

Abstract
α-helices within proteins are often terminated (capped) by distinctive configurations of the polypeptide chain. Two common arrangements are the Schellman motif and the alternative αL motif. Rose and coworkers developed stereochemical rules to identify the locations of such motifs in proteins of unknown structure based only on their amino acid sequences. To check the effectiveness of these rules, they made specific predictions regarding the structural and thermodynamic consequences of certain mutations in T4 lysozyme. We have constructed these mutants and show here that they have neither the structure nor the stability that was predicted. The results show the complexity of the protein-folding problem. Comparison of known protein structures may show that a characteristic sequence of amino acids (a sequence motif) corresponds to a conserved structural motif. In any particular protein, however, changes in other parts of the sequence may result in a different conformation. The structure is determined by sequence as a whole, not by parts considered in isolation.
Keywords: Helix capping, Schellman motif, protein folding, structure prediction, T4 lysozyme
Deciphering the code of amino acid sequences of proteins to predict their structures remains one of the prime challenges in biology. Much interest has focused on possible rules that might be used to predict from the amino acid sequence the structure of parts of a given protein. Such rules could be an important first step in predicting the overall structure of the protein. The existence of such rules would also imply that the folding of these regions of the protein must proceed locally (i.e., independent of tertiary contacts). As possible examples, statistical investigation of protein sequences has revealed that the positions and lengths of secondary structures can usually be predicted with a 70% accuracy (Rost and Sander 1993; Zhang and Zhang 2001). Also helix-stop or capping sequences that mark the boundaries of α-helices can be identified to a large extent from the amino acid sequence only (Schellman 1980; Presta and Rose 1988; Richardson and Richardson 1988; Dasgupta and Bell 1993; Aurora et al. 1994; Aurora and Rose 1998).
Capping interactions satisfy the hydrogen-bonding potential of the otherwise unsatisfied backbone amide and carbonyl groups that occur at the beginning and end of an α-helix. Different types of capping interactions can be distinguished for the amino- and carboxy-termini. Whereas the amino-terminal cap is usually formed by side-chain interactions with the helix backbone atoms of the first helical turn, the C-terminal cap is formed by interactions between the backbone of the final helical turn and the backbone atoms of a subsequent loop (Aurora et al. 1994; Aurora and Rose 1998). Two very common conformations of this sort are the Schellman motif (named for its discoverer, Charlotte Schellman) and the αL motif (Schellman 1980; Milner-White 1988; Aurora et al. 1994; Viguera and Serrano 1995; Sukumar and Gierasch 1997; Kallenbach and Gong 1999). These are shown in Figure 1 ▶, which also defines the nomenclature that is used. A typical Schellman motif has a glycine at the C` position, a polar residue or alanine at C1, and a stabilizing apolar interaction between a nonpolar side chain at C" and a nonpolar side chain at C3 (Aurora et al. 1994; Aurora and Rose 1998). In contrast, an αL cap retains the glycine at the C` position but the following residue is polar. In both capping conformations, the φ and ξ values of the C` residue have positive values that can most easily be adopted by glycine (Schellman 1980; Aurora et al. 1994). It has been suggested that glycine also permits maximal solvation of the peptide backbone, which could enhance local capping interactions (Serrano et al. 1992; Aurora et al. 1998; Thomas et al. 2001).
Fig. 1.
