

Abstract
Bacterial chemoreceptors signal across the membrane by conformational changes that traverse a four-helix transmembrane domain. High-resolution structures are available for the chemoreceptor periplasmic domain and part of the cytoplasmic domain but not for the transmembrane domain. Thus, we constructed molecular models of the transmembrane domains of chemoreceptors Trg and Tar, using coordinates of an unrelated four-helix coiled coil as a template and the X-ray structure of a chemoreceptor periplasmic domain to establish register and positioning. We tested the models using the extensive data for cross-linking propensities between cysteines introduced into adjacent transmembrane helices, and we found that many aspects of the models corresponded with experimental observations. The one striking disparity, the register of transmembrane helix 2 (TM2) relative to its partner transmembrane helix 1, could be corrected by sliding TM2 along its long axis toward the periplasm. The correction implied that axial sliding of TM2, the signaling movement indicated by a large body of data, was of greater magnitude than previously thought. The refined models were used to assess effects of inter-helical disulfides on the two ligand-induced conformational changes observed in alternative crystal structures of periplasmic domains: axial sliding within a subunit and subunit rotation. Analyses using a measure of disulfide potential energy provided strong support for the helical sliding model of transmembrane signaling but indicated that subunit rotation could be involved in other ligand-induced effects. Those analyses plus modeled distances between diagnostic cysteine pairs indicated a magnitude for TM2 sliding in transmembrane signaling of several angstroms.
Keywords: Bacterial chemotaxis, transmembrane receptors, conformational change, molecular modeling
Motile bacteria move toward nutrients and away from undesirable chemicals. This behavior, called chemotaxis, is mediated by a histidine kinase signaling pathway that can respond to a wide variety of compounds (for review, see Falke et al. 1997; Falke and Hazelbauer 2001). Each compound binds, directly or indirectly, to a specific transmembrane chemoreceptor at a site in the receptor periplasmic domain. Changes in ligand occupancy at these sites cause a change in the activity of a histidine kinase associated with the receptor cytoplasmic domain and thus cause an alteration in phosphorylation of a response regulator. This transmembrane signaling involves a conformational change within the receptor transmembrane domain (Falke and Hazelbauer 2001). In the current work, we used modeling approaches to investigate chemoreceptor transmembrane structure and conformational signaling.
Bacterial chemoreceptor structure
The most thoroughly studied chemoreceptors are those of Escherichia coli and Salmonella typhimurium. Structures have been determined by X-ray crystallography for receptor fragments: the entire periplasmic domain of the aspartate receptor, Tar, from both species (Milburn et al. 1991; Bowie et al. 1995; Yeh et al. 1996) and of most of the cytoplasmic domain of the serine receptor, Tsr, from E. coli (Kim et al. 1999). It has not been possible to obtain crystals of intact chemoreceptors. Instead, the most extensive information about transmembrane domain structure comes from disulfide scanning, patterns of oxidative cross-linking between introduced cysteines (Pakula and Simon 1992; Lee et al. 1994, 1995a; Hughson et al. 1997).
A model of the homodimeric chemoreceptor as an extended helical bundle (Fig. 1 ▶) can be created by combining the crystal structures of receptor fragments with information provided by disulfide scanning of the transmembrane and cytoplasmic domains of intact receptors (Kim et al. 1999; Falke and Hazelbauer 2001). The receptor can be considered to have three domains: a periplasmic domain that binds ligand, a cytoplasmic domain that binds and regulates the associated histidine kinase, and a transmembrane domain that couples the other two domains structurally and functionally. The periplasmic domain contains two membrane-distal four-helix bundles, one from each subunit. The two central helices (α1 and α4; see Fig. 1 ▶) from each bundle extend to the membrane. The transmembrane domain consists of four transmembrane helices, transmembrane helix 1 (TM1) and transmembrane helix 2 (TM2) from one monomer and TM1` and TM2` from the other.
Fig. 1.
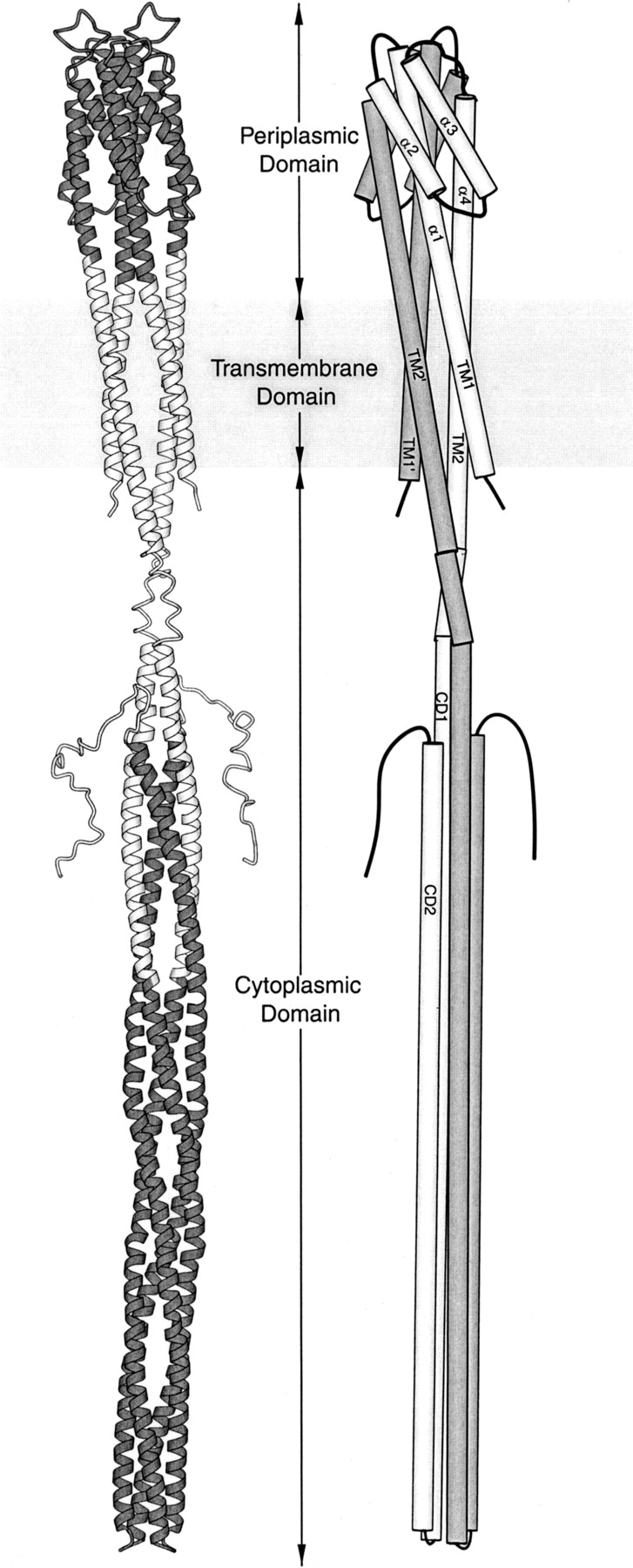
Models of chemoreceptor structure. In a ribbon diagram (left) of a homodimeric bacterial chemoreceptor, the portions of the model based on X-ray crystallography of receptor fragments are shaded. In a schematic diagram (right), helices are labeled, subunits are differentially shaded, and helix super-coiling is omitted. Each ∼60-kD subunit begins on the cytoplasmic side of the membrane, crosses the membrane as transmembrane helix 1 (TM1), becomes helix α1 of the periplasmic domain, turns to make the membrane-distal four-helix bundle, extends to the membrane as helix α4, crosses the membrane as transmembrane helix 2 (TM2), extends into the cytoplasm, and makes a membrane-distal turn to create the extended helical hairpin of cytoplasmic helices 1 and 2 (CD1 and CD2). The ligand-binding site is at the membrane-distal end of the periplasmic domain, and the kinase-interaction site is at the membrane-distal end of the cytoplasmic domain. This figure was made using Molscript (Kraulis 1991).
