

Abstract
The Strep-tag II is a nine-amino acid peptide that was developed as an affinity tool for the purification of corresponding fusion proteins on streptavidin columns. The peptide recognizes the same pocket of streptavidin where the natural ligand is normally bound so that biotin or its chemical derivatives can be used for competitive elution. We report here the crystal structures of the streptavidin mutants `1' and `2,' which had been engineered for 10-fold higher affinity towards the Strep-tag II. Both streptavidin mutants carry mutations at positions 44, 45, and 47, that is, in a flexible loop region close to the binding site. The crystal structures of the two apo-proteins and their complexes with the Strep-tag II peptide were refined at resolutions below 2 Å. Both in the presence and absence of the peptide, the lid-like loop next to the ligand pocket—comprising residues 45 through 52—adopts an `open' conformation in all four subunits within the asymmetric unit. The same loop was previously described to be disordered in the wild-type apo-streptavidin and to close over the pocket upon complexation of the natural ligand biotin. Our findings suggest that stabilization of the `open' loop conformation in the absence of a ligand abolishes the need for conformational rearrangement prior to the docking of the voluminous peptide. Because no direct contacts between the flexible part of the loop and the peptide ligand were detected, it seems likely that the higher affinity of the two streptavidin mutants for the Strep-tag II is caused by a predominantly entropic mechanism.
Keywords: Biotin, crystal structure, protein engineering, Strep-tag, streptavidin
The development of protein purification protocols that utilize affinity tags has been an important factor contributing to the recent increase in the number of protein structures determined by X-ray crystallography or NMR techniques. The oligo-His-tag is widely used in conjunction with metal chelate resins (Hochuli 1990). The Strep-tag was developed as an alternative tool (Schmidt and Skerra 1993, 1994) and has been successfully applied in our own and numerous other laboratories (for a review, see Skerra and Schmidt 2000).
The Strep-tag is a nine-amino acid peptide (sequence: AWRHPQFGG) with high specificity and affinity towards the protein reagent streptavidin. Its sequence was derived by selection from a genetic peptide library (Schmidt and Skerra 1993) and it was found to occupy the same pocket of streptavidin where biotin otherwise becomes complexed (Schmidt et al. 1996). Hence, a corresponding fusion protein can be eluted under gentle buffer conditions—which may be optimized for a given polypeptide and can even contain additives like thiol compounds or detergents—from an affinity column with immobilized streptavidin, just by applying a low concentration of biotin or related compounds as competitor (Skerra and Schmidt 2000). This method usually permits the efficient purification of a recombinant protein in a single step. Another advantage of the Strep-tag is its independence from metal ions in the purification, an aspect often of interest when metallo-proteins are to be studied or when paramagnetic impurities must be avoided for NMR. In addition, the His-tag has a tendency to cause precipitation of proteins after elution from the metal chelate column due to aggregation induced by heavy metal ions.
The original Strep-tag had to be fused to the C-terminus of the recombinant protein in order to maintain a free peptide carboxylate group, giving rise to a salt bridge with an Arg side chain of streptavidin (Schmidt et al. 1996). To also allow for application at the N-terminus of the protein to be purified or even when inserted amid the polypeptide chain, the modified Strep-tag II (sequence: NWSHPQFEK) was subsequently engineered (Schmidt et al. 1996). One initial drawback of the Strep-tag II was its lower affinity towards streptavidin (with a KD value of approximately 13 μM; Voss and Skerra 1997). Consequently, streptavidin itself was engineered for improved binding of the peptide by preparing a genetic library with the randomized amino acid positions 44 through 47. These residues were found in the vicinity of the C-terminus of the Strep-tag II in the crystallized complex with streptavidin (Schmidt et al. 1996). With the use of a colony screening assay, the streptavidin mutants `1' and `2' (referred to herein as SAm1 and SAm2, respectively) were selected and shown to possess a significantly higher affinity towards the Strep-tag II, with KD values close to 1 μM (Voss and Skerra 1997).
To investigate the structural reasons underlying the enhanced performance of this generally useful biochemical tool (Skerra and Schmidt 2000), we pursued crystallization of the engineered streptavidin mutants. Here we report the results of the X-ray crystallographic analysis of both apo-proteins and their complexes with the Strep-tag II peptide. The structures will be compared to those of the wild-type streptavidin with the bound Strep-tag II as well as to the natural streptavidin/biotin complex.
Results
Crystal structure determination and quality of the final model
Crystals were obtained at physiological pH, belonging to the space group P21 for the two apo-proteins and their peptide complexes (Table 1). Complete X-ray diffraction data sets were collected from single crystals and directly refined (see Materials and Methods) using atom coordinates from the isomorphous structure of the crystallized streptavidin mutant Tyr43→Phe described by Freitag et al. (1999; PDB accession code 1swu) as a starting model. The mutated loop region was manually remodeled to fit the new electron density, and the refinement was extended to resolutions below 1.9 Å in all cases, with good stereochemistry and reaching final R-factors below 20% (Table 2).
Table 1.
Data collection statistics for crystals of streptavidin mutants `1' and `2'
| Mutant | apo-SAm1 | SAm1•StrepII | apo-SAm2 | SAm2•StrepII |
| Protein | streptavidin mutant `1' | streptavidin mutant `1' in complex with Strep-tag II peptide | streptavidin mutant `2' | streptavidin mutant `2' in complex with Strep-tag II peptide |
| Crystallization conditions | 1.2 M (NH4)2SO4, 100 mM Na2HPO4, pH 7.3 | 1.3 M (NH4)2SO4, 100 mM Na2HPO4, pH 6.9 | 50 mH Na2HPO4, 50 mM Na-Citrate, 30% PEG 600, pH 7.3 | 1.2 M (NH4)2SO4, 100 mM Na2HPO4, pH 7.5 |
| Space group | P21 | P21 | P21 | P21 |
| a, b, c | 58.42 Å, 86.92 Å, 47.20 Å | 58.70 Å, 86.34 Å, 47.45 Å | 58.45 Å, 86.81 Å, 47.08 Å | 58.39 Å, 86.51 Å, 47.24 Å |
| β | 98.90° | 98.92° | 98.82° | 99.10° |
| Resolution | 1.9 Å | 1.7 Å | 1.7 Å | 1.8 Å |
| No. of total reflections | 52768 | 126949 | 178009 | 120275 |
| No. of unique reflections | 33067 | 49002 | 48419 | 41071 |
| Completeness | 89.2% (99.00 − 1.90 Å) | 94.8% (99.00 − 1.70 Å) | 93.8% (99.00 − 1.70 Å) | 94.5% (99.00 − 1.80 Å) |
| 85.5% (1.97 − 1.90 Å) | 90.3% (1.76 − 1.70 Å) | 89.7% (1.76 − 1.70 Å) | 93.2% (1.86 − 1.80 Å) | |
| Rmerge | 0.05 | 0.04 | 0.07 | 0.04 |
Table 2.