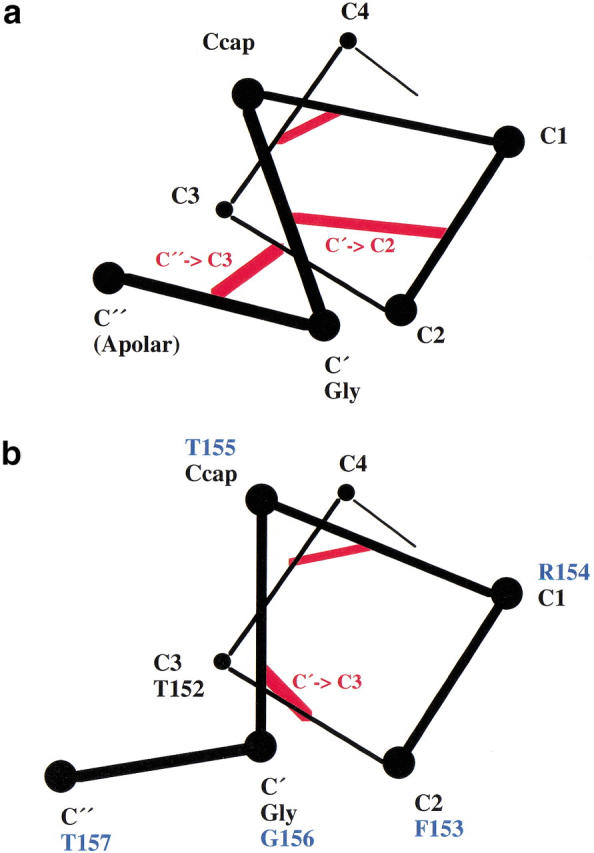
(a) Schematic illustration of the hydrogen-bonding interactions (red lines) in a Schellman motif. As noted by Aurora et al. (1994) such motifs have a preference for glycine at C` and an apolar residue at C". (b) Schematic illustration showing the backbone hydrogen-bonding interactions (red lines) in a typical αL capping motif, as well as the sequence of the subject motif in T4 lysozyme (in blue). Such motifs commonly have a glycine at the Cρ position and a polar residue at C". (In her original article, Schellman [1980] proposed the name αL for the conformation shown in panel a. Aurora et al. [1994] renamed this the Schellman motif and used αL to designate the conformation shown in panel b.)
In T4 lysozyme, residues Thr 152-Gly 156 form a typical αL helix cap at the C terminus of helix J (residues 143–155; Nicholson et al. 1991). It is known that the replacement of Thr 157 at the C" position with nonpolar residues destabilizes the protein (Alber et al. 1987). Using their rules for helix capping, Aurora et al. (1994) argued that apolar substitutions at Thr 157 would be expected to switch the conformation from αL to a Schellman motif but that this was prevented in T4 lysozyme by the presence of the bulky side chain of Trp 158. They went on to predict that mutation of Trp 158 to a nonaromatic residue would permit formation of a Schellman motif and, as well, that the Thr157 → Ile mutant would then lose its observed temperature-sensitive phenotype. We test these predictions here.
Results
Structure and stability of the mutant lysozymes
Mutant T157I crystallized isomorphously with wild-type. Difference maps and electron density maps phased on the wild-type structure (Eriksson et al. 1993) both indicated that the structure of the mutant was very similar to WT* (see Materials and Methods; data not shown). Following refinement, the coordinates of T157I and WT* could be superimposed with a root-mean-square discrepancy of 0.19 Å for all atoms. Within the capping region, the backbone atoms of residues 152–157 differed from wild-type by 0.1 Å, indicating that the capping geometry in the native structure is retained in the mutant.
Mutant W158L crystallized nonisomorphously with wild-type in space group P212121 with four molecules per asymmetric unit. The four molecules display large-scale changes in structure. Most notably, the size of the active-site cleft widens and, in comparison to the wild-type enzyme (or to mutant T157L), the hinge-bending angle (Zhang et al. 1995) varies from +14.3° to +22.3° (Fig. 2a ▶ ). Within the carboxy-terminal and amino-terminal domains, however, the changes in structure are much more modest. In particular, no distinctive changes are seen in the conformation of the helix cap at the site of the mutation (Fig. 2b ▶). Based on the superposition of carboxy-terminal domains, the atoms of residues Arg 145-Lys 162 of the mutant superimpose on wild-type with a discrepancy of ∼0.49 Å. Essentially, the same value (0.34 Å–0.60 Å) is obtained if the C-terminal domains of the four mutant molecules are compared among themselves. If just the capping residues themselves are superimposed (i.e., residues 152–157) the discrepancy for the backbone atoms ranges from 0.19 Å to 0.42 Å. This shows that the αL capping motif present at this site in wild-type lysozyme is retained in the mutant W158L.
Fig. 2.