The conformational change of transmembrane signaling
Ligand binding does not alter the dimeric state of a chemoreceptor (Milligan and Koshland 1991), and receptors can signal even if subunit dissociation is prohibited by a covalent cross-link across the dimer interface (Falke and Koshland 1987; Scott and Stoddard 1994; Chervitz et al. 1995; Lee et al. 1995b). Thus, transmembrane signaling must involve a conformational change within the dimeric structure. Many lines of evidence indicate that this conformational change is subtle (for review, see Falke and Hazelbauer 2001), and thus, high-resolution structural studies by X-ray crystallography are appropriate approaches for identifying its essential nature. However, such studies have been possible only with a receptor fragment, the isolated periplasmic domain of TarS, the aspartate receptor of S. typhimurium (Milburn et al. 1991; Yeh et al. 1996). Comparisons of aspartate-bound and aspartate-free structures by the model-independent approach of distance-difference analysis identify two different conformational shifts, depending on the presence of an engineered, inter-subunit disulfide (Chervitz and Falke 1996; Yeh et al. 1996; Björkman et al. 2001; Falke and Hazelbauer 2001). When the truncated ends of helices α1 and α1` are held in their natural proximity by an engineered cross-link, the largest motion seen on ligand binding is an axial sliding of α4 relative to the rest of the receptor. In the absence of that cross-link, ligand binding causes several changes, the largest of which is a rotation of one subunit relative to the other. A significant body of experimental data implicates axial sliding of α4/TM2 in conformational signaling (Falke and Hazelbauer 2001), but the full significance of the different ligand-induced conformational changes observed in crystals of different forms of the periplasmic fragment is yet to be understood.
Modeling the transmembrane domain
What are the structural relationships among the four helices of the chemoreceptor transmembrane domain? How does transmembrane signaling alter those relationships? Addressing these questions requires a detailed structure for the transmembrane domain. In the absence of an atomic-resolution structure, an attractive alternative is to create a molecular model using available experimental information. In previous work, the relative dispositions of the transmembrane helices were modeled by using patterns of oxidative cross-linking between introduced cysteines for TarE (Pakula and Simon 1992) and Trg (Lee et al. 1994; Lee and Hazelbauer 1995), or by extending the helices in the X-ray structure of the periplasmic domain of TarS (Milburn et al. 1991; Scott and Stoddard 1994; Chervitz and Falke 1995), but these models were not analyzed in detail. In the current study, we extended modeling efforts by creating models based on X-ray structures, testing and refining those models with information derived from oxidative cross-linking, and using the refined models to analyze conformational signaling.
Results
Modeling the chemoreceptor transmembrane domain
We constructed molecular models of chemoreceptors Tar and Trg that consisted of their respective periplasmic and transmembrane domains. We began with coordinates of the periplasmic fragment of TarS (1LIH; Milburn et al. 1991) and used the coiled-coil structure of a mutant leucine zipper that forms a four-helix bundle (Harbury et al. 1993) as a template for extending the four long helices of the periplasmic domain to create the transmembrane helices. This strategy for modeling the transmembrane domain was based on the approach of Scott and Stoddard (1994), who used coordinates of a natural two-helix leucine zipper as the template for a model of TM1 and TM1`. For Tar, we retained residues of TarS in the periplasmic domain and replaced side-chains of the leucine zipper with side-chains of TarS. For Trg, we aligned the sequences of Trg and TarS (see Materials and Methods) and replaced the side-chains of both the leucine zipper and the TarS periplasmic domain with side-chains of Trg. Studies to be described elsewhere (W-C. Lai, M.L. Peach, T.P. Lybrand, and G.L. Hazelbauer, in prep.) indicate that the periplasmic domain of TarS provides a valid template for the periplasmic domain of Trg, even though sequence relationship is marginal. Both models were refined by correcting steric clashes and asymmetry between corresponding side-chains in the two subunits and by limited energy minimization to relax the structure and remove remaining steric overlaps.
Testing and refining: The subunit interface
In the initial versions of our models, the position, orientation, and register of the transmembrane helices were determined by the X-ray structures, independent of experimental data about the transmembrane helices. To refine and validate the models, it was important to determine the degree to which they were consistent with experimental characterization of the two transmembrane domains. The most extensive experimental probing of transmembrane structure in chemoreceptors has been determination of propensities for oxidative cross-linking between cysteine pairs at homologous positions in the two subunits. These determinations have been made quantitatively for every position in the transmembrane segments of Trg (Lee et al. 1994) and qualitatively for almost every position in TarE (Pakula and Simon 1992), the receptor from E. coli that has 70% residue identity to TarS in the periplasmic and transmembrane domains.
Because cross-linking propensity is a function of the distance between reactive sulfhydryls (Careaga and Falke 1992), we compared α-carbon distances in our models to experimental cross-linking propensities at homologous positions in TM1 and TM2 (Fig. 2 ▶). Our models corresponded to experimental data in terms of both general patterns and specific features. Experimentally, there was more extensive cross-linking across the TM1-TM1` interface than across the TM2-TM2` interface for both Trg and Tar. In the models of Trg and Tar, the TM1-TM1` pair was significantly closer than the TM2-TM2` pair. Experimentally, local maxima of cross-linking identified the rotational orientation of each helix relative to its homologous partner in the other subunit. This was the same orientation identified by residue pairs separated by the shortest distances in both chemoreceptor models. For Trg, in which cross-linking propensities were quantified (Lee et al. 1994; Hughson et al. 1997), the distinct local maxima corresponded well to positions that in the model were in closest proximity across the subunit interface. Not all positions in the modeled interface showed extensive cross-linking, probably because oxidation is influenced by factors in addition to the distance between residues.
Fig. 2.

Patterns of homologous cross-linking and Cα-Cα` distance for cysteines introduced into transmembrane helices of chemoreceptors. Patterns of cross-linking between cysteines introduced at a single position in the receptor polypeptide, and thus present at two homologously placed positions in the two subunits of the receptor homodimer, are shown for transmembrane helix 1 (TM1) of Trg (A) and TarE (B) and for transmembrane helix 2 (TM2) of Trg (C) and TarE (D). Values were plotted as a function of residue position, starting with the periplasmic end of each transmembrane helix on the left. Cα-Cα` distances in angstroms (solid squares, right-hand axes) were plotted on an inverted scale so that upward peaks corresponded to shorter distances and thus greater probabilities of cross-linking. Values for extent of oxidative cross-linking (left-hand axes) for Trg (open circles) were averages of in vivo and in vitro data with two different oxidizing reagents (Hughson et al. 1997). Qualitative extents of cross-linking in Tar (bars) were from Pakula and Simon (1992).
Because the four long helices of the leucine zipper template were aligned individually with the corresponding helices in the periplasmic domain, which does not have a perfect coiled-coil structure (Scott et al. 1993), the resulting model did not maintain its coiled-coil organization over the entire length of the transmembrane domain. TM1 and TM2 remained in proximity within a subunit, but the distances separating TM1 and TM1` or TM2 and TM2` increased from the periplasmic to the cytoplasmic side of the domain (Fig. 2 ▶). In essence, the coiled-coil became partially unwound. This modeled unwinding corresponded to experimental observations. In the quantitative determinations of cross-linking propensities along the TM1-TM1` interface of intact Trg, the maxima in the periodic relationship between cross-linking and residue position decrease from periplasm to cytoplasm (Fig. 2A ▶), indicating a gradual separation of the subunits across the width of the membrane. For cross-linking across the TM1-TM1` interface of Tar, which was assessed only qualitatively, there is a general pattern of decreased propensity from periplasm to cytoplasm (Fig. 2B ▶), consistent with subunit splaying. Evidence for subunit splaying in Tar is provided by effects of TM1-TM1` cross-links on Tar function; cross-links near the periplasm-membrane boundary allow function, but those near the membrane-cytoplasm boundary do not (Chervitz et al. 1995).