Refinement statistics for streptavidin mutants `1' and `2'
| Mutant | apo-SAm1 | SAm1•StrepII | apo-SAm2 | SAm2•StrepII |
| R-factor/RFree | 17.0/22.9 | 17.5/21.7 | 19.1/22.8 | 17.8/22.1 |
| Protein residues | 478 | 480 | 478 | 480 |
| Protein residues visible | A16–A135 | A16–A136 | A16–A135 | A16–A136 |
| B15–B134 | B15–B134 | B15–B134 | B15–B134 | |
| C16–C134 | C15–C134 | C16–C134 | C15–C134 | |
| D16–D134 | D16–D134 | D16–D134 | D16–D134 | |
| Ligand residues | — | 24 | — | 22 |
| Ligand residues visible | E3–E8 | E3–E7 | ||
| F4–F9 | F4–F8 | |||
| G4–G9 | G4–G9 | |||
| H4–H9 | H4–H9 | |||
| No. of solvent molecules | 212 | 240 | 195 | 167 |
| R.m.s. deviations from ideality of bond lengths | 0.016 Å | 0.018 Å | 0.018 Å | 0.017 Å |
| R.m.s. deviations from ideality of bond angles | 2.868° | 2.773° | 2.548° | 2.679° |
| Average B-values: | ||||
| whole molecule | 27.5 Å2 | 27.3 Å2 | 25.1 Å2 | 24.4 Å2 |
| residues 46–51 | 48.6 Å2 | 43.2 Å2 | 47.4 Å2 | 41.8 Å2 |
| peptide ligand | — | 33.1 Å2 | — | 35.1 Å2 |
| PDB accession code | 1kff | 1kl3 | 1kl4 | 1kl5 |
The resulting structural models of the streptavidin mutants `1' and `2' are made up of one tetramer in the asymmetric unit with 119 to 120 amino acid residues per polypeptide chain, depending on the subunit (full length of the recombinant core streptavidin: 127 residues; Schmidt and Skerra 1994). The binding-site loop itself (residues 45 through 52; for mutations see Table 3) appears well ordered in all four subunits of the two mutants, both in the presence and absence of the ligand (Fig. 1 ▶). In contrast, the same loop was found to be disordered in a large number of streptavidin structures deposited in the PDB (for a compilation see Table 4). In the electron densities from the cocrystals with the Strep-tag II, at least 5 amino acids of the peptide ligand (residues 4 through 8) are clearly visible in all four subunits (Table 2, Fig. 1B ▶) and were thus included in the structural model. Lys9 could only be placed in some of the subunits (Table 2) and usually showed weak electron density for its side chain. Spurious density for the N-terminal residues of the peptide ligand could not be fitted unambiguously.
Table 3.
Amino acid sequences of the binding-site loop of wild-type streptavidin and its mutants
| Residue | 44a | 45 | 46 | 47 |
| wtSA | Glu | Ser | Ala | Val |
| SAm1b | Val | Thr | Ala | Arg |
| SAm2b | Ile | Gly | Ala | Arg |
a Numbering according to the mature full length protein (cf. Schmidt and Skerra 1994).
b Voss and Skerra (1997).
Fig. 1.

(A) 2Fo-Fc difference Fourier electron density of the binding-site loop in apo-SAm1 (subunit A) contoured at 1 σ. The binding-site loop is well ordered in the absence of the peptide ligand. (B) 2Fo-Fc difference Fourier electron density of the bound peptide in SAm1•StrepII (subunit A) contoured at 1 σ. Apart from the flanking residues, that is, Asn1 and Trp2 as well as Lys9, the Strep-tag II peptide is ordered and placed unambiguously in the electron density.
Table 4.