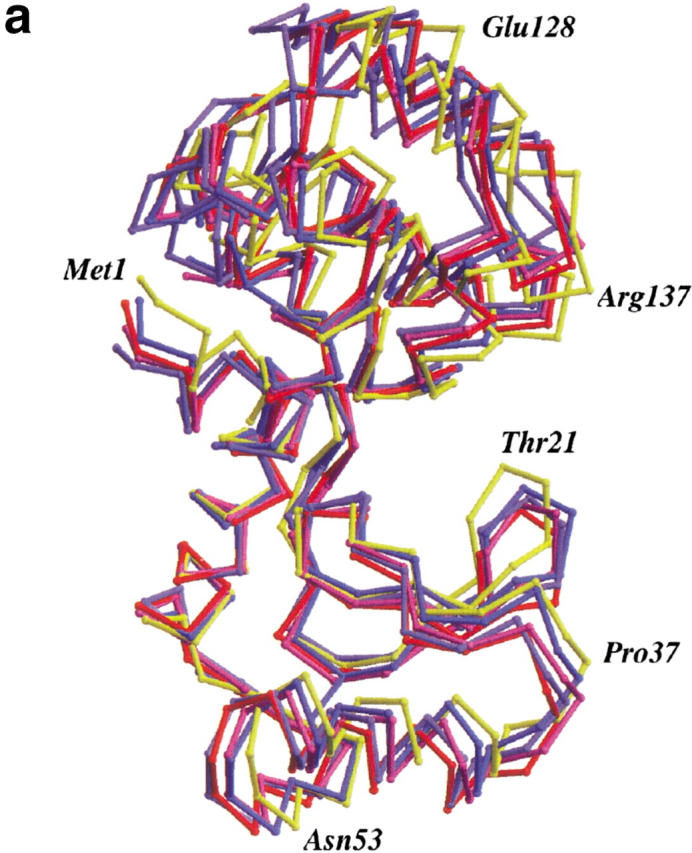
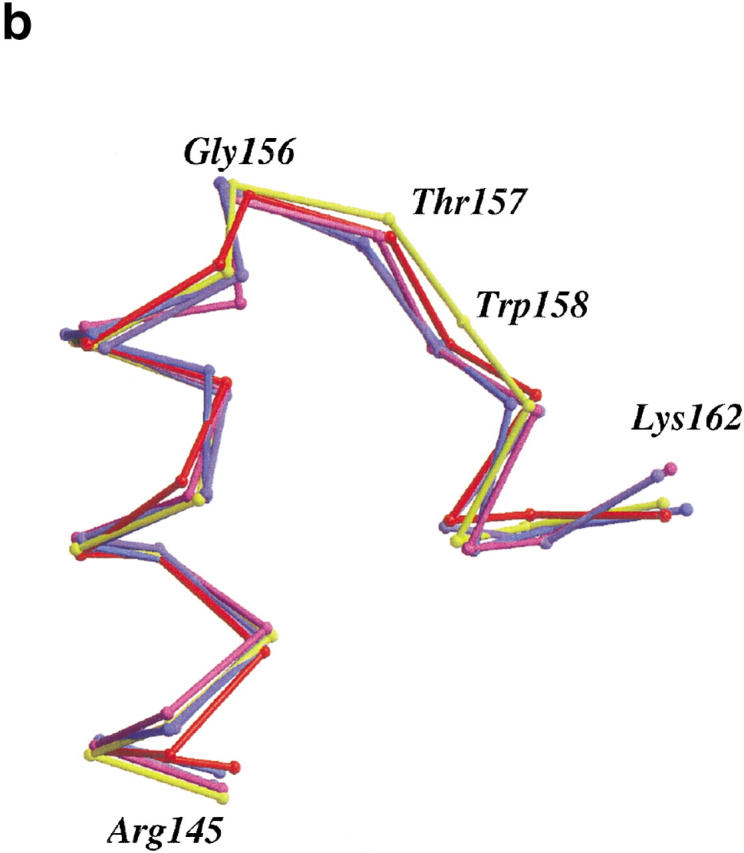
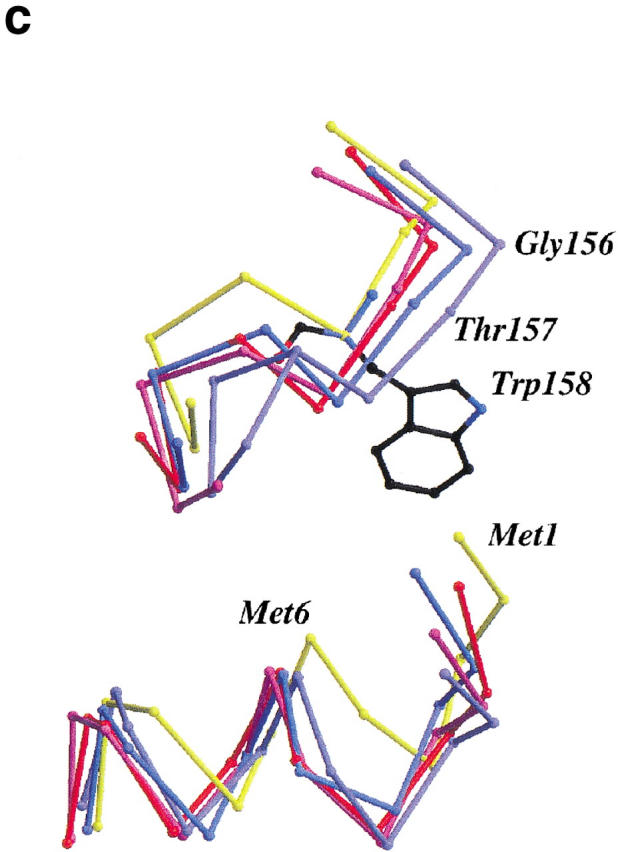
(a) Backbone of wild-type T4 lysozyme (yellow) with the four molecules in the asymmetric unit of mutant W158L (red, light purple, purple, and blue) overlaid. The structures were superimposed based on the long helix between residues 60 and 80 that connects the N- and C-terminal domains. Compared to wild-type T4 lysozyme, the active site of the mutant proteins has widened by 14° to 22°. Figures prepared using MOLSCRIPT (Kraulis 1991; Merritt and Bacon 1997). (b) Superposition of the backbone traces of residues 145–162 of T4 lysozyme mutant T157I (yellow) and the four independent molecules of mutant W158L (red, light purple, purple, and blue). This segment includes helix J (residues 143–155) and the αL capping motif (residues 152–157). Only minor rearrangements occur within the segment. (c). Comparison of the backbone structure of wild-type and W158L lysozyme in the vicinity of the substitution. Wild-type (backbone, yellow; Trp158 side chain, black) and the four copies of mutant W158L (red, light purple, purple, and blue) were superimposed as in panel a). The substitution of the indole ring results in an increased separation between the amino terminus of helix A (residues 1–6) and the residues in the vicinity of the C terminus of helix J (residues 155–162).
The thermodynamic analysis of the mutants reveals that at pH 5.42 the unfolding behavior can be modeled as a two-state transition. The stability of mutant T157I decreased by 2.05 kcal/mole relative to the wild-type enzyme; whereas, mutant W158L is destabilized by 1.75 kcal/mole. Each of these values is typical for single-site substitutions of the enzyme (Grütter et al. 1987; Matthews 1993). The double-mutant T157I/W158L is destabilized by 3.55 kcal/mole.
Discussion
Predictions of stability