Testing and refining: Positioning within a subunit
In contrast to the good agreement between our models and experiment for the TM1-TM1` and TM2-TM2` interfaces, there was a disparity for the TM1-TM2 interface between modeled positions of key residues and proximities indicated by cross-linking. Screening of random and engineered combinations of TM1 and TM2 cysteines in both Trg and Tar has identified certain pairs that cross-link readily (Pakula and Simon 1992; Lee et al. 1994; Beel and Hazelbauer 2001). These cysteines are likely to be among the most closely positioned across the TM1-TM2 interface. The left-hand portions of Figures 3 and 4 ▶ ▶ show these TM1-TM2 pairs in the initial models of Trg and Tar, respectively. In contrast to the TM1-TM1` cysteine pairs, none of the extensively cross-linked TM1-TM2 pairs in Trg and only two of six pairs in Tar were directly opposite each other. All the rest were significantly displaced, in the same direction and to roughly the same extent. For each displaced pair, the TM2 residue was shifted toward the cytoplasm relative to its optimal position opposite the corresponding TM1 residue. These displacements were not unique to models based on the 1LIH coordinates, which are for the TarS periplasmic fragment containing the disulfide between α1 and α1`. Very similar displacements were observed for models based on the coordinates of the TarS domain lacking the engineered disulfide (1VLS) or of the TarE periplasmic domain (2ASR), also devoid of cross-links. The systematic nature of this disparity argued that the two models should be adjusted by sliding TM2 along its long axis toward the periplasm, and such a sliding is plausible because there is much evidence that axial sliding of TM2 occurs readily (see Discussion). Thus, we modified our initial models of Tar and Trg by sliding TM2 along its axis toward the periplasm (Figs. 3, 4 ▶ ▶; right-hand diagrams). The shift that provided the best positioning of the cysteine pairs with high cross-linking propensities was 3.5 Å for Tar and 6.4 Å for Trg. These specific shifts also resulted in better alignments of the charged residues that bracket the two ends of TM1 and TM2 (Figs. 3, 4 ▶ ▶) and of aromatic and amidated residues commonly found in "aromatic belts" just inside the membrane boundaries (Reithmeier 1995; Wallin et al. 1997). This improved alignment of features independent of cross-linking lent substantial support to the validity of the direction and specific magnitudes of the shifts we made.
Fig. 3.
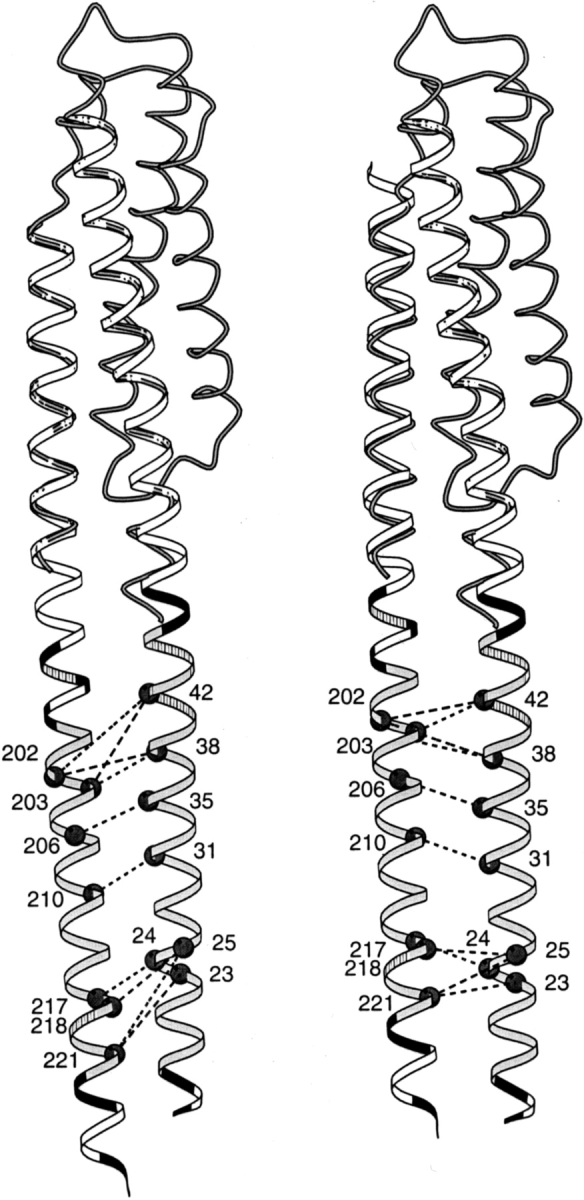
Positions of cysteine pairs with high propensity for cross-linking transmembrane helix 1 (TM1) and transmembrane helix 2 (TM2) of Trg. Initial (left) and revised (right) positions of TM2 relative to TM1 are diagrammed with cysteine pairs, showing high cross-linking propensities labeled with dark circles and joined by dashed lines. The cross-links shown are those in Lee et al. (1994), excluding one pair that showed very modest cross-linking on later quantification and four pairs created after that study that show cross-linking propensities within the range of the initial set (Beel and Hazelbauer 2001). The shaded coil shows the backbone structure of the monomer of the periplasmic domain (1LIH; Milburn et al. 1991) that was used to align the helices of the extended coiled coil template (ribbon). (Right) The sliding of α4/TM2 toward the periplasm has been emphasized by shifting only the coiled coil template, not the coil representing the original position of helix α4. The hydrophobic core of the transmembrane domain is shaded in light gray, positions of aromatic and amidated residues at the edges of the hydrophobic core are striped, and charged residues (including histidines) are black. This figure was made using MolScript (Kraulis 1991).
Fig. 4.
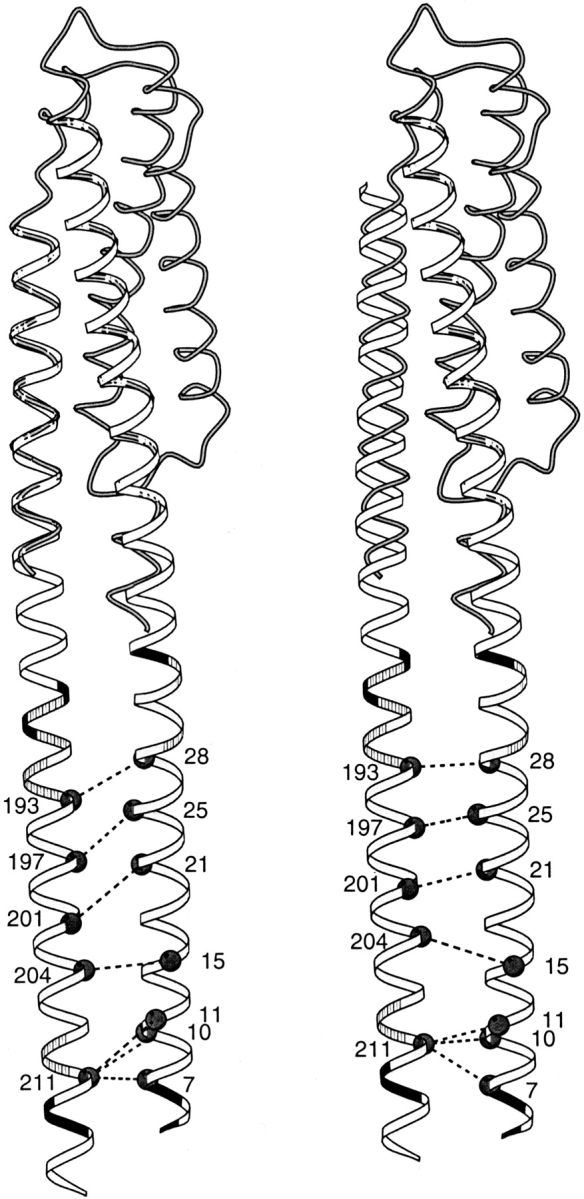
Positions of cysteine pairs with high propensity for cross-linking transmembrane helix 1 (TM1) and transmembrane helix 2 (TM2) of Tar. Initial (left) and revised (right) positions of TM2 relative to TM1 are diagrammed with cysteine pairs, showing high cross-linking propensities (Pakula and Simon 1992). Details and symbols as for Figure 3 ▶.