Conformations of the binding-site loop in streptavidin structures from this study and as deposited at the PDB
| Subunit | |||||||
| Structurea | A | B | C | D | Bound ligand | pH | Mutations |
| apo-SAm1 | Ob | O | O | O | — | 7.3 | E44V/S45T/V47R |
| SAm1•StrepII | O | O | O | O | Strep-tag II peptide | 6.9 | E44V/S45T/V47R |
| apo-SAm2 | O | O | O | O | — | 7.3 | E44I/S45G/V47R |
| SAm2•StrepII | O | O | O | O | Strep-tag II peptide | 7.5 | E44I/S45G/V47R |
| 1df8 | C | biotin | 4.5 | S45A | |||
| 1pts | C | D | FSHPQNT | na | |||
| 1rst | O | Strep-tag peptide | 7.0 | ||||
| 1rsu | O | Strep-tag II peptide | 7.0 | ||||
| 1sld | O | Cyclo-Ac-CHPQFC-NH2 | 7.5 | ||||
| 1sle | O | O | Ac-CHPQGPPC-NH2 | 5.0 | |||
| 1slf | C | C | 2 • (NH4)2SO4 | 5.6 | |||
| 1slg | O | O | Ac-FCHPQNT | 5.6 | |||
| 1sre | C | C | HABA | na | |||
| 1srf | C | C | 3-t-Butyl-HABA | na | |||
| 1srg | C | C | 3-Methyl-HABA | na | |||
| 1srh | C | C | 3`5`-Dimethoxy-HABA | na | |||
| 1sri | C | C | 3`5`-Dimethyl-HABA | na | |||
| 1srj | C | C | Naphtyl-HABA | na | |||
| 1stp | C | biotin | 7.8 | ||||
| 1str | O | O | Ac-CHPQNT-NH2 | 7.0 | |||
| 1sts | O | O | FCHPQNT-NH2 | 7.0 | |||
| 1swa | C | D(O) | D(O) | D(O) | — | 4.5 | |
| 1swb | C | D(O) | D(O) | D(O) | — | 7.5 | |
| 1swc | D(O) | O | D(O) | O | — | 4.5 | |
| 1swd | C | D(O) | D(O) | C | biotin | 4.5 | |
| 1swe | C | C | C | C | biotin | 4.5 | |
| 1swh | D | D | D(O) | D(O) | — | 4.5 | W79F |
| 1swj | D(O) | C | O | D(O) | — | 4.5 | W79F |
| 1swk | C | C | C | C | biotin | 4.5 | W79F |
| 1swl | C | D(O) | D | D | — | 7.0 | W108F |
| 1swn | C | D(O) | C | C | biotin | 7.0 | W108F |
| 1swo | C | D(O) | D(O) | D(O) | — | 7.5 | W120F |
| 1swp | C | C | C | C | biotin | 7.5 | W120F |
| 1swq | D | D(O) | D | D(O) | — | 7.5 | W120A |
| 1swr | C | C | C | C | biotin | 7.5 | W120A |
| 1sws | D(O) | O | D(O) | D(O) | — | 4.5 | D128A |
| 1swt | O | O | biotin | 4.5 | D128A | ||
| 1swu | C | D(O) | D(O) | D(O) | — | 4.5 | Y43F |
| 1vwa | O | O | FSHPQNT | 4.5 | |||
| 1vwb | O | Cyclo-Ac-CHPQFC-NH2 | 11.8 | ||||
| 1vwc | O | Cyclo-Ac-CHPQFC-NH2 | 2.0 | ||||
| 1vwd | O | Cyclo-Ac-CHPQFC-NH2 | 3.0 | ||||
| 1vwe | O | Cyclo-Ac-CHPQFC-NH2 | 3.6 | ||||
| 1vwf | O | Cyclo-Ac-CHPQGPPC-NH2 | 3.7 | ||||
| 1vwg | O | (Cyclo-Ac-CHPQGPPC-NH2)2 | 2.5 | ||||
| 1vwh | O | (Cyclo-Ac-CHPQGPPC-NH2)2 | 3.5 | ||||
| 1vwi | O | O | Cyclo-[5S-Valeramido-HPQGPPC]K-NH2 | 1.5 | |||
| 1vwj | O | O | Cyclo-[5S-Valeramido-HPQGPPC]K-NH2 | 2.5 | |||
| 1vwk | O | O | Cyclo-[5S-Valeramido-HPQGPPC]K-NH2 | 3.0 | |||
| 1vw1 | O | O | Cyclo-[5S-Valeramido-HPQGPPC]K-NH2 | 3.5 | |||
| 1vwm | O | Cyclo-Ac-CHPQFC-NH2 | 4.2 | ||||
| 1vwn | O | Cyclo-Ac-CHPQFC-NH2 | 4.8 | ||||
| 1vwo | O | Cyclo-Ac-CHPQGPPC-NH2 | 2.9 | ||||
| 1vwp | O | Cyclo-Ac-CHPQGPPC-NH2 | 2.5 | ||||
| 1vwq | O | Cyclo-[5S-Valeramido-HPQGPPC]K-NH2 | 2.5 | ||||
| 1vwr | O | Cyclo-[5S-Valeramido-HPQGPPC]K-NH2 | 3.5 | ||||
| 2iza | C | — | 2.0 | ||||
| 2izb | C | — | 3.1 | ||||
| 2izc | C | — | 2.0 | ||||
| 2izd | C | — | 3.0 | ||||
| 2ize | C | — | 3.1 | ||||
| 2izf | C | C | biotin | 4.0 | |||
| 2izg | C | C | biotin | 2.0 | |||
| 2izh | C | C | biotin | 10.4 | |||
| 2izi | C | C | biotin | 2.5 | |||
| 2izj | C | C | biotin | 3.5 | |||
| 2izk | C | C | glycoluril | 2.6 | |||
| 2izl | C | C | 2-iminobiotin | 7.3 | |||
| 2rta | C | C | — | 3.0 | |||
| 2rtb | C | C | — | 3.3 | |||
| 2rtc | C | C | — | 3.6 | |||
| 2rtd | C | C | biotin | 1.4 | |||
| 2rte | C | C | biotin | 1.9 | |||
| 2rtf | C | C | biotin | 2.0 | |||
| 2rtg | C | C | biotin | 2.4 | |||
| 2rth | C | C | glycoluril | 2.5 | |||
| 2rti | C | C | glycoluril | 2.5 | |||
| 2rtj | C | C | glycoluril | 2.5 | |||
| 2rtk | C | C | glycoluril | 2.6 | |||
| 2rtl | C | C | 2-iminobiotin-sulfate | 2.5 | |||
| 2rtm | C | C | 2-iminobiotin-sulfate | 3.5 | |||
| 2rtn | C | C | 2-iminobiotin | 2.0 | |||
| 2rto | C | C | 2-iminobiotin | 2.6 | |||
| 2rtp | C | C | 2-iminobiotin | 3.3 | |||
| 2rtq | C | C | 2-iminobiotin | 3.3 | |||
| 2rtr | C | C | 2-iminobiotin | 4.0 | |||
a For authors and references of the crystal structures, cf. the PDB deposition.
b O, `open' loop conformation; C, `closed' loop conformation; D, disordered loop conformation; D(O), disordered loop conformation, but positioning of neighboring residues indicates preference for the `open' loop conformation.