The mutation Thr157 → Ile was one of the earliest temperature-sensitive mutants of T4 lysozyme to be characterized and the apparent reasons for its destabilization have been discussed (Alber et al. 1987; Grütter et al. 1987). The early work was with the true wild-type (i.e., cysteine-containing) protein; whereas, the present study utilizes the structurally and thermodynamically similar pseudo-wild-type (i.e., cysteine-free) protein (see Materials and Methods).
As shown in Table 1 the thermodynamic analysis confirms that T157I is indeed a temperature-sensitive mutant, with melting temperature 5.7°C less than wild-type. The mutation W158L also destabilizes the protein by roughly the same amount.
Table 1.
Thermodynamic data
| Protein | ΔTm (°C) | ΔH (kcal/mole) | ΔΔG (kcal/mole) |
| WT* | 0.00 | 130 | 0.00 |
| T157I | −5.7 | 118 | −2.05 |
| W158L | −4.6 | 115 | −1.75 |
| T157I/W158L | −10.3 | 98 | −3.55 |
Tm is the melting temperature and ΔTm the change in Tm relative to wild type (WT*; see Materials and Methods), the Tm of which is 65.3°C in this buffer. ΔH is the enthalpy of unfolding at Tm and ΔΔG the change in stability relative to wild type, evaluated at an isotherm of 60°C. The uncertainty of ΔΔG was estimated from the standard deviation of ΔG° for WT* to be ±0.15 kcal/mole.
According to the prediction of Aurora et al. (1994), when the mutation Thr157 → Ile is introduced into the W158L protein, it should lose its temperature-sensitive phenotype. This is not the case. When T157I is combined with W158L to achieve the double mutant there is an additional loss of stability of 1.8 kcal/mole (i.e., 3.55 minus 1.75 kcal/mole). This is essentially the same as the amount by which the mutation Thr157 → Ile destabilizes the native protein (2.05 kcal/mole).
Predictions of structure
It was previously shown in the context of the cysteine-containing protein that the mutation T157I causes small adjustments in side chains at the site of the substitution and very little change in the backbone (Alber et al. 1987; Grütter et al. 1987). The present analysis, based on the pseudo-wild-type protein, confirms this result.