Ligand-induced conformational changes
Many studies of ligand-induced conformational changes in chemoreceptors have used oxidative cross-links between cysteines introduced into adjacent helical segments (for review, see Falke and Hazelbauer 2001). Several studies determined whether signaling was blocked by specific cross-links (Scott and Stoddard 1994; Chervitz and Falke 1995; Chervitz et al. 1995; Lee et al. 1995b). We investigated the degree to which these engineered disulfides constrained the two different ligand-induced conformational changes observed in crystal structures of the periplasmic domain: α4/TM2 axial sliding and subunit rotation. We constructed models of cross-linked receptors by replacing residues with cysteines in a disulfide linkage. We modeled conformational changes by gradually shifting receptor structure from one conformation to another in many incremental steps. To assess the constraint an introduced disulfide would impose over the course of a conformational change, we calculated at each step a value for the potential energy of the disulfide, based on deviations from ideal values of bond lengths, bond angles, and dihedral angles.
Modeling axial sliding of α4/TM2 was straightforward: The helix was translated along its long axis from its position in our refined model to a displacement of 10 Å in either direction. Modeling subunit rotation was more complex because there were two sets of coordinates for unoccupied/occupied pairs of periplasmic domains, and the two unoccupied structures differed by a rotation substantially greater than the rotational differences between either unoccupied/occupied pair. We quantified the differences by determining the root mean square displacements of the rotating subunit relative to its position in the structure of the unoccupied, cross-linked periplasmic domain of TarS(1LIH). Analysis of the comparable subunit from the structure of the unoccupied periplasmic domain of TarE revealed a root mean square displacement equal to approximately half the displacement between the two unoccupied structures of TarS, and so that structure was included in our plots. Rotational translations between unoccupied and occupied structures were in approximately the same direction and magnitude for both pairs and are shown as dark lines. Rotational translations between the three different kinds of periplasmic domains were not in a single direction and are shown as gray lines. These latter translations may not represent natural transitions but serve to illustrate conformational differences that can be achieved, at least in receptor fragments.
Constraint on conformational changes by inter-helical cross-links
Lee et al. (1995b), examined effects on Trg signaling of disulfide cross-links formed in vivo in intact chemotactically functional cells. Two TM1-TM2 cross-links eliminated tactic response, but four between α1/TM1 and α1`/TM1` had no discernible effect. We used our models to investigate the effect of the two conformational changes on the potential energy of these six disulfide cross-links. As expected, because the potential energy of a disulfide cross-link depends only on the position of the backbone atoms to which it is directly connected, the energy of TM1-TM2 intra-subunit cross-links did not change as a function of rotation between subunits, and the energy of the TM1-TM1` inter-subunit cross-links did not change as a function of TM2 sliding within a single subunit (data not shown). The potential energies of the two TM1-TM2 disulfides as a function of TM2 axial sliding are plotted in Figure 5A ▶. For both disulfides, potential energy is at a minimum near the null position of TM2, at −0.38 Å and −1.12 Å for the 42–202 and 42–203 disulfides, respectively, and rises sharply as TM2 is displaced toward the cytoplasm. For the 42–202 disulfide, there is a similar rise for displacement toward the periplasm, whereas the potential energy of the 42–203 disulfide remains close to the minimum for displacements toward the periplasm up to ∼4 Å. Thus, formation of either signal-blocking disulfide would cause a minimal shift in the helical register but would constrain sliding of TM2 toward the cytoplasm. This implies that these disulfides block signaling by providing a barrier to axial sliding of TM2 toward the cytoplasm, not by causing it to slide. The 42–203 disulfide would not perturb the null position of TM2 in any way. The relatively high potential energy of the 42–202 disulfide reflects the separation of the cysteines and the need for helical rotation to bring the residues within the normal length of a disulfide bond. However, the 42–202 cross-link forms very readily, implying that such rotation is frequent (Chervitz and Falke 1995) and not an unusual distortion.
Fig. 5.

Energies of inter-helical disulfides in chemoreceptor Trg as a function of ligand-induced conformational changes. Values for potential energy of disulfide bonds in models of oxidatively cross-linked Trg periplasmic plus cytoplasmic domains were calculated at incremental steps of two ligand-induced conformational changes. (A) Energies of signal-blocking transmembrane helix 1 (TM1)–transmembrane helix 2 (TM2) disulfides between the indicated positions over the course of a sliding displacement of TM2 along its long axis. The kink in the 42–202 curve reflects a switch in dihedral angle from one conformation to another. (B) Energies of signal-allowing TM1-TM1` disulfides between the indicated positions over the course of rotation of one subunit from one to another orientation in the crystal structures of the periplasmic domain. The abscissa shows the total root mean square distance traveled by the moving subunit. Positions of the crystal structures are indicated along the top. XL S.t. indicates cross-linked Salmonella structure; WT S.t., wild-type Salmonella structure; and asp, aspartate.
Figure 5B ▶ shows plots of the potential energy, using the same energy scale as in Figure 5A ▶, of the Trg α1/TM1–α1`/TM1` cross-links as a function of subunit rotation over the course of a movement of one subunit, from its starting position in the crystal structure of the unoccupied cross-linked TarS periplasmic domain, to its positions in the occupied cross-linked TarS structure, the unoccupied TarE structure, the unoccupied wild-type TarS structure, and the occupied wild-type TarS structure. It is striking that with the exception of the 32–32` disulfide, these inter-subunit movements had little or no effect on the potential energy of the inter-subunit disulfides. These cross-links would not provide significant constraint on rotation between an unoccupied and an occupied conformation.
In an in vitro study of Tar, 11 cysteine substitutions in α1/TM1 preserved the ability of the receptor to activate kinase, and cross-links across the subunit interface at seven positions allowed signaling (Chervitz et al. 1995). The potential energies of these 11 disulfides as a function of subunit rotation are plotted in Figure 6B ▶. The potential energies of the seven disulfides that allowed signaling (75, 72, 68, 65, 61, 36, and 29) were quite low, reflecting the close apposition of the two helices. There was little change in energy on subunit rotation from an unoccupied to an occupied state. Thus, these cross-links would not constrain subunit rotation over the range investigated. Disulfides across the subunit interface at positions 11, 8, 7, and 4 drastically reduced the signaling ability of the receptor (Chervitz et al. 1995). The potential energies of these disulfides were quite high, reflecting distortions that disrupted receptor function. This is consistent with the greater distances between TM1 and TM1` near the cytoplasm, the splaying noted above.
Fig. 6.

Energies of interhelical disulfides in chemoreceptor Tar as a function of ligand-induced conformational changes. (A) Transmembrane helix 1 (TM1)–transmembrane helix 2 (TM2) disulfides that lock the receptor in a kinase-activating (on) or kinase-inhibiting state (off). (B) TM1-TM1` disulfides that allow (allow) or block (block) signaling. Details and symbols as for Figure 5 ▶. The kink in the 36–183 curve reflects a switch in dihedral angle from one conformation to another.
