

Abstract
We analyzed the energetic importance of residues surrounding the hot spot (the P1 position) of bovine pancreatic trypsin inhibitor (BPTI) in interaction with two proteinases, trypsin and chymotrypsin, by a procedure called molecular shaving. One to eight residues of the structural epitope, composed of two extended and exposed loops, were mutated to alanine(s). Although truncation of the side chains of residues surrounding the P1 position to methyl groups caused a decrease in ΔGden values up to 6.4 kcal mole−1, it did not influence the overall conformation of the inhibitor. We found that the replacement of up to six residues with alanines was fully additive at the level of protein stability. To analyze the influence of the structural epitope on the association energy, we determined association constants for BPTI variants and both enzymes and applied the additivity analysis. Shaving of two binding loops led to a progressive drop in the association energy, more pronounced for trypsin (decrease up to 9.6 kcal mole−1) than chymotrypsin (decrease up to 3.5 kcal mole−1). In the case of extensively mutated variants interacting with chymotrypsin, the association energies agreed very well with the values calculated from single mutational effects. However, when P1-neighboring residues were shaved to alanine(s), their contribution to the association energy was not fully removed because of the presence of methyl groups and main chain–main chain intermolecular hydrogen bonds. Moreover, the hot spot had a different contribution to the complex stability in the fully shaved BPTI variant compared with the wild type, which was caused by perturbations of the P1–S1 electrostatic interaction.
Keywords: Additivity, bovine pancreatic trypsin inhibitor, protein-protein recognition, serine proteinase
Specific protein–protein recognition plays a crucial role in a great number of vital processes. Because structural information about different complexes is growing rapidly, deducing the rules that determine specificity and strength of protein–protein associations is a particularly attractive problem at this time (Jones and Thornton 1996; Otlewski and Apostoluk 1997; Lo Conte et al. 1999).
Proteins form stable complexes via numerous noncovalent interactions. Protein–protein interfaces are quite large (usually 600–1200 Å2) and typically involve 12–25 amino acid residues of each interacting protein. In several model studies of protein–protein recognition, it was shown by double or multiple additivity cycle analysis that the contribution of individual residues is often independent of other residues forming a structural epitope (Wells 1990). The additivity assumption was tested extensively for the Kazal family of protein inhibitors of serine proteinases using 92 natural and 191 recombinant variants with all coded amino acids introduced at contact positions (Lu et al. 2001). A conclusion of that study was that 90% of natural sequences that contain up to six changes at contact positions show full or at least partial additivity. Clear exceptions from the additivity rule are residues on the structural epitope that are in direct contact (Wells 1990) or affect each other through a long–range electrostatic effect (Schreiber and Fersht 1995).
Alanine scanning mutagenesis studies (Wells 1991) on many model protein–protein systems showed that the functional roles of residues at a structural epitope are not equal. Mutation to alanine of less than a quarter of the contacting residues causes a significant decrease in the association energy (above 1 kcal mole−1), whereas the remainder of the interacting area plays a passive role (Bogan and Thorn 1998). The location of this hot spot is typically central and occluded from the bulk solvent by passive residues.
The two above-mentioned phenomena, additivity of residual free energies and presence of the hot spot residue(s), suggest that it should be possible to truncate side chains of passive residues without affecting global association energy. Indeed, Jin and Wells (1994), in a unique study on human growth hormone (hGH)–monoclonal antibody recognition, showed that introduction of up to 16 alanines in the passive zone of hGH (a procedure called molecular shaving) caused only marginal reduction of the affinity. The hot spot of hGH is relatively large, composed of five residues and clearly provides contact area, sufficient to outweigh the loss of surrounding side chains caused by alanine shaving. The question arises whether a more localized hot spot, for example, composed of one or two residues, could provide enough energy to stabilize the complex when the surrounding side chains are shaved.
In this study, we analyzed a single residue hot spot—the P1 position (according to Schechter and Berger 1967) of bovine pancreatic trypsin inhibitor (BPTI). We chose an interaction of BPTI with two bovine proteinases: trypsin and chymotrypsin. High-resolution crystal structures are available for trypsin (Huber et al. 1974; Helland et al. 1999) and chymotrypsin (Scheidig et al. 1997) complexes with this inhibitor. A variant of BPTI, containing Lys15Arg and Met52Leu mutations, was used as the template for alanine shaving and further as the pseudo wild-type reference. The former mutation was introduced to increase the association constant for some of the most extensively mutated variants, as for both enzymes Arg is preferred about threefold over the naturally occurring Lys (Krowarsch et al. 1999). The latter mutation was made to allow CNBr cleavage of a fusion protein during the purification procedure.