Aurora et al. (1994) predicted that the mutation W158L would allow the switch from an αL to a Schellman motif at this site, but as shown in Figure 2b ▶, this does not occur. Although the W158L substitution does result in some large-scale changes in the structure of the protein, the backbone conformation in the immediate vicinity of the mutation, and including the helix-capping region, changes very little.
Aurora et al. (1994) reasoned that presence of the bulky side chain of Trp 158 prevented the formation of a Schellman motif by mutant T157I. The side chain of Trp 158 is wedged between Val 94, a loop that includes residues 152–162 and residues 1–6 of the amino-terminal α-helix. As shown in Figure 2c ▶, when Trp 158 is replaced by leucine, the surrounding residues actually move farther apart and expose a hydrophobic cleft on the surface of the protein, presumably contributing to the loss in stability.
We were not able to obtain the crystal structure of the double-mutant T157I/W158L. It may be noted, however, that the loss of stability of the double mutant (3.55 kcal/mole) is close to the sum of the stability losses of the constituent single mutants (3.80 kcal/mole). Such additivity is the hallmark of, and is most simply explained by, noninteraction between the two sites. Therefore, although the possibility cannot be excluded, it seems unlikely that the structure containing both mutations would undergo a structural rearrangement to form a Schellman motif. In this context it might be noted that the mutant lysozyme G156D has been analyzed (Gray and Matthews 1987) and shown to maintain the αL motif. Consideration of sequence alone would suggest that Gly 156 (Fig. 1 ▶) is essential for formation of either an αL or a Schellman motif, but the mutant structure shows that the surrounding interactions are sufficient to maintain αL motif even when an aspartic acid is substituted at the C` position.
Inferences for protein design
To illustrate the structural rearrangements that would be required to permit the introduction of the predicted Schellman conformation in T4 lysozyme, 20 representative Schellman motifs derived from the Protein Data Bank were superimposed on the αL conformation at the end of helix J. The result is shown in Figure 3 ▶. As can be seen, the designed structural change would require a movement of Gly 156 toward that occupied by the glycine residue in a typical Schellman conformation. To permit such a change, the loop that includes residues 157, 158, and 159 would have to move toward Val 94. This would cause steric clashes between the loop backbone and the valine side chain even if the conformational variability seen in the superimposed Schellman conformations is taken into account. A possible way to relieve these steric clashes would be via a rotation of helix J, but this would require the disruption of other favorable interactions. It might also be noted that the carbonyl groups in the last turn at the C terminus of helix J interact with the guanidinium group of Arg 95 (Fig. 3 ▶). The N atom of this residue forms a hydrogen bond to the carbonyl oxygen of residue 154 and the NH atom forms another hydrogen bond to the carbonyl oxygen of residue 154. Substitution of Arg 95 with alanine substantially destabilizes the protein (Baldwin et al. 1998), suggesting that the interactions of the guanidinium are important for stability. These stabilizing interactions would be interrupted most likely if a Schellman cap were to be introduced.
Fig. 3.

Stereo diagram showing the backbone structure of 20 Schellman conformations (green) superimposed on the αL capping motif at the C terminus of helix J of T4 lysozyme. The superpositions were based on the helix residues only and had an average root-mean-square discrepancy of <0.2 Å. The Protein Data Bank codes and residue ranges (in parentheses) for the selected Schellman motifs are 351c (1–8), 2act (70–78), 5cpa (51–59), 2cts (33–41), 2cts (150–159), 2cts (282–290), 2cts (301–309), 2cts (351–362), 2cts (405–413), 1gd1 (101–109), 3grs (32–40), 3grs (231–239), 3grs (281–289), 3grs (443–451), 1lz1 (5–13), 3lzm (40–48), 1mbo (139–147), 9pap (68–76), 4pep (133–141), 4pep (96–104). Trp 158 and the residues it contacts—Met 1, Asn 2, Val 94, Arg 95, and Gly 156—are displayed in a ball-and-stick representation.