Cross-links that lock receptors in a signaling state
Certain cross-links between α1/TM1–α4/TM2 in Tar lock the receptor into a kinase-activating (lock-on) or kinase-inhibiting (lock-off) state, corresponding to an unoccupied or ligand-bound receptor state, respectively (Chervitz and Falke 1995,Chervitz and Falke 1996). Figure 6A ▶ shows plots of potential energy versus α4/TM2 displacement for these four disulfides. The lock-on cross-links 39–183 and 25–197 would be at minimum-energy positions with TM2 very close to the null position, at −0.12 Å and −0.50 Å, respectively, whereas the minimum-energy positions for the lock-off disulfides 39–179 and 43–176 would be with TM2 shifted toward the cytoplasm by 6.1 Å and 3.8 Å, respectively. This is consistent with the helical sliding model of transmembrane signaling.
Distances between diagnostic cysteines
Oxidative cross-linking has been used diagnostically to assess ligand-induced shifts between helices (Hughson and Hazelbauer 1996; Beel and Hazelbauer 2001). In these experiments, rates of disulfide formation for cysteine pairs spanning the TM1-TM1` or TM1-TM2 interface of Trg were assayed in vivo in the presence and absence of saturating ligand. We used our model of Trg to determine distances between the α-carbons at the positions of the diagnostic cysteine pairs as a function of the incremental conformational changes of TM2 axial sliding or of subunit rotation (Fig. 7 ▶). TM2 sliding did not change α-carbon distances across the TM1-TM1` interface (Fig. 7C ▶), and subunit rotation from a ligand-free to a ligand-bound state resulted in only modest changes in α-carbon distance across the TM1-TM1` interface. At the TM1-TM2 interface, distances were not affected by subunit rotation but were strongly dependent on TM2 sliding. Experimentally, ligand had no substantial effect on rates of cross-linking for the four TM1-TM1` cysteine pairs, but significantly changed rates for all TM1-TM2 pairs, increasing two and reducing two others (Hughson and Hazelbauer 1996; Beel and Hazelbauer 2001). Specifically, the rates of cross-linking between cysteines at positions 42–202 or 42–203 decreased in the presence of ligand, and the rates for cysteine pairs 38–202 and 38–203 increased. The plots in Figure 7A ▶ indicate that an axial sliding of TM2 toward the cytoplasm would account for all of those experimentally observed changes.
Fig. 7.

Distances between the Cα atoms of diagnostic, inter-helical cysteine pairs in chemoreceptor Trg as a function of ligand-induced conformational changes. Distances between Cα atoms of cysteine pairs in the model of the Trg periplasmic plus cytoplasmic domains were calculated at incremental steps of two ligand-induced conformational changes. (A) Transmembrane helix 1 (TM1)–transmembrane helix 2 (TM2) disulfides between the indicated positions over the course of sliding displacements of TM2 along its long axis. Ligand occupancy increased the rates of formation for two disulfides (incr) and decreased the rates for the other two (decr). (B) TM1-TM2 disulfides between the indicated positions over the course of rotation of one subunit from one to another orientation in the crystal structures of the periplasmic domain. (C) TM1-TM1` disulfides between the indicated positions over the course of sliding displacements of TM2 along its long axis. (D) TM1-TM1` disulfides between the indicated positions over the course of rotation of one subunit from one to another orientation in the crystal structures of the periplasmic domain. Labels as in Figure 5B ▶.
Discussion
Models refined by experimental data
High-resolution structural information is not available for transmembrane domains of bacterial chemoreceptors, so we constructed molecular models of this four-helix domain. First, we used coordinates of an unrelated four-helix coiled coil as a template for the transmembrane domain, positioning this template by fusing it with the X-ray structure of the periplasmic domain of chemoreceptor TarS. There was significant experimental data about transmembrane structure for the closely related receptor TarE, as well as for the distantly related receptor Trg. Thus, we created a model of the periplasmic plus transmembrane domains of both Tar and Trg.
We tested the two first-stage models with data of propensities for cross-linking between cysteines introduced into transmembrane segments. We found that many aspects of the models corresponded with experimental observations, but there was one striking disparity, the register of TM2 relative to its partner TM1. However, the disparity could be corrected by a simple change in the first-stage models, a sliding of TM2 toward the periplasm several angstroms along its long axis. The implication of this correction is that the position of TM2 captured in crystals of periplasmic domain fragments is shifted toward the cytoplasm relative to the average conformation of the intact membrane-embedded receptor, the state probed in cross-linking studies. This is plausible because a large body of experimental data indicates that axial sliding of a mobile TM2 relative to a static TM1 occurs during ligand-induced signaling in chemoreceptors (Falke and Hazelbauer 2001); such sliding must have a low energy barrier because it can be induced by single residue substitutions near the ligand-binding site (Beel and Hazelbauer 2001), and the distance between bracketing charged residues allows a range of TM2-TM1 registers (Figs. 3, 4 ▶ ▶). With a low barrier to TM2 sliding, the helical register could be altered as a consequence of truncating the receptor to create the periplasmic domain or by conditions of crystallization, for instance, an ion occupying the ligand-binding site (Chi et al. 1997). Thus, we suggest that crystallization of the periplasmic domains of TarS and TarE captured receptor fragments shifted toward the signaling conformation. This implies there is a greater difference between the axial position of α4/TM2 in the ligand-free and ligand-bound conformations than that revealed by comparison of X-ray structures of aspartate-free and aspartate-bound fragments of the TarS periplasmic domain (Chervitz and Falke 1996), and thus, that the magnitude of ligand-induced axial sliding is greater than the value of ∼1.5 Å deduced from analysis of those crystal structures.
Effects of cross-links on signaling
An assumption in investigations of effects of disulfide cross-links on receptor signaling was that cross-links that blocked signaling would identify helical interfaces that were shifted by the conformational change, whereas cross-links that allowed signaling would identify interfaces that were essentially static. Thus, different effects on specific interfaces could distinguish between alternative notions of what moved in conformational signaling. However, there could be complications if disulfide cross-links across a static interface deformed receptor structure sufficiently to block signaling or if the conformational change of signaling was sufficiently modest to occur within the limits imposed by specific disulfide bonds. We used our chemoreceptor models to explore these issues, creating disulfides that had been used experimentally and calculating an energy parameter for those bonds to compare the degree to which the bonds would (1) strain and thus deform the structure of our refined models and (2) constrain each of the two ligand-induced conformational changes observed in crystal structures of the periplasmic domain. We found that α1/TM1–α4/TM2 cross-links that blocked signaling would cause little deformation of our modeled structures. In contrast, α1/TM1–α1`/TM1` cross-links that disrupted receptor function would also strain and deform the receptor, whereas the several α1/TM1–α1`/TM1` cross-links that allowed signaling would cause little strain and thus little disruption of the receptor. These observations support and strengthen the conclusions of the original studies (Chervitz et al. 1995; Lee et al. 1995b) that conformational signaling involves a shift between the transmembrane helices within a single subunit, not a shift between the two subunits. However, it was interesting to discover that the several inter-subunit α1/TM1–α1`/TM1` cross-links that allowed signaling not only caused little strain in the structure of the unoccupied receptor but also would not constrain subunit rotations that relate crystal structures of ligand-occupied to unoccupied forms of the periplasmic domain of TarS. Thus, signaling by receptors with α1/TM1–α1`/TM1` cross-links does not in itself address the functional significance of subunit rotation.