Previously, we showed that truncation of the P1 Arg 15, to Gly decreases the Ka for trypsin 3 × 109-fold. That is, it removes 68% of the total free energy of association. The effect of the Arg15Gly substitution on interaction with chymotrypsin is much smaller, a 3.16 × 103-fold decrease, equivalent to a 41% loss of the association energy. In addition, kinetic and thermodynamic data for binding of 15 single alanine mutants of BPTI comprising all residues that are in contact with both enzymes are available (Castro and Anderson 1996). Interaction of the pseudo wild-type BPTI with trypsin (Ka = 4.5 × 1013 M−1) is over five orders of magnitude stronger than that with chymotrypsin (Ka = 2.5 × 108 M−1) (Krowarsch et al. 1999). The strongest contribution to association energy for the BPTI–trypsin complex seems to come from the electrostatic interaction between the P1 Lys 15, and Asp 189 in the S1 pocket of the enzyme. From the comparison of crystal structures of trypsin complexes with Lys 15 and Met 15 BPTI variants and their binding energies, we could estimate an over 4.4 × 105-fold increase in the association constant due to the presence of the Lys 15 side chain positive charge (Krowarsch et al. 1999). Thus, because of the presence of this extremely favorable electrostatic complementarity, the P1 position serves as the true hot spot of trypsin–BPTI recognition. There is no equivalent electrostatic interaction in the chymotrypsin complex and the association energy is much weaker. In this study, we analyzed the energetics of complex formation with trypsin and chymotrypsin using 10 mutants of BPTI. In these mutants, one to eight proteinase-contacting residues were converted to alanine(s) with the aim of revealing the energetic role of the P1 residue in alanine shaved mutants.
Results
Alanine mutations
The analysis of the crystal structures of bovine trypsin–BPTI (Huber et al. 1974; Helland et al. 1999) and bovine chymotrypsin–BPTI (Capasso et al. 1997; Scheidig et al. 1997) complexes shows that there are 13 residues of the inhibitor forming <4 Å contacts with the enzyme. Those residues form a structural epitope in the form of two extended and exposed loops, the primary (Thr 11–Ile 19) and the secondary one (Gly 36–Arg 39; Fig. 1A ▶). In addition, Val 34 is the only residue in both complexes that makes contacts in the range of 4.0–5.0 Å. Individual residue contacts below 4.0 Å together with a residue change in the solvent accessible surface area (ASA) accompanying complex formation are summarized in Table 1. For both complexes, contact numbers and distribution of van der Waals contacts among inhibitor residues are similar, which is a reflection of similar folds and sequences of both enzymes.
Fig. 1.
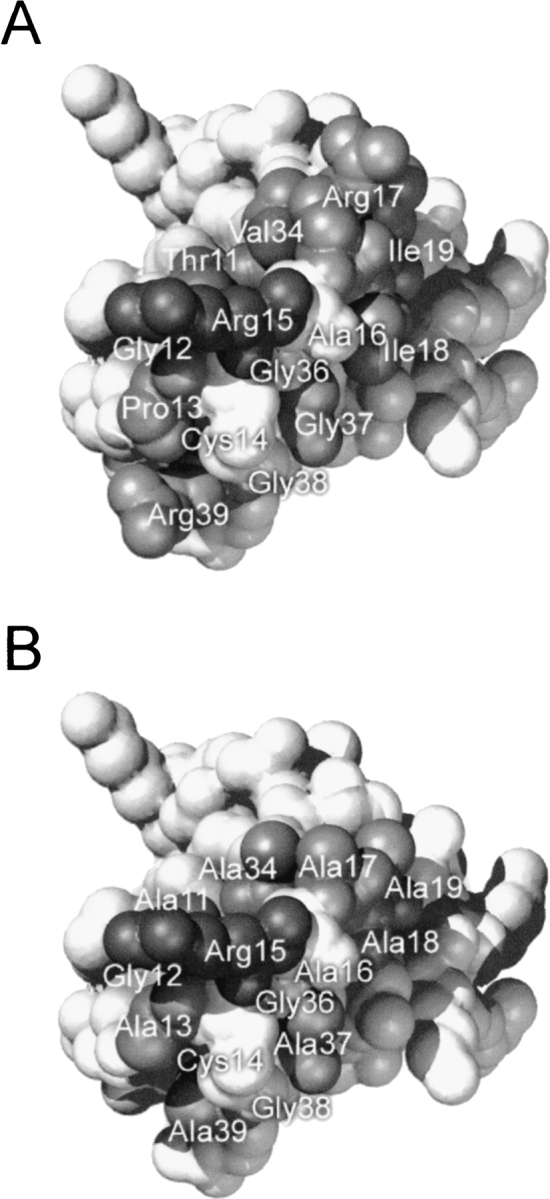
Space-filling model of the pseudo wild-type BPTI (A) and A11A13R15A17A18A19A34A37A39 mutant (B) surface in contact with trypsin. Residues replaced in this work are highlighted and shaded and other contact residues (Gly 12, Cys 14, Ala 16, Gly 36, and Cys 38) are highlighted but not shaded.