To the extent that these results apply to proteins in general, they strongly suggest that the αL and Schellman helix-capping motifs cannot be routinely interchanged by altering the amino acid sequence within the motif itself. Changes from one motif to another require shifts in the protein backbone of up to ∼3.5 Å (typically ∼1.5 Å for the Cα atom at C` and 3.5 Å for that at C" [Fig. 1 ▶]). These may perturb stabilizing interactions that exist in the reference protein or introduce unfavorable interactions. The presence of such favorable or unfavorable interactions cannot be inferred from the amino acid sequence alone; it requires knowledge of the three-dimensional structure. Studies of peptide fragments that include helix-capping motifs suggest that capping interactions can occur in solution (Sukumar and Gierasch 1997; Das et al. 2001) although they do not appear to be strong determinants of structure (Viguera and Serrano 1995; Wang and Shortle 1997; Kallenbach and Gong 1999).
In terms of the use of rules to predict helix-capping motifs, the approach suggested by Aurora et al. (1994) is a useful first step. In common with related approaches, however, such as the prediction of the locations of α-helices, the rules are not perfect. It strongly suggests that the structures of proteins are determined by a hierarchy of interactions. The amino acid sequence within a short segment of the polypeptide chain can strongly influence its structure, but the conformation of the segment in the folded protein can be dictated by interactions with residues in other parts of the polypeptide chain.
Materials and methods
Mutagenesis
Mutants were derived from a cysteine-free version of T4 lysozyme (WT*; Matsumura and Matthews 1989) using the method of Kunkel (1985) on a single-stranded M13 phage DNA T4 lysozyme template plasmid (Poteete et al. 1991). The mutants constructed were Thr157 → Ile (T157I) and Trp158 → Leu (W158L), as well as the double-mutant T157I/W158L. The mutated T4 lysozyme-containing fragment of the M13 plasmid was excised with BamHI and HindIII and ligated into the pHS1403 expression vector. Plasmids were subsequently transformed into Escherichia coli RR1 cells. All mutants were expressed and purified as described (Poteete et al. 1991). Protein activities were monitored using a halo-assay (Matsumura and Matthews 1989) and found to be essentially the same as wild-type.
Crystallization
Protein concentrations used for crystallization were ∼15 mg/ml in 20 mM HEPES buffer at pH 7.0. Initial crystallization conditions were screened using the Hampton Crystal Screen II. Crystals were obtained for mutants T157I and W158L. T157I crystallized isomorphously to wild-type T4 lysozyme in space group P3221 in 1.8 M phosphate at pH 7.8. Mutant W158L crystallized using 30% polyethylene glycol 4000, 0.1 M PIPES at pH 7.0 and 0.2 M Li2SO4 in space group P212121 with four molecules in the asymmetric unit. No crystals were obtained for the double mutant.
Data collection
X-ray data (Table 2) were collected at room temperature on a San Diego Multiwire System area detector (Hamlin 1985). The structure of W158L was solved by molecular replacement using the programs AMore (Navaza 1994) and the direct-rotation search function of CNS (DeLano and Brünger 1995; Brünger et al. 1998). Successful searches for three of the molecules in the asymmetric unit were carried out with different T4 lysozyme mutants that have been shown to adopt different hinge-bending angles between the amino- and carboxy-terminal domains (Zhang et al. 1995). The fourth molecule was localized using difference-Fourier electron density maps.
Table 2.
X-ray data collection and refinement statistics
| Mutant | W158L | T157I |
| Space group | P212121 | P3221 |
| Cell dimensions | ||
| a (Å) | 74.3 | 61.0 |
| b (Å) | 77.2 | 61.0 |
| c (Å) | 138.2 | 97.4 |
| Number of molecules per asymmetric unit | 4 | 1 |
| Resolution (Å) | 2.6 | 1.8 |
| Rsym (%) | 5.7 | 4.0 |
| Completeness (%) | 78 | 87 |
| R factor (%) | 23.5 | 19.6 |
| R free (%) | 31.5 | 24.5 |
| Δbond lengths (Å) | 0.022 | 0.012 |
| Δbond angles (°) | 2.2 | 1.6 |
| PDBa code | 1JQU | 1JOZ |
a (PDB) Protein Data Bank.
Refinement
The refinement of all mutants was carried out with TNT (Tronrud et al. 1987; Tronrud 1997). Model building was performed with program O (Jones and Kjeldgaard 1997). The accuracy of the final model was assessed with the programs WHATIF and PROCHECK (Vriend 1990; Laskowski et al. 1993). Hinge-bending angles were determined with EDPDB (Zhang and Matthews 1995).
Thermodynamic analysis
Thermodynamic measurements were made in 0.1 M NaCl, 1.4 mM HOAc, 8.6 mM NaOAc at pH 5.42. In this buffer, all mutant proteins appeared to melt in a two-state manner at protein concentrations between 0.01 and 0.03 mg/ml. ΔΔG, the free energy of unfolding of the mutant relative to wild-type, was estimated at 60°C, assuming the change in the heat capacity on unfolding, ΔCp, to be 2.5 kcal/mole-K.
