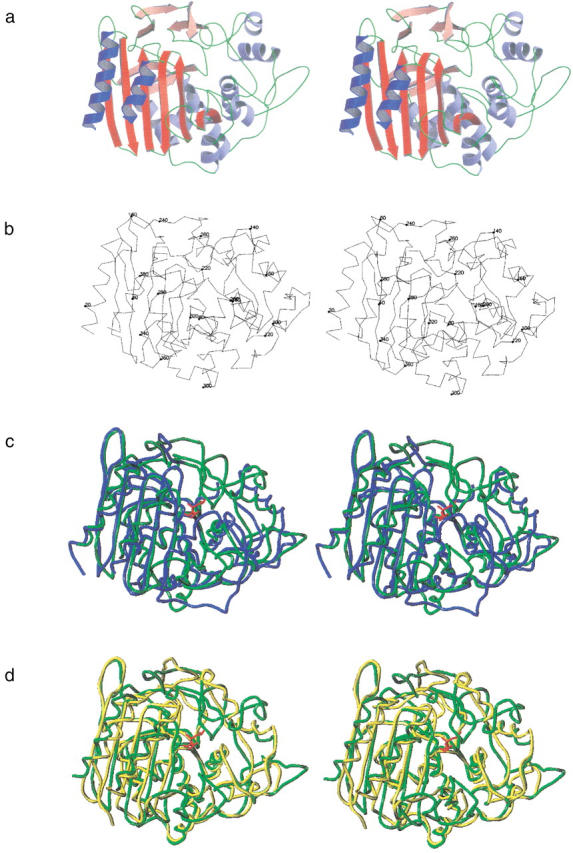
Chain fold of EstB from B. gladioli. (a) Stereoscopic ribbons diagram with β-sheets in red and α-helices in blue. (b) Stereoscopic Cα-trace in the same orientation. (c) Superposition of EstB (green) with class C β-lactamase P99 from Enterobacter cloacae (1bls, blue; Lobkovsky et al. 1993), and (d) with d-alanyl-d-alanine carboxypeptidase from Streptomyces R61 (3pte, yellow; Kelly et al. 1989). The covalently attached phosphonate inhibitor of EstB is drawn in red. (a) and (b) were generated with MOLSCRIPT (Kraulis 1991) and rendered with Raster3D (Merritt and Bacon 1997); (c) and (d) were generated with program SYBYL (Tripos).
Two cis amino acids are found in both monomers: Pro236 adopts the cis conformation to allow a short connection between two adjacent β-chains; Tyr374 is located in the loop connecting the C-terminal helix with its preceding strand, which makes a sharp bend stabilized by hydrogen bonding between main-chain atoms (O373–NH233; O374–NH377).
Binding of diisopropyl-fluorophosphate
Figure 3a ▶ shows a difference density map of the active site for the complex between EstB and diisopropyl-fluorophosphate (DFP), indicating covalent attachment of the inhibitor molecule through Oγ of Ser75, presumably as the result of a nucleophilic substitution reaction that replaced fluorine by oxygen. Evidently, the phosphonyl oxygen of the phosphotriester hydrogen bonds to the main-chain NH groups of Ser75 and Val351 (see Fig. 3b ▶). Otherwise, the conformational consequences of inhibitor binding are minimal: the RMS differences of Cα-atoms between the native structure and the DFP derivative are 0.27 Å for molecules A and B.
Fig. 3.
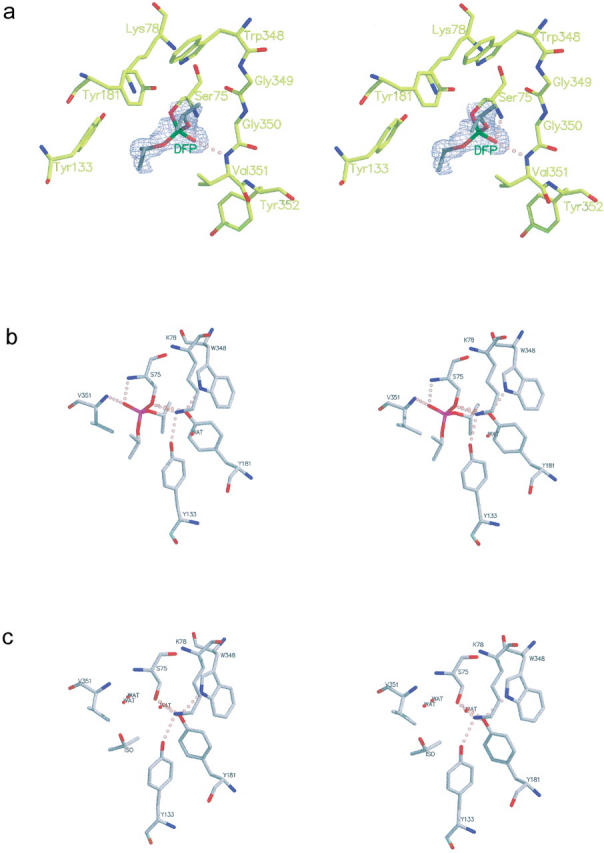
Difference density map (Fo–Fc, 3σ contours) of the active site of the EstB–DFP complex (a) and stereoscopic representations of the active-site residues of EstB with (b) and without (c) the covalently attached DPF inhibitor. Note that the feature labeled "ISO" represents a molecule of isopropanol observed near the active site of the native structure.
Comparison with other esterases
Esterases have been classified into eight families according to conserved sequence motifs and biological properties (Arpigny and Jaeger 1999). According to this classification EstB belongs to family VIII, which contains enzymes with approximately 380 residues showing sequence similarity to class C β-lactamases. The esterase/lipase consensus sequence Gly-Xaa-Ser-Xaa-Gly is found in some members of this family, but is not believed to contribute to catalysis. In addition, all known sequences of family VIII esterases share the lactamase/peptidase sequence motif Ser-Xaa-Xaa-Lys. To our knowledge, no three-dimensional (3D) structure of a family VIII esterase has been reported so far. Therefore, we will focus the structural comparisons of EstB to lactamases and peptidases rather than to esterases of other families.