Table 2 shows refinement statistics for the final models of the proteins in their apo-form and in complex with the Strep-tag II peptide. In a Ramachandran plot (Ramachandran and Sasisekharan 1968), no main chain dihedral angles of nonglycine residues lie outside the allowed regions (not shown). After a pairwise least squares superposition of Cα-atoms (LSQKAB; CCP4 1994), mutual r.m.s. deviations of the subunits within the tetramer lie in a range between 0.23 and 0.30 Å. Average B-values for the whole streptavidin subunits, their binding-site loops, and the peptide ligands are given in Table 2.
Overall topology
The Cα-atoms of the corresponding A subunits of SAm1•StrepII and of the conventional streptavidin•biotin complex (1swu; Freitag et al. 1999) can be superimposed with an overall r.m.s.d. of 0.52 Å (Fig. 2 ▶). The largest deviations in Cα-positions occur within the binding-site loop (up to more than 13 Å), which is `closed' in wtSA•biotin but `open' in our mutant structures. Further deviations larger then 0.5 Å can be seen at residue 26, which is part of the ligand-binding pocket, at residues 66 and 100, which are involved in crystal and tetramer contacts, respectively, and at the C-terminus of the polypeptide chain. The subunit structures of the streptavidin mutant `1' in the presence and absence of the peptide ligand can be superimposed with a significantly smaller Cα r.m.s.d. of 0.32 Å (Fig. 3A ▶).
Fig. 2.

Mutual distances of Cα-positions after pairwise superposition of the A chains from the tetrameric streptavidin crystal structures: SAm1•StrepII versus wtSA•biotin (1swu; Freitag et al. 1999), SAm1•StrepII versus wtSA•StrepII (1rsu; Schmidt et al. 1996), SAm1•StrepII versus apo-SAm1, apo-SAm2 versus apo-SAm1, and apo-SAm2 versus SAm2•StrepII.
Fig. 3.
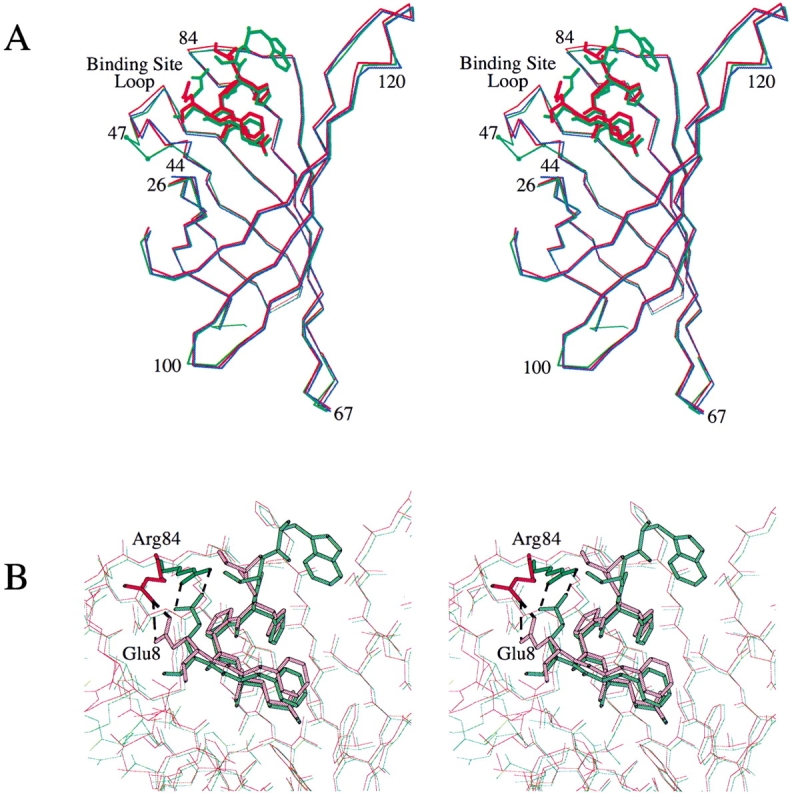
(A) Superposition of the Cα-backbones from the A chains of apo-SAm1 (blue) with SAm1•StrepII (red) and with the wtSA•StrepII complex (green, 1rsu). The residues that were mutated for the generation of SAm1 (44 to 47) are marked with small dots (green). Residues flanking this segment and other positions with large mutual deviations (cf. Fig. 2 ▶) are labeled. The deviations in positions 67, 100, and 120 are due to tetramer or crystal contacts. The binding-site loop assumes an `open' conformation in all structures, but has undergone partial rearrangement at the mutated positions with respect to wild-type streptavidin. (B) Superimposed binding sites of SAm1•StrepII (red) and wtSA•StrepII (green). The salt bridge between residues Arg84 of streptavidin and Glu8 of the peptide ligand has been proposed to be crucial for the complexation of the Strep-tag II by streptavidin (Schmidt et al. 1996). This salt bridge has shifted as a whole by 3 Å in SAm1•StrepII but remains intact. Residue Lys9 of the Strep-tag II peptide, as deposited in 1rsu (Schmidt et al. 1996), was omitted in both graphs because it exhibits only weak electron density and is probably disordered.
Interaction with the peptide ligand
When compared with the structure of wild-type streptavidin in complex with the Strep-tag II (PDB accession code 1rsu; Schmidt et al. 1996), the conformations of the peptide ligands are very similar (Fig. 3 ▶). Cα-atoms of the Strep-tag II complexed with SAm1 and of the same peptide ligand complexed with wtSA show an overall r.m.s.d. of 0.06 Å when directly superimposed.
In the crystal structure of wtSA•StrepII, the side chain of Lys9 from the peptide protrudes into the hydrophobic cleft formed by the side chain Pro4 of the ligand together with the side chains Tyr43, Tyr54, and Trp79 of streptavidin. However, reinspection of the electron density, calculated from the structure factors deposited along with 1rsu, revealed rarely significant density for this side chain (data not shown). Lys9 is at least partially disordered both in SAm1•StrepII and SAm2•StrepII, but 2Fo-Fc difference Fourier maps indicate that here its side chain should point into the solvent without specific contacts to protein residues. Lys9 was previously shown to significantly contribute to the binding of the Strep-tag II to streptavidin when covalently immobilized via its C-terminus during peptide screening experiments (Schmidt et al. 1996). However, the weak electron densities found for this residue do not permit the identification of a relevant interaction. The situation during complex formation with a C-terminally fused Strep-tag II may therefore be different from the one observed with the free peptide in our crystal structure.