Coordinates
Coordinates have been deposited in the Protein Data Bank with access codes 1JQU and 1JOZ (Table 2).
Acknowledgments
We thank Dr. Charlotte Schellman for helpful discussion, Sheila Snow and Hong Xiao for expert assistance in purifying and crystallizing the mutant proteins, and Drs. Larry Weaver and Dale Tronrud for help with data collection and refinement. This work was supported in part by NIH grant GM21967 to B.W.M.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.39802.
References
- Alber, T., Dao-pin, S., Wilson, K., Wozniak, J.A., Cook, S.P., and Matthews, B.W. 1987. Contributions of hydrogen bonds of threonine 157 to the thermodynamic stability of phage T4 lysozyme. Nature 330 41–46. [DOI] [PubMed] [Google Scholar]
- Aurora, R. and Rose, G.D. 1998. Helix capping. Prot. Sci. 7 21–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aurora, R., Srinivasan, R., and Rose, G.D. 1994. Rules for α-helix termination by glycine. Science 264 1126–1130. [DOI] [PubMed] [Google Scholar]
- Baldwin, E., Baase, W.A., Zhang, X-J., Feher, V., and Matthews, B.W. 1998. Generation of ligand binding sites in T4 lysozyme by deficiency-creating substitutions. J. Mol. Biol. 277 467–485. [DOI] [PubMed] [Google Scholar]
- Brünger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N.S., Read, R.J., Rice, L.M., Simonson, T., and Warren, G.L. 1998. Crystallography and NMR system (CNS): A new software system for macromolecular structure determination. Acta Cryst. D54 905–921. [DOI] [PubMed] [Google Scholar]
- Das, C., Shankaramma, S.C., and Balaram, P. 2001. Molecular carpentry: Piecing together helices and hairpins in designed peptides. Chemistry 7 840–847. [DOI] [PubMed] [Google Scholar]
- Dasgupta, S. and Bell, J.A. 1993. Design of helix ends. Amino acid preferences, hydrogen bonding and electrostatic interactions. Int. J. Peptide Protein Res. 41 499–511. [PubMed] [Google Scholar]
- DeLano, W.L. and Brunger, A.T. 1995. The direct rotation function: Rotational Patterson correlation search applied to molecular replacement. Acta Cryst. D51 740–748. [DOI] [PubMed] [Google Scholar]
- Eriksson, A.E., Baase, W.A., and Matthews, B.W. 1993. Similar hydrophobic replacements of Leu 99 and Phe 153 within the core of T4 lysozyme have different structural and thermodynamic consequences. J. Mol. Biol. 229 747–769. [DOI] [PubMed] [Google Scholar]
- Gray, T.M. and Matthews, B.W. 1987. Structural analysis of the temperature-sensitive mutant of bacteriophage T4 lysozyme, Gly 156 → Asp. J. Biol. Chem. 262 16858–16864. [DOI] [PubMed] [Google Scholar]
- Grütter, M.G., Gray, T.M., Weaver, L.H., Alber, T., Wilson, K., and Matthews, B.W. 1987. Structural studies of mutants of the lysozyme of bacteriophage T4. The temperature sensitive mutant protein Thr 157 → Ile. J. Mol. Biol. 197 315–329. [DOI] [PubMed] [Google Scholar]
- Hamlin, R. 1985. Multiwire area X-ray diffractometers. Methods Enzymol. 114 416–452. [DOI] [PubMed] [Google Scholar]
- Jones, T.A. and Kjeldgaard, M. 1997. Electron-density map interpretation. Methods Enzymol. 277 173–208. [DOI] [PubMed] [Google Scholar]
- Kallenbach, N.R. and Gong, Y. 1999. C-terminal capping motifs in model helical peptides. Bioorg. Med. Chem. 7 143–151. [DOI] [PubMed] [Google Scholar]
- Kraulis, P.J. 1991. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Cryst. 24 946–950. [Google Scholar]
- Kunkel, T.A. 1985. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc. Natl. Acad. Sci. 82 488–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski, R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Cryst. 26 283–291. [Google Scholar]
- Matsumura, M. and Matthews, B.W. 1989. Control of enzyme activity by an engineered disulfide bond. Science 243 792–794. [DOI] [PubMed] [Google Scholar]