Homomolgy with α/β-hydrolases of similar fold
β-Lactamases and transpeptidases show close tertiary structural homology. Their 3D structure consists of two structural domains, an all-α and an α/β one (Matagne et al. 1998). The active site is located in a groove between the two domains near the N-terminus of the first helix of the all-α domain. The α/β domain consists of a central β-sheet flanked by three helices on the one and one helix on the opposite face. There is some variation in the width of the central β-sheet, which varies between five (class A β-lactamases), nine (class C), and seven strands (DD-peptidase and class D). Another variable structural element is a 20–40-residues long peptide loop that borders the active site and is occasionally termed the ω-loop (Knox et al. 1996). This loop is generally longer, more twisted, and runs in opposite direction in the class C β-lactamases and in the DD-peptidases compared to class A lactamases, where it carries a presumably catalytic glutamate residue (Glu166) not present in the other two enzyme classes (Knox and Moews 1991; Lobkovsky et al. 1993; Knox et al. 1996).
We used the program DALI (Holm and Sander 1996) for a more quantitative comparison between EstB and members of the four classes of lactamases and peptidases. The most pronounced homology was obtained for the d-alanyl-d-alanine carboxypeptidase from Streptomyces R61 (transpeptidase, pdb entry 3pte; Kelly et al. 1989; Z-score 33.0) and the class C β-lactamase Gc1 (a natural mutant of the wild-type P99 enzyme) from Enterobacter cloacae (pdb-entry 1gce; Crichlow et al. 1999; Z-score 28.7). Similarities to β-lactamases of class A (pdb-entry 1btl; Jelsch et al. 1993; Z-score 14.4) and class D (pdb-entry 1e3u, Z-score 12.1) are less striking. The following discussion will, therefore, focus on the structural comparison between EstB and d-alanyl-d-alanine carboxypeptidase from Streptomyces R61 (Kelly et al. 1989) and class C-β-lactamase P99 from Enterobacter cloacae (Lobkovsky et al. 1993). Such a comparison is shown in Figure 2c and d ▶, based on a superposition of equivalent residues indicated by DALI. Although the overall fold of the three enzymes is obviously similar, there are significant differences affecting the accessibility of the active site from the surrounding solvent. While in class C lactamases the substrate could enter the active site from "above" (i.e., from the top of the page for the molecular orientation of Fig. 2 ▶), the analogous accession path is obstructed in the DD-peptidase and in EstB, leaving access to the active site from the "front" (i.e., from the viewer). Differences between the DD-peptidase and EstB mainly concern an additional β-sheet (residues 249 to 255) and a loop (residues 314 to 320), which cover part of the active site entrance in EstB, thus narrowing the access path to the active serine (Fig. 2d ▶).
Identification of catalytic residues and implications for catalysis
X-ray diffraction studies have revealed striking structural similarities between the active sites of Streptomyces R61 DD-peptidase and class A and class C β-lactamases (Dodson and Wlodawer 1998; Matagne et al. 1998). Conserved elements have been identified, and their relevance for substrate recognition and its catalytic conversion has been putatively assigned. Some of these elements are also observed in EstB, though in somewhat modified form.
The first element has the sequence Ser-Xaa-Xaa-Lys, and consists of the serine believed to act as catalytic nucleophile plus a lysine residue one helix turn downstream. This element is also observed in EstB. The second common element, a short loop (sometimes called the SDN-loop) in the all-α domain forms one wall of the catalytic cavity and consists of a Tyr-Xaa-Asn (Class C β-lactamases) or Ser-Xaa-Asn (class A β-lactamases) sequence motif. Side chains from the first and third residue of this motif point towards the active site. The analogous loop in EstB has the sequence Tyr-Ser-Leu, with the tyrosine hydroxyl group near the active site but the leucin residue pointing away. A tyrosine from an adjacent loop is observed at the equivalent position of the asparagine from the SDN loop.
The third element is formed by amino acids from the α/β-domain, and forms the opposite wall of the catalytic cavity. Typically, a Lys-Thr-Gly (KTG-box) sequence is observed at this location, but Lys may be replaced by His or Arg, and Thr may be replaced by Ser. A positively charged side chain followed by one bearing a hydroxyl group has been suggested to be universally conserved (Matagne et al. 1998). The third residue of the KTG box must be glycine, because any side chain would impair the approach of the substrate. In EstB, the space equivalent to the KTG-box is occupied by a peptide with sequence Trp-Gly-Gly.
A fourth element, the so-called Ω-loop bearing a negatively charged glutamate residue occurs in class A β-lactamases. This glutamate (E166) is believed to play an important role in the catalytic reaction of class A β-lactamases. The Ω-loop is also observed in class C β-lactamases, but there it does not contribute to the catalytic reaction. No counterpart to the Ω-loop can be identified in EstB.
Class A β-lactamases are most widespread and have been most thoroughly studied. Much less is known about class C β-lactamases and DD-peptidases. The following discussion of potential catalytic residues in EstB will therefore also include results from class A β-lactamases, although the three dimensional homology is not as high as it is to the other two enzyme classes.
There is no doubt that hydroxyl of the serine within the SxxK motif is the catalytic nucleophile, which eventually forms a tetrahedral intermediate with the substrate. The question, however, of which residue acts as a general base, is still a matter of debate. For class A β-lactamases Lys73 (from the SxxK motif; Herzberg 1991; Strynadka et al. 1992, 1996; Dodson and Wlodawer 1998) or Glu166 (located on the Ω-loop, Matagne et al. 1998; Maveyraud et al. 1998; Page and Laws 1998) are candidates. However, to function as a general base the pKa of Lys73 would have to be drastically decreased. This has been suggested to occur as a result of substrate binding (Swaren et al. 1995; Zawadzke et al. 1996), but was questioned on the basis of experimental (Damblon et al. 1996) and theoretical (Lamotte-Brasseur et al. 1999) evidence. On the other hand, the removal of Glu166 by mutagenesis results in long-lived acyl-intermediates (Escobar et al. 1991, 1994; Banerjee et al. 1998). In fact, it is quite possible that different amino acids act as general bases for the acylation and for the deacylation steps (Herzberg et al. 1991; Strynadka et al. 1992), possibly with the involvement of a conserved water molecule (Zawadzke et al. 1996; Massova and Mobashery 1998; Lamotte-Brasseur et al. 1999).