Table 1.
Number of noncovalent interactions formed by 13 residues comprising the binding epitope of BPTI, their ASA in free inhibitor, and ASA change accompanying complex formation with trypsin or chymotrypsin
| Trypsin | Chymotrypsin | |||||||
| vdWa | H..Hb | ASAc (Å2) | ASA changed | vdWa | H..Hb | ASAc (Å2) | ASA changed | |
| Thr 11 | 1 | 0 | 64 | 25% | 1 | 0 | 64 | 56% |
| Gly 12 | 1 | 0 | 20 | 21% | 2 | 0 | 18 | 88% |
| Pro 13 | 9 | 1 | 72 | 76% | 14 | 1 | 78 | 75% |
| Cys 14 | 16 | 1 | 55 | 100% | 9 | 0 | 48 | 100% |
| Arg 15 | 62 | 7 | 239 | 100% | 60 | 4 | 224 | 98% |
| Ala 16 | 15 | 0 | 46 | 100% | 17 | 0 | 55 | 100% |
| Arg 17 | 21 | 3 | 182 | 78% | 28 | 0 | 201 | 88% |
| Ile 18 | 2 | 0 | 65 | 89% | 1 | 0 | 67 | 52% |
| Ile 19 | 8 | 1 | 105 | 36% | 0 | 0 | 118 | 33% |
| Gly 36 | 2 | 0 | 1 | 100% | 2 | 0 | 1 | 100% |
| Gly 37 | 2 | 0 | 37 | 60% | 7 | 0 | 34 | 68% |
| Cys 38 | 3 | 0 | 32 | 88% | 2 | 0 | 36 | 91% |
| Arg 39 | 5 | 2 | 179 | 48% | 2 | 1 | 168 | 41% |
| Total | 147 | 15 | 1097 | 790 Å2 | 145 | 6 | 1112 | 808 Å2 |
All values were calculated using NACCESS (Hubbard and Thornton 1993) or HBPLUS program (McDonald and Thornton 1994) and structural data for respective trypsin–BPTI (Helland et al. 1999) and chymotrypsin–BPTI complexes (Scheidig et al. 1997); in case of Arg 15, values were calculated from data for APPI complexed with trypsin or chymotrypsin (Scheidig et al., 1997). Both ASA value sets for free inhibitor slightly differ, since they were calculated from inhibitor portion obtained from respective complexes (Helland et al. 1999; Scheidig et al. 1997).
a Number of intermolecular van der Waals contacts below 4 Å.
b Number of intermolecular hydrogen bonds defined according to standard parameters.
c ASA in free inhibitor calculated using 1.4 Å water radius.
d ASA change accompanying complex formation.
We prepared 10 mutants of BPTI in which alanines were progressively introduced into the primary and/or secondary binding loop (Table 2) except for two disulfide-bonded cysteine and two glycine positions. We did not substitute Gly 12 as its torsion angles are not compatible with amino acids containing β-carbons, and we showed previously that substitution of Gly 12 always caused a strong decrease in Ka and stability (Grzesiak et al. 2000b). Also, we did not introduce the Gly36Ala mutation because it is known to produce a very large, 4000-fold, decrease in Ka with trypsin (Castro and Anderson 1996). Hence, Gly36Ala, together with several other mutations, would lead to variants with unmeasurably low Ka values.