Similarly, the electron density for the side chain of Trp2, which was found to be an important residue in the initial selection of the Strep-tag from a random peptide library (Schmidt and Skerra 1993), was rather weak. Nevertheless, both residues may exert an effect on the conformation of the peptide in solution and thus indirectly contribute to the apparent strength of binding (see Discussion). Finally, side chains of Glu8 from the ligand and Arg84 from streptavidin have been displaced by about 3 Å in the SAm1•StrepII structure with respect to wtSA•StrepII (Fig. 3B ▶). The salt bridge between these residues has been proposed to be crucial for the binding of the Strep-tag II to streptavidin, particularly when used as an N-terminal fusion peptide (Schmidt et al. 1996), and is still intact.
Structure of the binding site
The backbone structure of the binding-site loop comprising residues 45 through 52 is almost identical between the mutants `1' and `2', both in the presence and absence of the ligand (Fig. 2 ▶). Only marginal side chain conformational changes can be observed between SAm1 and SAm2 or upon binding of the Strep-tag II (Fig. 4 ▶). In the crystallographic unit cell, the binding-site loop is located close to packing contacts. However, other crystal structures of streptavidin have been determined in the same space group (e.g., 1swa and 1swb, Freitag et al. 1997; Table 4), where the binding-site loop is still disordered or even in the `closed' conformation. Furthermore, since the tetramer subunits are not related by crystallographic symmetry, all four loops form different contacts with their neighborhood. In subunit C, the closest distance between any of the residues from ValC44 to ArgC53 and a symmetry-related molecule is 5.0 Å (i.e., between ArgC53 and LysB134*). Since noncrystallographic symmetry restraints were also not used during the refinement process, crystal contacts alone cannot be made responsible for the consistently ordered conformation of the loop in the `open' state.
Fig. 4.

(A) Superposition of the ligand-binding sites of SAm1•StrepII (red) and wtSA•StrepII (green, 1rsu). The bound peptide ligands are depicted in corresponding light colors. The labeling of amino acid residues refers to SAm1•StrepII. Partial movement of the binding-site loop towards the hydrophobic cleft lined by residues Tyr43, Tyr54, and Trp79 as well as Pro5 from the peptide ligand results in a shift of Thr45 by 1.9 Å. (B) Superposition of the ligand-binding sites of SAm1•StrepII (red) and apo-SAm1 (blue). The bound peptide ligand of SAm1•StrepII is shown in light red. The conformation of the binding-site loop (residues 45 through 52) of SAm1 is nearly the same in the presence and absence of the peptide ligand. The two binding-site loops show an r.m.s.d. of 0.3 Å following Cα superposition of the protomers. (C) Superposition of the ligand-binding sites of apo-SAm1 (blue) and apo-SAm2 (cyan). The conformation of the binding-site loop is almost identical between the two mutants, both in the absence and presence of the peptide ligand (cf. Fig. 2 ▶). (D) Superposition of binding-site loops in wtSA•biotin (green, 1swd) and SAm1•StrepII (red). The biotin ligand of wild-type streptavidin is shown as ball and sticks (light green), whereas the peptide ligand in SAm1•StrepII is depicted as Cα trace (light red). Unlike Val47 in wild-type streptavidin, Arg47 in SAm1 cannot participate in a hydrophobic interaction with Leu25, and hence the loop is sterically forced into an `open' conformation. The side chain of Arg47 itself is partially disordered (cf. Fig. 1A ▶).
When compared with wtSA•StrepII, most of the loop exhibits precisely the same conformation in the SAm1•StrepII structure (Figs. 3A and 4A ▶ ▶). However, the mutated residues 45 to 47 at its N-terminal side show a local deviation. In particular, Thr45 has moved by 1.9 Å into the hydrophobic cleft lined by residues Tyr43, Tyr54, and Trp79, together with Pro5 from the peptide ligand. Residues Ala46 and Arg47 from the binding-site loop are displaced into the same direction. The side chain of Arg47 is partially disordered (Fig. 1A ▶). When comparing the structure of apo-SAm1 with that of SAm1•StrepII or even with that of apo-SAm2, the conformation of the binding-site loop is virtually indistinguishable (Fig. 4B,C ▶). Thus, it appears to be fixed in the two streptavidin mutants.
Discussion
Even though the amino acids at the positions 44, 45, and 47 of streptavidin (see Table 3) resulted from a selection experiment for improved binding of the Strep-tag II, these residues do not make structural contacts to the bound peptide. Thr45, which replaces a Ser residue in the streptavidin mutant `1', may be involved in a weak hydrophobic interaction with the side chain of Pro5 from the Strep-tag II (with a 4.2 Å distance between Cβ Pro5 and Cγ Thr45). However, the same position is occupied by a Gly residue in the streptavidin mutant `2', which possesses essentially the same ligand affinity and loop conformation. Consequently, direct contributions of the mutated side chains to the enhanced binding activity for the peptide are probably not involved.
When the crystal structure of the complex between streptavidin and biotin was first described (Hendrickson et al. 1989; Weber et al. 1989), the binding-site loop was found to be closed almost like a lid over the pocket harboring the small ligand. In contrast, the same loop was seen disordered in the crystal structure of apo-streptavidin (Weber et al. 1989). Therefore, this structural mechanism was postulated to play a role for the remarkably high affinity observed in this system (Kempner 1993). Alternative conformations of loops that are involved in substrate or ligand binding have frequently been observed elsewhere, for example, in the cases of rhamnose isomerase (Korndörfer et al. 2000) and triose-phosphate isomerase (Alber et al. 1987). In these structures, the loop is usually disordered in the apo-protein. Upon binding of the ligand the loop becomes ordered, closes the active site, and shields the ligand from solvent access (for a general review of flexible loops, see Kempner 1993).