In class C β-lactamases and in DD-peptidases, there is no counterpart to Glu166. Tyr150 or Tyr159 (DD-peptidase) respectively might act as the general base (Oefner et al. 1990; Lobkovsky et al. 1994; Dubus et al. 1996). The following residues are believed to be involved in catalysis (residue numbers correspond to the E. cloacae enzyme with the ones of the Streptomyces R61 DD-peptidase in parentheses; see Fig. 4 ▶): Ser64 (Ser62) as the nucleophile, Tyr150 (Tyr159) as the general base, Lys67 (Lys65) and Lys315 (His298) surrounding Tyr150 (Tyr159) and supplying positive charges to stabilize the anion form. Asn152 (Asn162) is interacting with Lys67 (Lys65) through a hydrogen bond, and simultaneously interacts with the substrate. Oxyanion stabilization of the acyl-enzyme intermediate is achieved by hydrogen bonding between the substrate's carbonyl oxygen and the main-chain NH groups of Ser64 (Ser62) and Ser318 (Thr301).
Fig. 4.
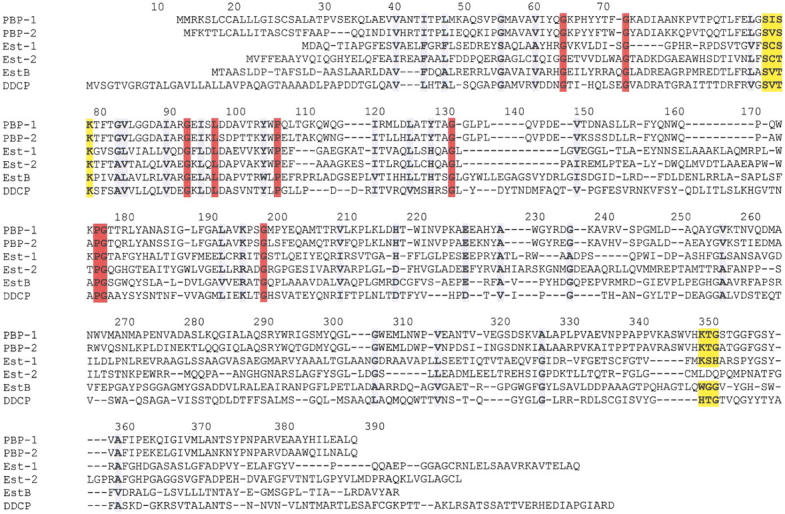
Multiple sequence alignment of several esterases, class C β-lactamases and a D,D-carboxypeptidase. PBP-1, Enterobacter cloacae β-lactamase P99 (Swissprot accession code Q59401); PBP-2, E. coli cephalosporinase (Q59398); Est-1 Arthrobacter globiformis esterase (Nishizawa et al. 1995); Est-2, Pseudomonas sp. strain LS107d2 esterase (McKay et al. 1992); EstB, Burkholderia gladioli esterase (Petersen et al. 2001); DDCP, Streptomyces R61 D,D-carboxypeptidase (Joris et al. 1988). Alignment was performed with program pileup from the GCG sequence analysis package (University of Wisconsin) using 2,3 penalty. The active-site residues and the residues of the Lys-Thr-Gly signature (for EstB the topologically equivalent residues) are highlighted with a yellow background. A red background denotes amino acids fully conserved in all six peptides, a gray one denotes well-conserved amino acids. The numbering corresponds to the amino acid position of the B. gladioli esterase.
The superposition of the active sites of the β-lactamase P99 from E. cloacae (Lobkovsky et al. 1994) with EstB is shown in Figure 5 ▶. It reveals a striking similarity between the active site architecture of the two enzymes, despite two amino acid exchanges, which places Nɛ1-Trp348 of EstB at approximately the same position as Nɛ-Lys315 of P99; a similar correspondence is found between Oη-Tyr133 (EstB) and Nδ-Asn152 (P99). Together with the sequence data (Fig. 4 ▶), these homologies lead to the assignment of structurally equivalent residues between P99 and EstB, as summarized in Table 3. Together with the result of the DFP-binding experiment and the mutagenesis data (no esterolytic activity of the Ser75Ala mutant; Petersen et al. 2001), this suggests the following functional significance for active-site residues in EstB: the Ser75-oxygen acts as nucleophile and attacks the ester-carbonyl of the substrate, which is activated by hydrogen bonds between its carbonyl oxygen and the main-chain NH groups of residues Ser75 and Val351. The nucleophilicity of Ser75 is enhanced by Tyr181, which acts as a general base and presumably is stabilized as the phenolate due to the proximity of the side chains of Lys78 and Trp348. The second step of enzyme catalysis—deacylation—has to involve the attack of the acyl enzyme intermediate by a water molecule. From the available data it is not obvious whether this step involves one of the water molecules observed within the active site cleft of the native structure (see Fig. 3c ▶), which would then be occluded upon substrate binding. Alternatively, the water for the deacylation step might be derived directly from solution. Either possibility was discussed for β-lactamases (Knox and Moews 1991; Lobkovsky et al. 1994).
Fig. 5.

Superposition of the active site residues of the class C β-lactamase from Enterobacter cloacae (PDB entry 1bls; Lobkovsky et al. 1994; drawn in dark gray) and of the esterase EstB from B. gladioli (light gray). Figure generated with MOLSCRIPT (Kraulis 1991) and rendered with Raster3D (Merritt and Bacon 1997).