- Matthews, B.W. 1993. Structural and genetic analysis of protein stability. Ann. Rev. Biochem. 62 139–160. [DOI] [PubMed] [Google Scholar]
- Merritt, E.A. and Bacon, D.J. 1997. Raster3D: Photorealistic molecular graphics. Methods Enzymol. 277 505–524. [DOI] [PubMed] [Google Scholar]
- Milner-White, E.J. 1988. Recurring loop motif in proteins that occurs in right-handed and left-handed forms. Its relationship with alpha-helices and beta-bulge loops. J. Mol. Biol. 199 503–511. [DOI] [PubMed] [Google Scholar]
- Nicholson, H., Anderson, D.E., Dao-pin, S., and Matthews, B.W. 1991. Analysis of the interaction between charged side-chains and the α-helix dipole using designed thermostable mutants of phage T4 lysozyme. Biochemistry 30 9816–9828. [DOI] [PubMed] [Google Scholar]
- Poteete, A.R., Dao-pin, S., Nicholson, H., and Matthews, B.W. 1991. Second-site revertants of an inactive T4 lysozyme mutant restore activity structuring the active site cleft. Biochemistry 30 1425–1432. [DOI] [PubMed] [Google Scholar]
- Presta, L.G. and Rose, G.D. 1988. Helix signals in proteins. Science 240 1632–1641. [DOI] [PubMed] [Google Scholar]
- Richardson, J.S. and Richardson, D.C. 1988. Amino acid preferences for specific locations at the ends of α-helices. Science 240 1648–1652. [DOI] [PubMed] [Google Scholar]
- Rost, B. and Sander, C. 1993. Prediction of protein secondary structure at better than 70% accuracy. J. Mol. Biol. 232 584–599. [DOI] [PubMed] [Google Scholar]
- Schellman, C. 1980. The αL conformation at the ends of helices. In Protein Folding (ed. R. Jaenicke), pp. 53–61. North-Holland Biomedical Press, Elsevier, Amsterdam.
- Serrano, L., Sancho, J., Hirshberg, M., and Fersht, A.R. 1992. α-helix stability in proteins. I. Empirical correlations concerning substitution of side-chains at the N and C-caps and the replacement of alanine by glycine or serine at solvent-exposed surfaces. J. Mol. Biol. 227 544–559. [DOI] [PubMed] [Google Scholar]
- Sukumar, M. and Gierasch, L.M. 1997. Local interactions in a Schellman motif dictate interhelical arrangement in a protein fragment. Fold. Des. 2 211–222. [DOI] [PubMed] [Google Scholar]
- Thomas, S.T., Loladze, V.V., and Makhatadze, G.I. 2001. Hydration of the peptide backbone largely defines the thermodynamic propensity scale of residues at the C` position of the C-capping box of α-helices. Proc. Natl. Acad. Sci. 98 10670–10675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronrud, D.E. 1997. TNT refinement package. Methods Enzymol. 277 306–319. [DOI] [PubMed] [Google Scholar]
- Tronrud, D.E., Ten Eyck, L.F., and Matthews, B.W. 1987. An efficient general-purpose least-squares refinement program for macromolecular structures. Acta Cryst. A43 489–503. [Google Scholar]
- Viguera, A.R. and Serrano, L. 1995. Experimental analysis of the Schellman motif. J. Mol. Biol. 251 150–160. [DOI] [PubMed] [Google Scholar]
- Vriend, G. 1990. A molecular modeling and drug design program. J. Mol. Graph. 8 52–56. [DOI] [PubMed] [Google Scholar]
- Wang, Y. and Shortle, D. 1997. Residual helical and turn structure in the denatured state of staphylococcal nuclease: Analysis of peptide fragments. Fold. Des. 2 93–100. [DOI] [PubMed] [Google Scholar]
- Zhang, C.T. and Zhang, R. 2001. A refined accuracy index to evaluate algorithms of protein secondary structure prediction. Proteins 43 520–522. [DOI] [PubMed] [Google Scholar]
- Zhang, X-J. and Matthews, B.W. 1995. EDPDB: A multi-functional tool for protein structure analysis. J. Appl. Cryst. 28 624–630. [Google Scholar]
- Zhang, X-J., Wozniak, J.A., and Matthews, B.W. 1995. Protein flexibility and adaptability seen in 25 crystals forms of T4 lysozyme. J. Mol. Biol. 250 527–552. [DOI] [PubMed] [Google Scholar]