Cross-links that lock receptor signaling
Using the crystal structure of the TarS periplasmic domain (Milburn et al. 1991) to determine the positions of the cysteine pairs of ligand-insensitive lock-on and lock-off disulfides, it appeared that formation of kinase-activating, lock-on cross-links would move α4/TM2 ∼1 Å toward the periplasm, and formation of kinase-inhibiting, lock-off cross-links would move the helix ∼1 Å toward the cytoplasm (Chervitz et al. 1995; Lee et al. 1995b). In our refined model of Tar, lock-on cross-links would shift the helix only very slightly toward the periplasm, and the lock-off cross-links would result in a 4- to 6-Å axial sliding toward the cytoplasm (Fig. 6A ▶). The potential energy profiles of lock-on disulfides, which prohibit ligand-induced inhibition of kinase, indicate that those cross-links would cause little restraint on axial sliding of α4/TM2 for as much as 2 Å toward the cytoplasm, but would provide increasing restraint for larger movements. This implies that the natural ligand-induced shift should be >2 Å.
Diagnostic cross-links
Effects of ligand occupancy on rates of cross-linking between diagnostic cysteine pairs (Hughson and Hazelbauer 1996; Beel and Hazelbauer 2001) had been interpreted using a simple model of the transmembrane domain in which helices were placed in register by eye to account for patterns of disulfide cross-linking (Lee et al. 1994, 1995a; Hughson et al. 1997). Reanalysis with our refined model of Trg (Fig. 7A ▶) confirmed the original conclusion that ligand-induced signaling is axial sliding of α4 toward the cytoplasm and showed that a sliding of 3 to 4 Å would account for increased rates of cross-linking for the cysteine pairs 38–202 and 38–203 and decreased rates for the pairs 42–202 and 42–203.
None of the diagnostic cysteine pairs showed significant disulfide formation in the absence of added oxidation catalyst. This is consistent with our modeled Cα-Cα distances (Fig. 7A ▶), which are all >3.8- to 6.8-Å Cα-Cα distances for disulfide bonds in proteins of known structure (Srinivasan et al. 1990). Formation of disulfides in the presence of catalyst indicates that there must be fluctuations in the separation of helices to bring the sulfhydryls within reactive distance. The modeled Cα-Cα distances for the 38–203 and 42–203 cysteine pairs approach those for disulfides, so that only modest movements would be required. The modeled Cα-Cα distances for the 38–202 and 42–202 cysteine pairs are significantly greater than those for disulfides, implying that substantial helical movement would be necessary for disulfide formation. The movement is most likely a twisting of TM2, which would bring residue 202 closer to residue 42. Similar large twisting movements have in observed in studies of α4–α4` cross-linking (Chervitz et al. 1995).
Ligand-induced conformational changes
The two different forms of periplasmic fragments of TarS crystallized with and without aspartate revealed two different ligand-induced conformational changes: axial sliding of helix α4 within a subunit and monomer rotation between subunits. Results from the current work and from many previous studies (for review, see Falke and Hazelbauer 2001) identify helical sliding as a crucial conformational change in chemoreceptor transmembrane signaling. However, our modeling of the effects of inter-subunit cross-links that allow receptor signaling indicated that subunit rotation could occur in receptors containing many of these cross-links. Thus, this rotation could play a role in receptor function.
The difference in the subunit interaction angle is much greater between the three available unoccupied structures of the Tar periplasmic domain than between either of the unoccupied/occupied pairs of structures. This indicates that the angle of subunit packing is influenced by the crystal environment and that large fluctuations of the inter-subunit angle may occur spontaneously in the truncated periplasmic domain. Our results indicate that even a large rotation could be accommodated by a disulfide bond at some positions across the subunit interface. However, for both apo-holo structure pairs, the direction and magnitude of the small change in inter-subunit rotation angle on ligand binding is similar, consistent with this rotation being part of receptor function. Rotation cannot be sufficient for transmembrane signaling, because α1/TM1–α4/TM2 cross-links that block signaling do not block subunit rotation. However subunit rotation could participate in the strong negative cooperativity showed by chemoreceptors (Biemann and Koshland 1994; Lin et al. 1994; Yeh et al. 1996). Ligand-binding sites span the subunit interface, and occupancy at one of the two equivalent sites drastically reduces affinity for ligand at the second, nonoverlapping site. This must involve an inter-subunit allosteric change that could easily be mediated by subunit rotation. Thus, it seems plausible that in the intact receptor both ligand-induced conformational changes observed in crystal structures of receptor fragments would occur and have functional roles.
Materials and methods
Sequence alignment
TarS and Trg are closely related in sequence for the carboxyl 60% that constitutes their cytoplasmic domains, but they show few residue matches in the periplasmic and transmembrane domains. To optimize alignment of those minimally related sequences, we first identified a family of related chemoreceptor sequences (Gracy et al. 1993). The first iteration of a PSI-BLAST search (Altschul et al. 1997) with the periplasmic and transmembrane domains of Trg (residues 14–229), using the nonredundant National Center for Biotechnology Information (NCBI) database and the BLOSUM-62 matrix with the default gap penalty of 11 (Henikoff and Henikoff 1992), found Trg sequences with strict significance (e < 0.001) and the S. typhimurium aspartate receptor TarS (e = 0.002) and citrate receptor Tcp (e = 0.074) with lower significance. Including these sequences in the next iteration produced additional significant alignments: the aspartate and serine receptors from E. coli, TarE and Tsr, and Enterobacter aerogenes, Tas and Tse. Including all of these sequences in a third iteration found the E. coli dipeptide receptor Tap. In subsequent iterations no new sequences occurred, indicating that the periplasmic and transmembrane domains of these eight receptors are indeed homologous with similar structures.
In a second step, we used the crystal structures of the periplasmic domains of TarS and TarE. The periplasmic plus transmembrane sequences of those two proteins were aligned using the program AMPS (Alignment of Multiple Protein Sequences; Barton and Sternberg 1987b), and the alignment was edited to indicate the location of periplasmic loops. The remaining six sequences in the receptor family were aligned using a 100-fold greater gap penalty in helical regions. This had the effect of confining all gaps to loop regions, where they are most likely to occur (Barton and Sternberg 1987a). The resulting alignment was consistent with additional structural features. For the transmembrane helices, there was alignment of the hydrophobic cores, charged residues at the cytoplasmic boundaries, and a band of aromatic residue at the periplasmic ends. Periplasmic residues identified as important for interaction with respective binding protein ligands (Gardina et al. 1992; Yaghmai and Hazelbauer 1992) were at identical or adjacent positions.
Model construction
The next step in the modeling process was to decide which crystal structure of TarS provided the best template for the periplasmic domain of the receptor. The two sets of high-resolution structures in the Protein Data Bank (PDB) are 1LIH/2LIG, the apo and holo forms of a mutant receptor with a cysteine at residue 36 that forms a disulfide bond across the subunit interface of the dimer, and 1VLS/1VLT, the wild-type apo and holo structures. The principle difference between the two apo structures 1VLS and 1LIH is a ∼12.6° rotation of the subunits relative to one another (Yeh et al. 1993). This rotation affects the inter-subunit interface, so that in the wild-type structures, the membrane-proximal ends of the helices α1 and α1` diverge, with residues 36 and 36` facing away from one another, at a Cα-Cα distance of 20.6 Å. It is unclear whether this is because of differences in crystal packing or because of the absence of the transmembrane domains. However, studies have shown that a cross-link between 36–36` forms very readily and does not affect ligand binding or receptor signaling (Falke and Koshland 1987; Scott and Stoddard 1994; Chervitz et al. 1995), which implies that these residues should be close to one another in the intact receptor. For this reason, we chose the cross-linked crystal structure 1LIH as a template for the periplasmic domain.