Table 3.
Equivalent residues between the β-lactamase P99 and the esterase EstB
| P99 | EstB |
| Ser64 | Ser75 |
| Lys67 | Lys78 |
| Asn152 | Tyr133 |
| Tyr150 | Tyr181 |
| Lys315 | Trp348 |
| Thr316 | Gly349 |
| Gly317 | Gly350 |
| Ser318 | Val351 |
Esterolytic versus β-lactamase activity
Despite the structural similarity between EstB, DD-peptidases, and β-lactamases, it is remarkable that EstB shows no significant β-lactamase activity towards a variety of β-lactams, including nitrocefin, penicillin G and V, cephalothin, cephaloridine, and cephalosporin C (Petersen et al. 2001). Of specific relevance is the result of the EstB catalyzed hydrolysis of 7-aminocephalosporinic acid (7-ACA). Analysis of the reaction products by HPLC revealed 100% cleavage of the ester group, but no products resulting from lactam hydrolysis (Petersen et al. 2001).
This shift in reactivity could be due to at least two reasons: first, the above exchange of two catalytic residues, together with slight structural changes, might have altered the inherent reactivity of the enzyme in such a way that it lost its ability to attack the (compared to an ester slightly less reactive) amide. We currently have no data available (e.g., experimental pKa values of residues forming the active site) to substantiate such a proposition. A second explanation for the shift in reactivity could invoke steric reasons, implying that—in going from P99 to EstB—the shape of the active site has changed in such a way that attack of the nucleophile on the (more bulky) β-lactam is prevented due to steric inaccessibility.
The relevance of the second possible cause for the observed difference in reactivity is already indicated by the significant differences in the overall shape of the active site cavity between EstB and P99 (Fig. 6 ▶), which contrasts with the close similarity in the 3D arrangement of catalytic residues (Fig. 5 ▶). In both enzymes, the active site is located in a tunnel running approximately parallel to the central β-sheet. While (in the orientation of Fig. 6a and b ▶) the tunnel of the P99 enzyme widens towards the top of the figure, forming a potential site of entrance for a substrate, access to the active site of EstB along this entrance is curbed by two strands of the ω-loop, which narrow the tunnel. Instead, it appears that the active site of EstB is connected to the outside through the second entrance of the tunnel, which runs approximately towards the viewer in the orientation of Figure 6 ▶. Here, a helix of P99 is replaced by two loops, which widens the entrance to the active site. These differences between the two enzymes lead to drastically different overall shapes of the active site tunnel, which also appears narrower (specifically near the active site) in EstB compared to P99.
Fig. 6.
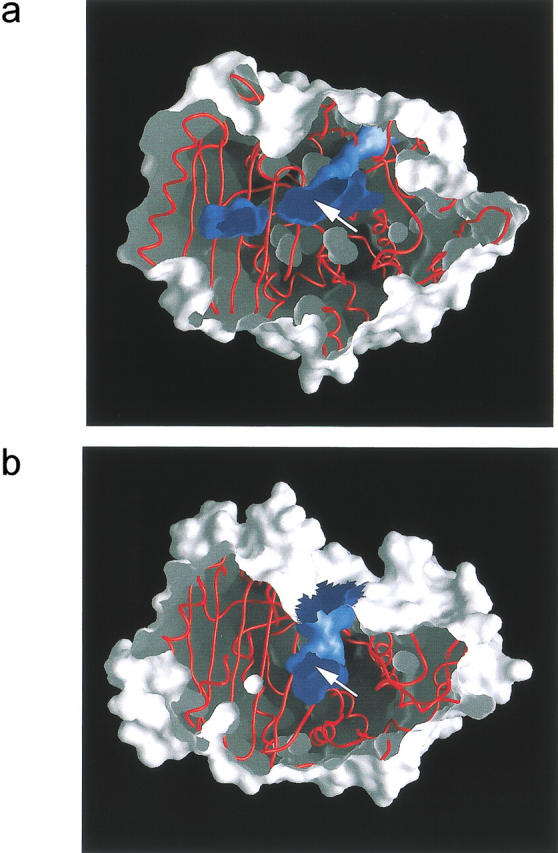
Section through surface representations of the esterase EstB from B. gladioli (a) and the β-lactamase P99 from E. cloacae (b). Both enzymes are in corresponding orientations, and the surface is cut approximately at the height of the active site, the approximate location of which is indicated by an arrow. Outer surfaces are drawn in shades of light gray, inner surfaces in dark blue. Figure produced with GRASP (Nicholls 1993).
To probe the effect of these differences in the active-site environment on the enzyme selectivity, we have performed a docking experiment with 7-ACA, and modeled the tetrahedral intermediate obtained by nucleophilic attack of Ser75 on the carbonyl carbon atoms of the ester (Fig. 7a ▶) and the β-lactam (Fig. 7b ▶) groups. In generating these complexes, the constraint was imposed that the oxygen atom of the tetrahedral intermediate should remain within hydrogen-bonding distance from the putative oxyanion hole formed by two main-chain NH groups of Ser75 and Val351. Although the tetrahedral intermediate for ester cleavage (Fig. 7a ▶) fits the active site well (no distance between protein and substrate shorter than 2.7 Å), the corresponding intermediate for hydrolysis of the β-lactam (Fig. 7b ▶) would require considerable conformational rearrangements of the protein to be accomodated within the (otherwise too narrow) active-site tunnel (closest distance between substrate and residue Tyr133: 1.38 Å). Because the close structural similarity between native EstB and its complex with DFP argues against pronounced flexibility of this part of the molecule, we conclude that the enzyme's preference of esterolytic versus β-lactamase activity is primarily due to steric factors.
Fig. 7.