Indeed, the binding-site loop (residues 45 through 52) is the conformationally most variable motif in the 83 different structures of streptavidin or its mutants deposited in the PDB (see Table 4). In an attempt to classify the various conformations of this loop, so-called `open' and `closed' states have been distinguished (Weber et al. 1989). As in the protein structures mentioned above, in the absence of a ligand the binding-site loop of streptavidin has been proposed to generally adopt an `open' conformation (Freitag et al. 1997). Still, in the structures deposited it can be seen that the loop may adopt `open' and `closed' conformations even within the same crystal structure (e.g., 1swa and 1swj; Freitag et al. 1997), although in the absence of a ligand it is mostly disordered (Table 4).
In the unique apo-streptavidin structure where the loop is ordered and in the `closed' conformation in all subunits (1slf; Katz 1995), two sulfate ions are actually localized in the binding pocket. Another series of apo-structures where the loop is `closed' has been crystallized at acidic pH (e.g., 2iza and 2izb; Katz 1997), which renders their physiological significance unclear. In the presence of the natural ligand biotin, the binding-site loop always assumes the `closed' conformation (e.g., 1stp; Weber et al. 1989 or 1swd; Freitag et al. 1997), whereas in the presence of a peptide ligand the loop is always `open' (e.g., 1rst; Schmidt et al. 1996). Therefore, the mutants `1' and `2,' which were obtained from a combinatorial mutagenesis experiment (Voss and Skerra 1997), would be the first engineered versions of streptavidin where the binding-site loop is well ordered and exhibits an `open' conformation in all of its four subunits, notably at physiological pH and in the absence of any ligand.
Taken together, the streptavidin mutants `1' and `2' differ from the wild-type core streptavidin in three amino acid positions (Table 3). These substitutions effect a decrease in the KD for the binding of the Strep-tag II from 13.0 ± 1.3μM to 1.37 ± 0.08 μM and 1.02 ± 0.04μM, respectively (Voss and Skerra 1997). Only the substitution Val47→Arg is strictly conserved in both streptavidin mutants. When the loop is closed, the Val47 side chain of wild-type streptavidin is positioned such that it faces towards the hydrophobic side chain of Leu25 (Fig. 4D ▶). The side chain of an Arg residue cannot be accommodated in the same orientation due to its larger size and to the highly polar character. Thus, a loop conformation where Arg47 is pointing into solution is probably favorable. Arg47 is not involved in specific ionic interactions or hydrogen bonds or in crystal packing contacts with symmetry-related molecules; rather, its side chain is disordered.
The `open' loop conformation may furthermore be stabilized by the substitution Glu44→Val. This hydrophobic residue might be partially shielded from solvent by the main chain atoms of Gly48 and the side chain of Arg53. In SAm1•StrepII, Val44 has an average accessible surface area of 9 Å2 (SURFACE; CCP4 1994). A simple substitution of Glu44 by Val in the crystal structure of 1swb (Freitag et al. 1997), where the loop is in the `closed' conformation, would lead to a much higher hypothetical accessible surface area of 28 Å2 for this residue. This effect should be even more pronounced in the mutant `2,' where an Ile residue occupies the corresponding position.
To understand the influence of the loop conformation on the ligand affinity, a thermodynamic consideration may be applied. Even though streptavidin constitutes a stable tetramer in solution, its four ligand-binding sites are independent and structurally remote from each other, including the corresponding flexible loops, so that it may be treated here to behave generally like a monomeric protein. Let us assume a simple association/dissociation equilibrium between such a protein, occurring in an active state A, and its ligand L, characterized by the dissociation constant:
 |
Let us further assume that the active state A corresponds to the wild-type streptavidin monomer with the `open' loop conformation, which participates in a dynamic equilibrium with a population of monomers X that have loop conformations in any other conformation (e.g., `closed') being sterically incompatible with the complexation of the ligand (i.e., the Strep-tag II):
 |
If we cannot distinguish between these two conformational states of the protein by means of physicochemical or spectroscopic methods, as is in fact the case for the system treated here, a ligand-binding experiment may just reveal an `apparent' dissociation constant according to the following Law of Mass equation:
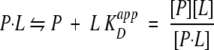 |
Here P constitutes the sum of the protein in both states: [P] = [A] + [X].
Since [P•L] must equal [A•L] from above, the following equation can be deduced:
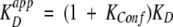 |
This means that the measurable apparent dissociation constant is raised by a factor (1 + KConf) compared with the intrinsic KD of the complexation-competent conformation A (here the `open' loop form of streptavidin) as a result of its equilibrium with the complexation-incompetent conformation X (here the closed-loop or related conformation). If this equilibrium is far on the side of the A state, KConf is small and this factor approaches unity; that is, the true KD will be measured. However, if the equilibrium is on the side of the X state, KConf becomes significantly larger than 1 and the apparent KD is increased to an appreciable extent.
For example, KConf = 9 (i.e., just 10% of the free protein is in the active state A) would result in KDapp = 10 • KD. This number should roughly reflect the situation for the complex formation between the wild-type streptavidin with its flexible loop and the Strep-tag II peptide, where the apparent dissociation constant was measured to be approximately ten-fold higher than for the two streptavidin mutants with the loop fixed in the `open' conformation. By the way, a similar theoretical consideration would in turn apply to distinct conformational states of the peptide ligand in solution and a corresponding indirect effect on the binding strength. Hence, it appears that the stabilization of a flexible loop in a conformation that is `competent' for the binding of the cognate ligand or substrate would be a general means to increase the affinity for this target via an entropic mechanism.
The same interpretation would be valid for the `induced fit' phenomenon, which is well known in the structural biology of antibody–antigen reactions (Stanfield and Wilson 1994). In this case, some of the six hypervariable loops, predominantly CDR-H3, are found in a different conformation when complexed with the antigen than in the crystal structure of the antibody fragment alone. Again, an equilibrium may be postulated between the two conformations, whereby the latter is preferentially populated in the absence of the antigen. Although it has been speculated that induced fit may be a general requirement for tight antigen binding, the affinity should actually be diminished by this phenomenon according to the argument given above. Of course, it could be the case that there is no alternative solution provided by the immune system that would lead to an equally good structural fit in the absence of an induced conformation. Nevertheless, if some somatic mutation could be acquired during maturation of the immune response that would effect fixation of the loop conformation in a productive manner, this in itself should result in an improved antigen affinity.