Table 2.
Alanine mutants of BPTI used in the study
| Lys15Arg | R15 | 80%a |
| Pro13Ala, Lys15Arg | A13R15 | 70%a |
| Thr11Ala, Pro13Ala, Lys15Arg | A11A13R15 | 60%a |
| Lys15Arg, Arg17Ala | R15A17 | 70%a |
| Lys15Arg, Val34Ala | R15A34 | 70%a |
| Lys15Arg, Val34Ala, Gly37Ala, Arg39Ala | R15A34A37A39 | 40%a |
| Pro13Ala, Lys15Arg, Arg17Ala, Ile18Ala, Ile19Ala | A13R15A17A18A19 | 40%a |
| Thr11Ala, Pro13Ala, Lys15Arg, Arg17Ala, Ile18Ala, Ile19Ala | A11A13R15A17A18A19 | 30%a |
| Pro13Ala, Lys15Arg, Arg17Ala, Ile18Ala, Ile19Ala, Val34Ala | A13R15A17A18A19A34 | 30%b |
| Thr11Ala, Pro13Ala, Lys15Arg, Arg17Ala, Ile18Ala, Ile19Ala, Val34Ala | A11A13R15A17A18A19A34 | 30%b |
| Thr11Ala, Pro13Ala, Lys15Arg, Arg17Ala, Ile18Ala, Ile19Ala, Val34Ala, Gly37Ala, Arg39Ala | A11A13R15A17A18A19A34A37A39 | 30%b |
Alanine mutations introduced into the primary binding loop are shown in bold and those in the secondary binding loop are in italic. All mutants contain additional Met52Leu substitution. Refolding yield is shown in the third column.
a Refolding conditions: 1 × 10−4 M BTPI, 4 M urea, 20 mM Tris, pH 8.7, 6 × 10−4 M GSH, 3 × 10−4 M
GSSG, 3 mM EDTA, 1 h, 47°C.
b Refolding conditions: 1 × 10−5 M BPTI, 0.4 M urea, 20 mM Tris, pH 8.7, 6 × 10−4 M GSH, 3 × 10−4 M GSSG, 3 mM EDTA, 1 h, 37°C.
In a free inhibitor, side chains of mutated residues are at least 40% solvent exposed (Arg 15, Arg 17, and Arg 39 in >75%) and do not form many interactions with the remainder of the inhibitor. The substituted residues form only one hydrogen bond, Thr 11 Oγ1–Val 34 O (2.64 Å), and seven van der Waals contacts, Ile 18 Cδ1–Gly 37 Cα (3.59 Å), Val 34 Cγ1–Arg 17 Cα (3.74 Å), Thr 11 Cβ–Tyr 10 C (3.67 Å), Val 34 Cγ2–Phe 33 C (3.74 Å), Val 34 Cβ–Phe 33 C (3.63 Å), Val 34 Cγ2–Ile 19 Cδ1 (3.48 Å) and Ile 19 Cγ2–Thr 32 Cβ (3.72 Å). Thus, alanine mutation(s) are not expected to break many stabilizing interactions.
The sequences of the mutants are shown in Table 2. All the variants were refolded from inclusion bodies to their native structures under conditions enabling correct pairing of disulfide bonds. For those that contain more than three alanine substitutions and are much less stable (see below), we had to apply more gentle refolding conditions (Table 2).
Association of alanine mutants with trypsin and chymotrypsin
The association constant values for interaction of all mutants with trypsin or chymotrypsin are shown in Table 3. We decided to determine Ka values at pH 8.2 to enable direct comparison with the single alanine mutants of Castro and Anderson (1996). However, the Ka values could be directly measured at pH 8.2 only for three trypsin and seven chymotrypsin mutant complexes. Partial digestion of the inhibitor observed upon incubation precluded direct Ka measurements for the remaining ones. To obtain Ka values at pH 8.2 for all mutant trypsin complexes, we measured Ka values at pH 5.5, at which the complex is still strong but no digestion of the inhibitor was observed due to decreased activity of the enzyme. As the ratio of Ka values at pH 8.2 and pH 5.5 was similar for all three mutants for which direct measurements were possible (70, 62.5, and 70, see Table 3), the remaining Ka values at pH 8.2 were calculated using the average ratio factor of 68. We also found similar factors, 19.7, 19.7, and 19.1, respectively, for the three chosen mutants in interaction with chymotrypsin. Therefore, we calculated the Ka value for A11A13R15A17A18A19A34A37A39 at pH 8.2 using an average factor of 19.5. The analysis of alanine mutation effects presented below will be based on Ka values at pH 8.2 because they allow direct and extensive comparison with the alanine scan data of Castro and Anderson (1996).