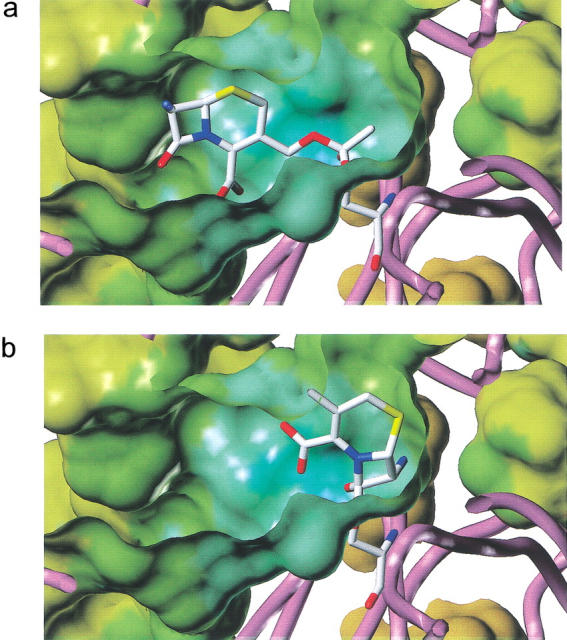
Modeling the tetrahedral intermediate formed by the attack of the nucleophile Ser75 of EstB on the carbonyl functions of the ester (a) and β-lactam (b) of 7-ACA. In generating these complexes, the constraint was imposed that the oxygen atom of the tetrahedral intermediate should remain within hydrogen-bonding distance from the putative oxyanion hole. The figure illustrates that the tetrahedral intermediate for ester cleavage (a) fits the active site well, whereas the corresponding intermediate for hydrolysis of the β-lactam (b) leads to steric clashes between protein and substrate. Figure produced with Sybyl, Vers. 6.4 (Tripos).
Materials and methods
Crystallization, data collection, and processing
The EstB esterase was expressed in E. coli, purified, and crystallized (hanging-drop vapor diffusion from 0.1 M HEPES buffer pH 7.5, 10% 2-propanol, and 20 % PEG 4000 at 4°C) to a final size of about 0.3 × 0.3 × 0.8 mm as described previously (Wagner et al. 1997). Crystals belong to the trigonal space group P3221 with cell dimensions of a = b = 83.4 and c = 194.6 Å. Heavy atom derivatives were prepared by soaking the crystals in the mother liquor containing millimolar amounts of the heavy atom compound. Soaking times varied between several hours and several days. Cryocooling was performed by dumping the crystals into liquid nitrogen after soaking them for a few seconds in a cryoprotectant containing mother liquor plus 20% v/v glycerol. Diffraction data were then collected at 100 K on a Siemens rotating anode generator equipped with a Siemens multiwire area detector on a three-circle goniometer and a locally constructed gas-stream cryostat (CuKα radiation, graphite monochromator, λ = 1.54 Å). A 0.1° oscillation angle was choosen for all data collections (with the exception of the DFP derivative—see below). Because a long crystal-to-detector distance (15 cm) had to be chosen due to the long crystallographic c-axis, data collection took several weeks for each crystal, leading to some incomplete heavy-atom data sets due to ice formation. Frames were processed with the XDS software (Kabsch 1988a, 1988b). We collected a native data set to 1.8-Å resolution (used to 2.0 Å) and eight derivative data sets, each to about 3.0-Å resolution.
For the complex between EstB and DFP, about 1 μL of DFP was added directly to the 10 μL crystallization drop and was allowed to react for several days. Data of the frozen crystals were collected at the EMBL-beamline X11 at DESY in Hamburg (Germany). The beamline was equipped with a MAR-345 imaging-plate scanner and an Oxford Cryostream cooler, and the following parameters were chosen for data collection: wavelength λ = 0.9076 Å, crystal-detector distance 300 mm, oscillation angle Δφ = 0.3°.
Phase determination
The first mercury sites from the p-chlormercuriphenylsulfonic acid monosodium salt (PCMB) were found with the program HEAVY (Terwilliger et al. 1987). All other heavy atom sites were determined by cross-phasing using the phases of the mercury derivative. It turned out that EMP and PCMB, the uranyl and PIP derivative, the gold and the platinium derivative pairwise share the same heavy-atom sites. Phasing was therefore based on seven derivatives with together eight different sites. Heavy-atom parameters were refined using MLPHARE (Otwinowski et al. 1991). From anomalous data of the mercury and uranyl derivatives, the space group could be unambiguously assigned as P3221, but no anomalous data were used for phase refinement. At this stage, the figure of merit using 3.0 Å resolution data was 0.60. The MIR phases were enhanced by a solvent flattening and histogram matching procedure using the program DM (Cowtan 1994), improving the figure of merit to 0.78. All phase refinement was carried out using the CCP4 program suite (Bailey 1994). The noncrystallographic symmetry (NCS) axis was determined from the heavy-atom sites. Statistics for heavy-atom refinement are given in Table 1.
Model building and crystallographic refinement
At this stage, the first secondary structure elements could be assigned and were modeled with the program O (Jones et al. 1991). A mask was created using MAMA (Kleywegt et al. 1994) and the noncrystallographic two-fold axis was determined and refined with RAVE (Kleywegt et al. 1994). Another cycle of solvent flattening, histogram matching and symmetry averaging (DM) improved the figure of merit to 0.82, and the map showed most of the protein backbone. At that stage, an initial model was built consisting of 343 residues in molecule A and 349 in molecule B. This partially traced model was then used for phase combination with SIGMAA (Read 1986), and the subsequent DM procedure was performed without symmetry averaging, followed by several cycles of model rebuilding and phase calculation. Although the first 14 and the last residue in each molecule were still invisible, electron density for the remainder of both molecules could now be seen. This model was refined with X-PLOR (Brünger 1992b), starting with data between 10 and 3.0 Å and gradually adding higher resolution shells. Both molecules were refined independently without using the NCS. A bulk solvent correction was applied, and water molecules were gradually included at positions where they may form hydrogen bonds. A grouped B-factor was calculated before positional refinement. Extensive model rebuilding was carried out between each refinement sep. In the last cycles of refinement, no sigma cutoff was applied. Rfree (Brünger 1992a) was monitored during the refinement using a set of reflections (5% of the unique data) chosen by X-plor.