We used a four-helix coiled coil structure of a mutant leucine zipper (1GCL in the PDB; Harbury et al. 1993) to extend helices α1, α4, α1`, and α4` of the crystal structure of the cross-linked periplasmic domain of TarS and thus create a backbone structure for a periplasmic plus transmembrane domain of a chemoreceptor. We extended the 30-residue helices in 1GCL by stacking them along their axes, preserving the pattern of hydrophobic residues and thus the coiling structure. We aligned each extended helical backbone to the membrane-distal end of a corresponding helix in the periplasmic domain. Because the membrane-proximal ends of the periplasmic helices are somewhat disordered in the crystal structure, we merged the leucine zipper helices with the periplasmic helices approximately midway along their length, at residues 51 and 171. Both receptors have a small N-terminal cytoplasmic sequence (residues 1–4 in Tar and 1–14 in Trg) unnecessary for transmembrane signaling or chemotaxis (Chen and Koshland 1995), so we began the models at residue 4 for Tar and residue 14 for Trg. To neutralize the charged helical ends, we capped them with acetyl (Ace) and methyl amide (Nme) terminal residues.
We placed Tar and Trg side-chains onto separate backbone templates with the program SCWRL (Side-Chains with a Rotamer Library), which uses a backbone-dependent rotamer library (Dunbrack and Martin 1993) to position side-chains in their most likely rotamer, based on the conformation of the backbone at that residue, followed by a combinatorial search to remove steric conflicts (Bower et al. 1997). For the Tar structure, we retained the crystal structure placement of the residues 51 through 171. For Trg, we deleted the periplasmic loops and replaced all residues. Because all gaps in the sequence alignment of Trg and TarS occurred in loops, both proteins could be fit on the same helical backbone structure. We built new loops for Trg by adding the loop residues in an extended conformation to the tops of the helices, with half of the loop on each helix, followed by limited molecular dynamics using AMBER (Case et al. 1997) at low temperature with a restraint between loop ends to bring them together and close the loop. Large steric clashes and asymmetry between corresponding side-chains in the two subunits were corrected by manual rotation of side-chain dihedral angles, and we performed limited energy minimization (200 steps) in vacuo using AMBER (Case et al. 1997) to relax the structures slightly and remove any remaining small steric overlaps. We checked for reasonable side-chain and backbone torsion angles using the program WhatIf (Vriend 1990).
Models of cysteine-substituted, disulfide cross-linked receptors were created by fixing the positions of the backbone atoms, substituting cysteine side-chains at the two designated positions, rotating the cysteine side-chains into the most favorable conformer, and linking the sulfur atoms with a new bond.
Modeling conformational changes
We simulated sliding of α4/TM2 by displacing its long-axis position in a series of 0.125-Å incremental steps. We simulated the inter-subunit rotations using a method related to targeted energy minimization (Engels et al. 1992), in which the distance between the starting structure and the target structure was reduced in a series of small steps by translating each atom in the direction of the target by a fraction of the total distance at each step. Models for the target structures were built using the same method outlined above, but with the cross-linked holo TarS crystal structure (2LIG), the wild-type apo TarS crystal structure (1VLS), the wild-type holo TarS crystal structure (1VLT), and the apo TarE crystal structure (2ASR) as templates. The target structures were aligned to the starting structure through the backbone atoms of the central helical residues in one subunit (47–70, 90–107, 120–139, and 155–172). For both conformational changes, after each step the backbone atoms of the helix were fixed in place, and the disulfide linkage was energy minimized to remove any unnecessary conformational strain.
We calculated the potential energy of the introduced disulfide using the Discover module of the program Insight II from Biosym/Molecular Simulations Inc. and a simplified version of the all-atom AMBER force field (Weiner et al. 1984, 1986). This force field describes the potential energy of a protein as the sum of quadratic terms for bond length and bond angle deviations from ideality, a trigonometric term for dihedral angle rotation, and a Lennard-Jones potential for van der Waals interactions. We did not include nonbonded terms for hydrogen bonding or for electrostatic interactions. The potential energy of the disulfide was calculated at each step as the sum of the bond, angle, and dihedral terms. Because the backbone atoms were fixed during the minimization, the final potential energies at each step are somewhat higher than they would be if we allowed small distortions of the helix to accommodate the disulfide. However, this method allows the accurate comparison of energies between different disulfide pairs across different receptors.
Acknowledgments
This work was supported in part by grants from the National Institutes of Health (GM29963 to G.L.H. and NS33290 to T.P.L.) and ZymoGenetics Corp.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.4640102.
References
- Altschul, S.F., Madden, T.L., Schäffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton, G.J. and Sternberg, M.J.E. 1987a. Evaluation and improvements in the automatic alignment of protein sequences. Protein Eng. 1 89–94. [DOI] [PubMed] [Google Scholar]
- ———. 1987b. A strategy for the rapid multiple alignment of protein sequences. J. Mol. Biol. 198 327–337. [DOI] [PubMed] [Google Scholar]
- Beel, B.D. and Hazelbauer, G.L. 2001. Signalling substitutions in the periplasmic domain of chemoreceptor Trg induce or reduce helical sliding in the transmembrane domain. Mol. Microbiol. 40 824–834. [DOI] [PubMed] [Google Scholar]
- Biemann, H.P. and Koshland, Jr., D.E. 1994. Aspartate receptors of Escherichia coli and Salmonella typhimurium bind ligand with negative and half-of-the-sites cooperativity. Biochemistry 33 629–634. [DOI] [PubMed] [Google Scholar]
- Björkman, A.M., Dunten, P., Sandgren, M.O.J., Dwarakanath, V.N., and Mowbray, S.L. 2001. Mutations that affect ligand binding to the Escherichia coli aspartate receptor. J. Biol. Chem. 276 2808–2815. [DOI] [PubMed] [Google Scholar]
- Bower, M.J., Cohen, F.E., and Dunbrack, Jr., R.L. 1997. Prediction of protein side-chain rotamers from a backbone-dependent rotamer library: A new homology modeling tool. J. Mol. Biol. 267 1268–1282. [DOI] [PubMed] [Google Scholar]
- Bowie, J.U., Pakula, A.A., and Simon, M.I. 1995. The three-dimensional structure of the aspartate receptor from Escherichia coli. Acta Crystallog. D 51 145–154. [DOI] [PubMed] [Google Scholar]
- Careaga, C.L. and Falke, J.J. 1992. Thermal motions of surface a-helices in the d-galactose chemosensory receptor. J. Mol. Biol. 226 1219–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case, D.A., Pearlman, D.A., Caldwell, J.W., Cheatham III, T.E., Ross, W.S., Simmerling, C.L., Darden, T.A., Merz, K.M., Stanton, R.V., Cheng, A.L., Vincent, J.J., Crowley, M., Ferguson, D.M., Radmer, R.J., Seibel, G.L., Singh, U.C., Weiner, P.K., and Kollman, P.A. 1997. AMBER. University of California, San Francisco, CA.