Table 3.
Association constants Kadetermined for the interaction of alanine mutants of BPTI with bovine β-trypsin and α-chymotrypsin at pH 8.2 and 5.5, 22°C
| Trypsin | Chymotrypsin | |||||
| BPTI mutant | Ka (M−1) pH 5.5 | Ka (M−1) pH 8.2 | effect | Ka (M−1) pH 5.5 | Ka (M−1) pH 8.2 | effect |
| R15 | — | 4.5 × 1013a | — | 1.27 × 107 | 2.5 × 108 | — |
| A13R15 | 7.2 × 109 | 4.5 × 1011 | 1.0 × 102 ↓ | 1.22 × 107 | 2.4 × 108 | 1 |
| A11A13R15 | 6.0 × 109 | 4.2 × 1011 | 1.1 × 102 ↓ | 1.15 × 107 | 2.2 × 108 | 1.1 ↓ |
| R15A17 | 4.1 × 1011 | 2.8 × 1013b | 1.6 ↓ | — | 1.8 × 108 | 1.4 ↓ |
| R15A34 | 6.5 × 1011 | 4.4 × 1013b | 1.0 | — | 2.2 × 108 | 1.1 ↓ |
| R15A34A37A30 | 7.4 × 108 | 5.2 × 1010 | 8.7 × 102 ↓ | — | 6.0 × 106 | 41.7 ↓ |
| A13R15A17A18A19 | 5.9 × 108 | 4.0 × 1010b | 1.1 × 103 ↓ | — | 1.5 × 106 | 1.7 × 102 ↓ |
| A11A13R15A17A18A19 | 6.1 × 108 | 4.1 × 1010b | 1.1 × 103 ↓ | — | 3.7 × 106 | 67.6 ↓ |
| A13 R15A17A18A19A34 | 3.5 × 106 | 2.4 × 108b | 1.9 × 105 ↓ | — | 2.0 × 105 | 1.3 × 103 ↓ |
| A11A13R15A17A18A19A34 | 3.6 × 106 | 2.4 × 108b | 1.9 × 105 ↓ | — | 3.7 × 106 | 67.6 ↓ |
| A11A13R15A17A18A19A34A37A39 | 5.1 × 104 | 3.4 × 106b | 1.3 × 107 ↓ | 3.4 × 104 | 6.6 × 105a | 3.8 × 102 ↓ |
Inhibition effects were compared with the pseudo wild-type BPTI. Downward arrow indicates a decrease in the association constant value upon mutations. Estimated error of Ka values is ±20%.
a From Krowarsch et al. (1999).
bKa values calculated from the equation Ka,pH 8.2 = Ka,pH 5.5 × 68 (see text).
cKa values calculated from the equation Ka,pH 8.2 = Ka,pH 5.5 × 19.5 (see text).
Generally, introduction of alanine(s) into the contact region led to a progressive decrease in the association energy with trypsin and chymotrypsin (Table 3). For the most extensively mutated variant compared with the pseudo wild type, the effect was much more pronounced for trypsin (1.3 × 107-fold decrease). In the case of chymotrypsin binding, it was only 3.8 × 102-fold weaker. For single alanine mutants, R15A17 and R15A34, no decrease in binding constants was observed for both enzymes. However, a single substitution, Pro13Ala, produced very different effects—it was neutral for chymotrypsin and highly deleterious for trypsin.
Multiple mutants, consisting of more than two alanine substitutions, behaved differently with the two proteinases. Generally, these six mutants were worse inhibitors, however, Ka decreases were always more pronounced for trypsin than chymotrypsin. Additionally, the ratio of decreases changed from mutant to mutant. For example, the ratio is the smallest for A13R15A17A18A19 (1.1 × 103 ÷ 1.7 × 102 = 6.7) and the highest for A11A13R15A17A18A19A34A37A39 (1.3 × 107 ÷ 3.8 × 102 = 3.5 × 104). In other words, the ratio of trypsin to chymotrypsin association constants is 1.8 × 105 for the pseudo wild-type BPTI, but, for the most extensive A11A13R15A17A18A19A34A37A39 mutant, it is only 5.2.