The structure of the DFP derivative was refined against the synchrotron data, starting with the molecular-replacement solution obtained with the the native structure. After rigid body refinement, a Fo–Fc density map showed clear density near Ser75, interpretable as a DFP molecule covalently attached to the Oγ of Ser75 (see Fig. 3a ▶). Subsequent refinement cycles were performed with program SHELX (Sheldrick 1993). The refinement using data from 15 to 1.8 Å converged at R = 0.176 and Rfree = 0.235, with 571 water molecules.
Molecular modeling
7-ACA was manually docked to EstB as the tetrahedral intermediate formed by nucleophilic attack of the Ser75 oxygen atom on the carbonyl carbon atoms of the ester and the β-lactam groups (Fig. 7a,b ▶, respectively). The manual search for the conformation with minimum steric interference between protein and 7-ACA was based on the stereochemistry of the DFP derivative with the constraint that the oxygen atom of the tetrahedral intermediate should remain within hydrogen-bonding distance from the putative oxyanion hole formed by two main-chain NH groups of Ser75 and Val351. No energy minimization was performed.
Coordinates
Coordinates of the native structure and of the DFP derivative have been deposited at the Brookhaven Protein Data Bank (code 1ci8 and 1ci9), and are available for immediate distribution.
Acknowledgments
We acknowledge support by the Österreichischer Fonds zur Förderung der wissenschaftlichen Forschung through the Spezialforschungsbereich Biokatalyse and through project 11599 (C.K. and U.G.W.). The synchrotron data for the DFP derivative were collected at the X11 beamline of the EMBL-Hamburg outstation at DESY. We acknowledge help from the EMBL staff and from Andrea Schmidt and Oliver Sauer in collecting these synchrotron data. We also thank Fritz Winkler and Karl Gruber for suggestions and help in the interpretation of structural results, as well as Jean-Pierre Samama and Laurent Maveyraud for providing us with the coordinates of their class D lactamase.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.33002.
References
- Arpigny, J.L. and Jaeger, K.E. 1999. Bacterial lipolytic enzymes: Classification and properties. Biochem. J. 343 177–183. [PMC free article] [PubMed] [Google Scholar]
- Bailey, S. 1994. The CCP4 suite—Programs for protein crystallography. Acta Crystallogr. D50 760–763. [DOI] [PubMed] [Google Scholar]
- Banerjee, S., Pieper, U., Kapadia, G., Pannell, L.K., and Herzberg, O. 1998. Role of the Ω-loop in the activity, substrate-specificity, and structure of class-A β-lactamase. Biochemistry 37 3286–3296. [DOI] [PubMed] [Google Scholar]
- Brünger, A.T. 1992a. The free R-value: A novel statistical quantity for assessing the accuracy of crystal structures. Nature 355 472–474. [DOI] [PubMed] [Google Scholar]
- Brünger, A.T. 1992b. X-PLOR, a system for X-ray crystallography and NMR, version 3.2. Yale University Press, New Haven, CT.
- Carson, M. 1997. Ribbons. In Methods in enzymology (eds. R.M. Sweet and C.W. Carter), pp. 493–505. Academic Press, New York.
- Cowtan, K. 1994. An automated procedure for phase improvement by density modification. Joint CCP4 and ESF-EACBM Newsletter on Protein Crystallography 31 34–38. [Google Scholar]
- Crichlow, G.V., Kuzin, A.P., Nukaga, M., Mayama, K., Sawai, T., and Knox, J.R. 1999. Structure of the extended-spectrum class C β-lactamase of Enterobacter cloacae GC1, a natural mutant with a tandem tripeptide insertion. Biochemistry 38 10256–10261. [DOI] [PubMed] [Google Scholar]
- Damblon, C., Raquet, X., Lian, L.Y., Lamottebrasseur, J., Fonze, E., Charlier, P., Roberts, G.C.K., and Frere, J.M. 1996. The catalytic mechanism of β-lactamases—NMR titration of an active-site lysine residue of the TEM-1 enzyme. Proc. Natl. Acad. Sci. 93 1747–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson, G. and Wlodawer, A. 1998. Catalytic triads and their relatives. Trends Biochem. Sci. 23 347–352. [DOI] [PubMed] [Google Scholar]
- Dubus, A., Ledent, P., Lamotte-Brasseur, J., and Frere, J.M. 1996. The roles of residues Tyr150, Glu272, and His314 in class C β-lactamases. Proteins 25 473–485. [DOI] [PubMed] [Google Scholar]
- Escobar, W.A., Tan, A.K., and Fink, A.L. 1991. Site-directed mutagenesis of β-lactamase leading to accumulation of a catalytic intermediate. Biochemistry 30 10783–10787. [DOI] [PubMed] [Google Scholar]
- Escobar, W.A., Tan, A.K., Lewis, E.R., and Fink, A.L. 1994. Site-directed mutagenesis of glutamate-166 in β-lactamase leads to a branched path mechanism. Biochemistry 33 7619–7626. [DOI] [PubMed] [Google Scholar]
- Faber, K. 1997. Biotransformations of non-natural compounds: State of the art and future developments. Pure Appl. Chem. 69 1613–1632. [Google Scholar]
- Faber, K. 2000. Biotransformations in organic chemistry, 4th ed. Springer, Berlin.