In conclusion, this study demonstrates that the introduction of three amino acid substitutions into a flexible loop that is part of the streptavidin binding site lowers its conformational entropy by freezing it in an `open' state. Thus, without making direct contacts to the artificial Strep-tag II ligand, its binding becomes considerably favored. This observation illustrates that it is not sufficient to consider the structural situation in the preformed biomolecular complex alone when attempting to improve molecular recognition.
Materials and methods
Protein crystallization
Recombinant core streptavidin mutants `1' and `2' (for amino acid replacements, see Table 3), comprising residues 13 to 139 of the mature protein, were overexpressed as inclusion bodies in E. coli using the expression vector pSA1 and refolded as described (Schmidt and Skerra 1994; Voss and Skerra 1997). The purified synthetic Strep-tag II peptide (Ac-Ser-Ala-Trp-Ser-His-Pro-Gln-Phe-Glu-Lys-NH2) was a gift from IBA (Göttingen, Germany). Sparse matrix crystallization screens (Jancarik and Kim 1991) were performed using the hanging-drop vapor diffusion technique (McPherson 1999). Three μL of protein solution (22 mg/mL in 5 mM Tris/HCl, pH 8.0) were mixed on a siliconized glass cover slip with 3 μL of the precipitant and equilibrated against 0.5 mL of the precipitant solution at 20°C. Typically, crystals of the streptavidin mutants were obtained after 4 to 5 days. Following optimization of the crystallization conditions, crystals of streptavidin mutants without the Strep-tag II peptide were obtained using (NH4)2SO4 or PEG 600 as precipitant. For cocrystallization experiments with the Strep-tag II peptide, 2 μL of the protein solution (11.5 mg/mL in 2.5 mM Tris, pH 8.0) were mixed with 1 μL of the peptide (8 mg/mL in H2O) and 2 μL of the reservoir solution. Precise crystallization conditions for the crystals described in this work are given in Table 1.
Data collection and processing
Crystals were mounted directly from the drop in thin wall glass capillary tubes (Müller, Schönwalde, Germany). Essentially complete X-ray diffraction data sets were collected from single crystals at room temperature on a mar345 imaging plate detector (X-RAY Research, Norderstedt, Germany) with CuKα radiation from an RU-300 rotating anode generator (Rigaku, Tokyo, Japan) monochromatized and focused through MaxFlux Confocal Multilayer Mirror Optics (Osmic, Troy, MI). Crystal-detector distances were 110 mm or 130 mm. The data were processed and scaled using the programs DENZO and SCALEPACK (Otwinowski and Minor 1997). Reduction to structure factor amplitudes was performed with TRUNCATE (French and Wilson 1978; CCP4 1994). Data collection statistics are given in Table 1.
Model building and refinement
The unit cell dimensions of the streptavidin crystals investigated here (Table 1) matched those of the crystallized streptavidin mutant Y43F described by Freitag et al. (1999) (PDB accession no. 1swu). Consequently, all solvent and ligand molecules were stripped from this model and, starting from these coordinates, rigid body refinement was directly carried out with data to 2.5 Å using the program CNS (Brunger et al. 1998). After 20 steps of rigid-body minimization of the whole streptavidin tetramer and 20 steps of refinement of the four individual chains in the asymmetric unit, the R-factor dropped from 35.2% (for apo-SAm1) and 38.1% (for SAm1•StrepII) to 31.3% and 32.8%, respectively. The loop from residues 45 to 52 was manually rebuilt using QUANTA (Oldfield 2001) in order to fit the 2Fo-Fc and Fo-Fc electron densities, which also confirmed the identity of the mutated side chains. Numbering of residues was adjusted to match that used in previous publications from our lab, thus referring to the mature full-length streptavidin instead of the truncated core protein (Schmidt and Skerra 1994). For apo-SAm2 and SAm2•StrepII, the refined structures of apo-SAm1 and SAm1•StrepII, respectively, were used as starting models. Refinement was continued with REFMAC5 (CCP4 1994; Murshudov et al. 1997). Several sessions of manual rebuilding alternated with further rounds of refinement against all data. Water molecules were positioned where stereochemically plausible and 2Fo-Fc and Fo-Fc difference Fourier maps showed densities of 1.0 and 3.0 σ, respectively. The Free-R factor (Brunger 1992) was used to monitor the refinement and solvent building process. However, it must be kept in mind that its significance is limited here due to the presence of noncrystallographic symmetry in the crystals and to the unavailability of the `free' data set from the 1swu model structure (Freitag et al. 1999) to allow for a corresponding selection. Noncrystallographic symmetry restraints were not used in the refinement process. Table 2 summarizes refinement statistics for the models presented in this work. Illustrations of molecular models and electron densities were prepared with MOLSCRIPT (Kraulis 1991) and MINIMAGE (Arnez 1994). The coordinates and structure factors of all four crystal structures have been deposited at the Protein Data Bank (Berman et al. 2000).
Acknowledgments
We thank J. Danzer for crystallization, M. Schlapschy for protein preparation and purification, and T.G.M. Schmidt (IBA, Göttingen) for providing samples of SAm2 and the synthetic Strep-tag II peptide.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
KD, equilibrium constant of dissociation
NMR, nuclear magnetic resonance
PDB, protein data bank
r.m.s.d., root mean square deviation
SAm1, streptavidin mutant `1'
SAm2, streptavidin mutant `2'
StrepII, Strep-tag II peptide
wtSA, recombinant wild-type core streptavidin
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.4150102.