- Chen, X. and Koshland, Jr., D.E. 1995. The N-terminal cytoplasmic tail of the aspartate receptor is not essential in signal transduction of bacterial chemotaxis. J. Biol. Chem. 270 24038–24042. [DOI] [PubMed] [Google Scholar]
- Chervitz, S.A. and Falke, J.J. 1995. Lock on/off disulfides identify the transmembrane signaling helix of the aspartate receptor. J. Biol. Chem. 270 24043–24053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 1996. Molecular mechanism of transmembrane signaling by the aspartate receptor: A model. Proc. Natl. Acad. Sci. 93 2545–2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chervitz, S.A., Lin, C.M., and Falke, J.J. 1995. Transmembrane signaling by the aspartate receptor: Engineered disulfides reveal static regions of the subunit interface. Biochemistry 34 9722–9733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi, Y.I., Yokota, H., and Kim, S.H. 1997. Apo structure of the ligand-binding domain of aspartate receptor from Escherichia coli and its comparison with ligand-bound or pseudoligand-bound structures. FEBS Lett. 414 327–332. [DOI] [PubMed] [Google Scholar]
- Dunbrack, Jr., R.L. and Martin, K. 1993. Backbone-dependent rotamer library for proteins. J. Mol. Biol. 230 543–574. [DOI] [PubMed] [Google Scholar]
- Engels, M., Jacoby, E., Krüger, P., Schlitter, J., and Wollmer, A. 1992. The T―R structural transition of insulin: pathways suggested by targeted energy minimization. Protein Eng. 5 669–677. [DOI] [PubMed] [Google Scholar]
- Falke, J.J. and Koshland, Jr., D.E. 1987. Global flexibility in a sensory receptor: A site-directed cross-linking approach. Science 237 1596–1600. [DOI] [PubMed] [Google Scholar]
- Falke, J.J. and Hazelbauer, G.L. 2001. Transmembrane signaling in bacterial chemoreceptors. Trends Biochem. Sci. 26 257–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falke, J.J., Bass, R.B., Butler, S.L., Chervitz, S.A., and Danielson, M.A. 1997. The two-component signaling pathway of bacterial chemotaxis: A molecular view of signal transduction by receptors, kinases, and adaptation enzymes. Annu. Rev. Cell Dev. Biol. 13 457–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardina, P., Conway, C., Kossman, M., and Manson, M. 1992. Aspartate and maltose-binding protein interact with adjacent sites in the Tar chemotactic signal transducer of Escherichia coli. J. Bacteriol. 174 1528–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gracy, J., Chiche, L., and Sallantin, J. 1993. Improved alignment of weakly homologous protein sequences using structural information. Protein Eng. 6 821–829. [DOI] [PubMed] [Google Scholar]
- Harbury, P.B., Zhang, T., Kim, P.S., and Alber, T. 1993. A switch between two-, three-, and four-stranded coiled coils in GCN4 leucine zipper mutants. Science 262 1401–1406. [DOI] [PubMed] [Google Scholar]
- Henikoff, S. and Henikoff, J.G. 1992. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. 89 10915–10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughson, A.G. and Hazelbauer, G.L. 1996. Detecting the conformational change of transmembrane signaling in a bacterial chemoreceptor by measuring effects of disulfide cross-linking in vivo. Proc. Natl. Acad. Sci. 93 11546–11551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughson, A.G., Lee, G.F., and Hazelbauer, G.L. 1997. Analysis of protein structure in intact cells: Crosslinking in vivo between introduced cysteines in the transmembrane domain of a bacterial chemoreceptor. Protein Sci. 6 315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, K.K., Yokota, H., and Kim, S.H. 1999. Four-helical-bundle structure of the cytoplasmic domain of a serine chemotaxis receptor. Nature 400 787–792. [DOI] [PubMed] [Google Scholar]
- Kraulis, P.J. 1991. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 24 946–950. [Google Scholar]
- Lee, G.F. and Hazelbauer, G.L. 1995. Quantitative approaches to utilizing mutational analysis and disulfide crosslinking for modeling a transmembrane domain. Protein Sci. 4 1100–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, G.F., Burrows, G.G., Lebert, M.R., Dutton, D.P., and Hazelbauer, G.L. 1994. Deducing the organization of a transmembrane domain by disulfide cross-linking. J. Biol. Chem. 269 29920–29927. [PubMed] [Google Scholar]
- Lee, G.F., Dutton, D.P., and Hazelbauer, G.L. 1995a. Identification of functionally important helical faces in transmembrane segments by scanning mutagenesis. Proc. Natl. Acad. Sci. 92 5416–5420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, G.F., Lebert, M.R., Lilly, A.A., and Hazelbauer, G.L. 1995b. Transmembrane signaling characterized in bacterial chemoreceptors by using sulfhydryl cross-linking in vivo. Proc. Natl. Acad. Sci. 92 3391–3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, L.N., Li, J., Brandts, J.F., and Weis, R.M. 1994. The serine receptor of bacterial chemotaxis exhibits half-site saturation for serine binding. Biochemistry 33 6564–6570. [DOI] [PubMed] [Google Scholar]
- Milburn, M.V., Privé, G.G., Milligan, D.L., Scott, W.G., Yeh, J., Jancarik, J., Koshland, Jr., D.E., and Kim, S.H. 1991. Three-dimensional structures of the ligand-binding domain of the bacterial aspartate receptor with and without a ligand. Science 254 1342–1347. [DOI] [PubMed] [Google Scholar]
- Milligan, D.L. and Koshland, Jr., D.E. 1991. Intrasubunit signal transduction by the aspartate chemoreceptor. Science 254 1651–1654. [DOI] [PubMed] [Google Scholar]
- Pakula, A.A. and Simon, M.I. 1992. Determination of transmembrane protein structure by disulfide cross-linking: The Escherichia coli Tar receptor. Proc. Natl. Acad. Sci. 89 4144–4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reithmeier, R.A.F. 1995. Characterization and modeling of membrane proteins using sequence analysis. Curr. Opin. Struct. Biol. 5 491–500. [DOI] [PubMed] [Google Scholar]
- Scott, W.G. and Stoddard, B.L. 1994. Transmembrane signalling and the aspartate receptor. Structure 2 877–887. [DOI] [PubMed] [Google Scholar]
- Scott, W.G., Milligan, D.L., Milburn, M.V., Privé, G.G., Yeh, J., Koshland, Jr., D.E., and Kim, S.H. 1993. Refined structures of the ligand-binding domain of the aspartate receptor from Salmonella typhimurium. J. Mol. Biol. 232 555–573. [DOI] [PubMed] [Google Scholar]
- Srinivasan, N., Sowdhamini, R., Ramakrishnan, C., and Balaram, P. 1990. Conformations of disulfide bridges in proteins. Int. J. Peptide Protein Res. 36 147–155. [DOI] [PubMed] [Google Scholar]
- Vriend, G. 1990. WHAT IF: A molecular modeling and drug design program. J. Mol. Graphics 8 52–56. [DOI] [PubMed] [Google Scholar]
- Wallin, E., Tsukihara, T., Yoshikawa, S., von Heijne, G., and Elofsson, A. 1997. Architecture of helix bundle membrane proteins: An analysis of cytochrome c oxidase from bovine mitochondria. Protein Sci. 6 808–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner, S.J., Kollman, P.A., Case, D.A., Singh, U.C., Ghio, C., Alagona, G., Profeta, Jr., S., and Weiner, P. 1984. A new force field for molecular mechanical simulation of nucleic acids and proteins. J. Am. Chem. Soc. 106 765–784. [Google Scholar]
- Weiner, S.J., Kollman, P.A., Nguyen, D.T., and Case, D.A. 1986. An all atom force field for simulations of proteins and nucleic acids. J. Comput. Chem. 7 230–252. [DOI] [PubMed] [Google Scholar]
- Yaghmai, R. and Hazelbauer, G.L. 1992. Ligand occupancy mimicked by single residue substitutions in a receptor: Transmembrane signaling induced by mutation. Proc. Natl. Acad. Sci. 89 7890–7894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh, J.I., Biemann, H.P., Pandit, J., Koshland, Jr., D.E., and Kim, S.H. 1993. The three-dimensional structure of the ligand-binding domain of a wild-type bacterial chemotaxis receptor: Structural comparison to the cross-linked mutant forms and conformational changes upon ligand binding. J. Biol. Chem. 268 9787–9792. [PubMed] [Google Scholar]
- Yeh, J.I., Biemann, H.-P., Privé, G.G., Pandit, J., Koshland, Jr., D.E., and Kim, S.H. 1996. High-resolution structures of the ligand binding domain of the wild-type bacterial aspartate receptor. J. Mol. Biol. 262 186–201. [DOI] [PubMed] [Google Scholar]