- Govardhan, C.P. and Pratt, R.F. 1987. Kinetics and mechanism of the serine β-lactamase catalyzed hydrolysis of depsipeptides. Biochemistry 26 3385–3392. [DOI] [PubMed] [Google Scholar]
- Herzberg, O. 1991. Refined crystal-structure of β-lactamase from Staphylococcus-aureus Pc1 at 2.0-A resolution. J. Mol. Biol. 217 701–719. [DOI] [PubMed] [Google Scholar]
- Herzberg, O., Kapadia, G., Blanco, B., Smith, T.S., and Coulson, A. 1991. Structural basis for the inactivation of the P54 mutant of β-lactamase from Staphylococcus aureus PC1. Biochemistry 30 9503–9509. [DOI] [PubMed] [Google Scholar]
- Holm, L. and Sander, C. 1996. Mapping the protein universe. Science 273 595–603. [DOI] [PubMed] [Google Scholar]
- Jelsch, C., Mourey, L., Masson, J.M., and Samama, J.P. 1993. Crystal structure of Escherichia coli TEM1 β-lactamase at 1.8 A resolution. Proteins 16 364–383. [DOI] [PubMed] [Google Scholar]
- Jones, M. and Page, M.I. 1991. An esterase with β-lactamase activity. J. Chem. Soc. Chem. Commun. 1991 316–317. [Google Scholar]
- Jones, T.A., Zou, J.Y., Cowan, S., and Kjeldgaard, M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A47 110–119. [DOI] [PubMed] [Google Scholar]
- Joris, B., Ghuysens, J.M., Dive, G., Renard, A., Dideberg, O., Charlier, P., Frere, J.M., Kelly, J.A., Boyington, J.C., Moews, P.C., et al. 1988. The active-site-serine penicillin-recognizing enzymes as members of the streptomyces R61 DD-peptidase family. Biochem. J. 250 313–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch, W. 1988a. Automatic-indexing of rotation diffraction patterns. J. Appl. Crystallogr. 21 67–71. [Google Scholar]
- Kabsch, W. 1988b. Evaluation of single crystal X-ray diffraction data from a position sensitive detector. J. Appl. Crystallogr. 21 916–924. [Google Scholar]
- Kelly, J.A., Charlier, P., Dideberg, O., Duez, C., Dusart, J., Fraipont, C., Frere, J.M., Ghuysen, J.M., Joris, B., Knox, J.R., et al. 1986. On the origin of bacterial-resistance to penicillin—Comparison of a β-lactamase and a penicillin target. Science 231 1429–1431. [DOI] [PubMed] [Google Scholar]
- Kelly, J.A., Knox, J.R., Zhao, H.C., Frere, J.M., and Ghuysen, J.M. 1989. Crystallographic mapping of β-lactams bound to a D-alanyl-D-alanine peptidase target enzyme. J. Mol. Biol. 209 281–295. [DOI] [PubMed] [Google Scholar]
- Kleywegt, G.J., Jones, T.A., Hubbard, R., and Waller, D. 1994. Haloween . . . Masks and bones. In From map to final model (ed. S. Bailey), pp. 59–66. SERC Daresbury Laboratory, Warrington, UK.
- Knox, J.R. and Moews, P.C. 1991. β-Lactamase of Bacillus licheniformis 749/C. Refinement at 2 A resolution and analysis of hydration. J. Mol. Biol. 220 435–455. [DOI] [PubMed] [Google Scholar]
- Knox, J.R., Moews, P.C., and Frere, J.M. 1996. Molecular evolution of bacterial β-lactam resistance. Chem. Biol. 3 937–947. [DOI] [PubMed] [Google Scholar]
- Kraulis, P.J. 1991. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 24 946–950. [Google Scholar]
- Lamotte-Brasseur, J., Lounnas, V., Raquet, X., and Wade, R.C. 1999. Pk(A) calculations for class-A β-lactamases—Influence of substrate-binding. Protein Sci. 8 404–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski, R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK version 2.0. Programs to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26 283–291. [Google Scholar]
- Lobkovsky, E., Billings, E.M., Moews, P.C., Rahil, J., Pratt, R.F., and Knox, J.R. 1994. Crystallographic structure of a phosphonate derivative of the Enterobacter cloacae P99 cephalosporinase: Mechanistic interpretation of a β-lactamase transition-state analog. Biochemistry 33 6762–6772. [DOI] [PubMed] [Google Scholar]
- Lobkovsky, E., Moews, P.C., Liu, H.S., Zhao, H.C., Frere, J.M., and Knox, J.R. 1993. Evolution of an enzyme activity: Crystallographic structure at 2-A resolution of cephalosporinase from the ampC gene of Enterobacter cloacae P99 and comparison with a class A penicillinase. Proc. Natl. Acad. Sci. 90 11257–11261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massova, I. and Mobashery, S. 1998. Kinship and diversification of bacterial penicillin-binding proteins and β-lactamases. Antimicrob. Agents Chemother. 42 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matagne, A., Lamotte-Brasseur, J., and Frere, J.M. 1998. Catalytic properties of class-A β-lactamases—Efficiency and diversity. Biochem. J. 330 581–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maveyraud, L., Pratt, R.F., and Samama, J.P. 1998. Crystal-structure of an acylation transition-state analog of the TEM-1 β-lactamase—Mechanistic implications for class-A β-lactamases. Biochemistry 37 2622–2628. [DOI] [PubMed] [Google Scholar]
- McKay, D.B., Jennings, M.P., Godfrey, E.A., MacRae, I.C., Roger, P.J., and Beacham, I.R. 1992. Molecular analysis of an esterase-encoding gene from a lipolytic psychotrophic pseudomonad. J. Gen. Microbiol. 138 701–708. [DOI] [PubMed] [Google Scholar]
- Merritt, E.A. and Bacon, D.J. 1997. Raster3D: Photorealistic molecular graphics. Methods Enzymol. 277 505–524. [DOI] [PubMed] [Google Scholar]
- Nicholls, A.J. 1993. GRASP: Graphical representation and analysis of surface properties. Columbia University, New York.
- Nishizawa, M., Shimizu, M., Ohkawa, H., and Kanaoka, M. 1995. Stereoselective production of (+)-trans-chrysanthemic acid by a microbial esterase—Cloning, nucleotide-sequence, and overexpression of the esterase gene of Arthrobacter-globiformis in Escherichia-coli. Appl. Environ. Microbiol. 61 3208–3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oefner, C., Darcy, A., Daly, J.J., Gubernator, K., Charnas, R.L., Heinze, I., Hubschwerlen, C., and Winkler, F.K. 1990. Refined crystal structure of β-lactamase from Citrobacter freundii indicates a mechanism for β-lactam hydrolysis. Nature 343 284–288. [DOI] [PubMed] [Google Scholar]
- Okuda, H. 1991. Esterases. In A study of enzymes (ed. S.A. Kuby), pp. 563–577. CRC Press, Boca Raton, FL.