References
- Alber, T.C., Davenport, Jr., R.C., Giammona, D.A., Lolis, E., Petsko, G.A., and Ringe, D. 1987. Crystallography and site-directed mutagenesis of yeast triosephosphate isomerase: What can we learn about catalysis from a "simple" enzyme? Cold Spring Harbor Symp. Quant. Biol. 52 603–613. [DOI] [PubMed] [Google Scholar]
- Arnez, J.G. 1994. MINIMAGE: A program for plotting electron density maps. J. Appl. Crystallogr. 27 649–653. [Google Scholar]
- Berman, H.M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T.N., Weissig, H., Shindyalov, I.N., and Bourne, P.E. 2000. The Protein Data Bank. Nucleic Acids Res. 28 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger, A.T. 1992. Free R value: A novel statistic quantity for assessing the accuracy of crystal structures. Nature 335 472–474. [DOI] [PubMed] [Google Scholar]
- Brunger, A.T., Adams, P.D., Clore, G.M., Delano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.-S., Kuszewski, J., Nilges, N., Pannu, N.S., Read, R.J., Rice, L.M., Simonson, T., and Warren, G.L. 1998. Crystallography and NMR system (CNS): A new software system for macromolecular structure determination. Acta Crystallogr. A 54 905–921. [DOI] [PubMed] [Google Scholar]
- CCP4 1994. Collaborative Computing Project Number 4: The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D 50 760–763. [DOI] [PubMed] [Google Scholar]
- Freitag, S., Le Trong, I., Klumb, L., Stayton, P.S., and Stenkamp, R.E. 1997. Structural studies of the streptavidin binding loop. Protein Sci. 6 1157–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freitag, S., Le Trong, I., Klumb, L.A., Stayton, P.S., and Stenkamp, R.E. 1999. Atomic resolution crystal structure of biotin-free Tyr43Phe streptavidin: What is in the binding site? Acta Crystallogr. D 55 1118–1126. [DOI] [PubMed] [Google Scholar]
- French, S. and Wilson, K. 1978. On the treatment of negative intensity observations. Acta Crystallogr. A 34 517–524. [Google Scholar]
- Hendrickson, W.A., Pähler, A., Smith, J.L., Satow, Y., Merritt, E.A., and Phizackerley, R.P. 1989. Crystal structure of core streptavidin determined from multiwavelength anomalous diffraction of synchrotron radiation. Proc. Natl. Acad. Sci. 86 2190–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochuli, E. 1990. Purification of recombinant proteins with metal chelate adsorbent. Genet. Eng. 12 87–98. [DOI] [PubMed] [Google Scholar]
- Jancarik, J. and Kim, S.-H. 1991. Sparse matrix sampling: A screening method for crystallization of proteins. J. Appl. Crystallogr. 24 409–411. [Google Scholar]
- Katz, B.A. 1995. Binding to protein targets of peptidic leads discovered by phage display: Crystal structures of streptavidin-bound linear and cyclic peptide ligands containing the HPQ sequence. Biochemistry 34 15421–15429. [DOI] [PubMed] [Google Scholar]
- Katz, B.A. 1997. Binding of biotin to streptavidin stabilizes intersubunit salt bridges between Asp61 and His87 at low pH. J. Mol. Biol. 274 776–800. [DOI] [PubMed] [Google Scholar]
- Kempner, E.S. 1993. Movable lobes and flexible loops in proteins. Structural deformations that control biochemical activity. FEBS Lett. 326 4–10. [DOI] [PubMed] [Google Scholar]
- Korndörfer, I.P., Fessner, W.-D., and Matthews, B.W. 2000. The structure of rhamnose isomerase from Escherichia coli and its relation with xylose isomerase illustrates a change between inter- and intra-subunit complementation during evolution. J. Mol. Biol. 300 917–933. [DOI] [PubMed] [Google Scholar]
- Kraulis, P.J. 1991. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 24 946–950. [Google Scholar]
- McPherson, A. 1999. Crystallization of biological macromolecules. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Murshudov, G.N., Vagin, A.A., and Dodson, E.J. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D 53 240–255. [DOI] [PubMed] [Google Scholar]
- Oldfield, T.J. 2001. A number of real-space torsion-angle refinement rechniques for proteins, nucleic acids, ligands and solvent. Acta Crystallogr. D 57 82–94. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276 307–326. [DOI] [PubMed] [Google Scholar]
- Ramachandran, G.N. and Sasisekharan, V. 1968. Conformation of polypeptides and proteins. Adv. Protein Chem. 23 283–437. [DOI] [PubMed] [Google Scholar]
- Schmidt, T.G.M., Koepke, J., Frank, R., and Skerra, A. 1996. Molecular interactions between the Strep-tag affinity peptide and its cognate target, streptavidin. J. Mol. Biol. 255 753–766. [DOI] [PubMed] [Google Scholar]
- Schmidt, T.G.M. and Skerra, A. 1993. The random peptide library-assisted engineering of a C-terminal affinity peptide, useful for the detection and purification of a functional Ig Fv fragment. Protein Eng. 6 109–122. [DOI] [PubMed] [Google Scholar]
- Schmidt, T.G.M. and Skerra, A. 1994. One-step affinity purification of bacterially produced proteins by means of the "Strep-tag" and immobilized recombinant core streptavidin. J. Chromatogr. A 676 337–345. [DOI] [PubMed] [Google Scholar]
- Skerra, A. and Schmidt, T.G.M. 2000. Use of the Strep-tag and streptavidin for detection and purification of recombinant proteins. Methods Enzymol. 326 271–304. [DOI] [PubMed] [Google Scholar]
- Stanfield, R.L. and Wilson, I.A. 1994. Antigen-induced conformational changes in antibodies: A problem for structural prediction and design. Trends Biotechnol. 12 275–279. [DOI] [PubMed] [Google Scholar]
- Voss, S. and Skerra, A. 1997. Mutagenesis of a flexible loop in streptavidin leads to higher affinity for the Strep-tag II peptide and improved performance in recombinant protein purification. Protein Eng. 10 975–982. [DOI] [PubMed] [Google Scholar]
- Weber, P.C., Ohlendorf, D.H., Wendoloski, J.J., and Salemme, F.R. 1989. Structural origins of high-affinity biotin binding to streptavidin. Science 243 85–88. [DOI] [PubMed] [Google Scholar]