- Ollis, D.L., Cheah, E., Cygler, M., Dijkstra, B., Frolow, F., Franken, S.M., Harel, M., Remington, S.J., Silman, I., Schrag, J., Sussman, J.L., Verschueren, K.H.G., and Goldman, A. 1992. The α/β hydrolase fold. Protein Eng. 5 197–211. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z., Evans, P.R., and Leslie, A.G.W. 1991. Maximum-likelyhood refinement of heavy-atom parameters. In Isomorphous replacement and anomalous scattering (ed. W. Wolf), pp. 80–86. SERC Daresbury Laboratory, Warrington, UK.
- Page, M.I. and Laws, A.P. 1998. The mechanism of catalysis and the inhibition of β-lactamases. Chem. Commun 1998 1609–1617. [Google Scholar]
- Petersen, E.I. 1995. Molecular cloning of a novel esterase gene and genes encoding for a protocatechuate-3,4-dioxygenase from Pseudomonas marginata and characterization of their gene products expressed in Escherichia coli. PhD thesis, Institut für Biotechnologie, Technische Universität Graz, Austria.
- Petersen, E.I., Valinger, G., Sölkner, B., Stubenrauch, G., and Schwab, H. 2001. A novel esterase from Burkholderia gladioli which shows high deacetylation activity on cephalosporins is related to β-lactamases and DD-peptidases. J. Biotechnol. 89 11–25. [DOI] [PubMed] [Google Scholar]
- Pratt, R.F. and Govardhan, C.P. 1984. β-Lactamase-catalyzed hydrolysis of acyclic depsipeptides and acyl transfer to specific amino-acid acceptors. Proc. Natl. Acad. Sci. 84 1302–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read, R.A. 1986. Improved Fourier coefficients for maps using phases from partial structures with errors. Acta Crystallogr. A42 140–149. [Google Scholar]
- Samraoui, B., Artymiuk, P.J., Phillips, D.C., Sutton, B.J., Todd, R.J., and Waley, S.G. 1986. Tertiary structural similarity between a class-A β-lactamase and a penicillin-sensitive D-alanyl carboxypeptidase-transpeptidase. Nature 320 378–380. [DOI] [PubMed] [Google Scholar]
- Schlacher, A., Stanzer, T., Ospirian, I., Mischitz, M., Klingsbichel, E., Faber, K., and Schwab, H. 1998. Detection of a new enzyme for stereoselective hydrolysis of linalyl acetate using simple plate assays for the characterization of cloned esterases from Burkholderia gladioli. J. Biotechnol. 62 47–54. [DOI] [PubMed] [Google Scholar]
- Schulze, B. and Wubbolts, M.G. 1999. Biocatalysis for industrial production of fine chemicals. Curr. Opin. Biotechnol. 10 609–615. [DOI] [PubMed] [Google Scholar]
- Sheldrick, G.M. 1993. SHELXL-93, a program for the refinement of crystal structures from diffraction data. University of Göttingen, Göttingen, Germany.
- Strynadka, N.C.J., Adachi, H., Jensen, S.E., Johns, K., Sielecki, A., Betzel, C., Sutoh, K., and James, M.N.G. 1992. Molecular structure of the acyl-enzyme intermediate in β-lactam hydrolysis at 1.7 A resolution. Nature 359 700–705. [DOI] [PubMed] [Google Scholar]
- Strynadka, N.C.J., Martin, R., Jensen, S.E., Gold, M., and Jones, J.B. 1996. Structure-based design of a potent transition state analogue for TEM-1 α-lactamase. Nat. Struct. Biol. 3 688–695. [DOI] [PubMed] [Google Scholar]
- Stubenrauch, G. 1991. Klonierung von Esterase-Genen aus Pseudomonas marginata sowie Xanthomonas campestris, sowie detaillierte Analyse des Pseudomonas marginata EstA gens sowie dessen Genprodukt (Esterase EP10). PhD thesis, Technische Universität Graz, Austria.
- Swaren, P., Maveyraud, L., Guillet, V., Masson, J.M., Mourey, L., and Samama, J.P. 1995. Electrostatic analysis of TEM1 β-lactamase—Effect of substrate-binding, steep potential gradients and consequences of site-directed mutations. Structure 3 603–613. [DOI] [PubMed] [Google Scholar]
- Terwilliger, T.C., Eisenberg, D., and Kim, H.S. 1987. General method of determining heavy atom positions using the difference-patterson function. Acta Crystallogr. A43 1–5. [Google Scholar]
- Toone, E.J., Werth, M.J., and Jones, J.B. 1990. Active-site model for interpreting and predicting the specifity of pig liver esterase. J. Am. Chem. Soc. 112 4946–4952. [Google Scholar]
- Wagner, U.G., Sölkner, B., Petersen, E.I., Schlacher, A., Schwab, H., and Kratky, C. 1997. Crystallization and preliminary X-ray diffraction studies of the Pseudomonas marginata esterase EstB. Acta Crystallogr. D53 596–598. [DOI] [PubMed] [Google Scholar]
- Wharton, C.W. 1998. The serine proteinases. In Comprehensive biological catalysis (ed. M. Sinnott), pp. 345–379. Academic Press, London.
- Zawadzke, L.E., Chen, C.C.H., Banerjee, S., Li, Z., Wasch, S., Kapadia, G., Moult, J., and Herzberg, O. 1996. Elimination of the hydrolytic water molecule in a class-A β-lactamase mutant—Crystal-structure and kinetics. Biochemistry 35 16475–16482. [DOI] [PubMed] [Google Scholar]