Additivity of association energies
The Ka values for alanine mutants were converted into free energy of association values (ΔGa) and grouped in seven thermodynamic additivity cycles, denoted A–G (Fig. 2 ▶). In the cycle, the effects of the same alanine substitution(s) (ΔΔGa) in two different sequence contexts were compared. The key value of the cycle is │ΔΔΔGa│, an absolute value of a difference of two ΔΔGas, called an interaction or coupling energy (Wells 1990; LiCata and Ackers 1995). │ΔΔΔGa│ determines whether the effect of substitution is additive. For an ideal additivity cycle, │ΔΔΔGa│ should equal 0. In our cycles │ΔΔΔGa│ ranges from 0 to 2.9 kcal mole−1 for trypsin and 0.1 to 1.78 kcal mole−1 for chymotrypsin, suggesting that nonadditivity occurs in most of the considered cycles. However, the cycle comprises four independent Kas, each with an estimated error of ±20%, equivalent to ±0.1 kcal mole−1 error in ΔGa value at the 1σ level and, consequently, 0.8 kcal mole−1 as an acceptable additivity limit of │ΔΔΔGa│ values at the 2σ level (Qasim et al. 1997; Krowarsch et al. 1999). Cases in which │ΔΔΔGa│ is <0.8 kcal mole−1 will be called additive and >0.8 kcal mole−1 – nonadditive. Three cycles: A, B, and C are additive, and four cycles: D, E, F, and G are nonadditive for trypsin. In the case of chymotrypsin, two cycles (B and E) are additive and five cycles (A, C, D, F, and G) are nonadditive.
Fig. 2.

Thermodynamic additivity cycles between alanine mutants of BPTI used in this work. The values for trypsin interaction are shown above (bold) and those for chymotrypsin are below (grey). The influence of the same alanine substitution(s) is described by two ΔΔGa values equal to ΔGa, mutant with less Ala − ΔGa, mutant with more Ala ΔΔΔGa is a difference between ΔΔGa values calculated for the same alanine mutation(s) in the two compared sequence contexts.
Besides the analysis presented above, there is another way to estimate additivity effects in our system. Castro and Anderson (1996) reported Ka values for chymotrypsin association with 15 single mutants of BPTI in which all contact residues were mutated to alanine. Their data for trypsin are less useful here, because, for many of the single mutants, only a lower limit of Ka could be estimated. To combine both data sets, we applied identical experimental conditions of buffer composition, pH, and temperature. Table 4 compares experimental ΔGa values for chymotrypsin determined in this work and ΔGa,calc values calculated by Castro and Anderson (1996) assuming full additivity. The nonadditivity term is equal to a difference between the calculated and measured value: ΔΔGa,calc = ΔGa,calc − ΔGa.
Table 4.
Experimental (Δ Ga) and calculated (ΔGa,calc) values of free energy of binding for 10 alanine mutants and chymotrypsin at 22°C and pH 8.2
| ΔΔGa,calc error | ||||||
| BPTI mutant | ΔGa (kcal mole−1) | ΔGa,calca (kcal mole−1) | ΔΔGa,calcb (kcal mole−1) | kc | (kcal mole−1) | additivityd |
| A13R15 | −11.25 | −11.35 | −0.1 | 1 | ±0.40 | + |
| A11A13R15 | −11.20 | −11.15 | 0.05 | 2 | ±0.40 | + |
| R15A17 | −11.08 | −10.75 | 0.33 | 1 | ±0.40 | + |
| R15A34 | −11.20 | −11.24 | −0.04 | 1 | ±0.40 | + |
| R15A34A37A39 | −9.10 | −10.26 | −1.16 | 3 | ±0.57 | − |
| A13R15A17A18A19 | −8.29 | −9.34 | −1.05 | 4 | ±0.75 | − |
| A11A13R15A17A18A19 | −8.82 | −9.15 | −0.33 | 5 | ±0.94 | + |
| A13R15A17A18A19A34 | −7.12 | −9.31 | −2.19 | 5 | ±0.94 | − |
| A11A13R15A17A18A19A34 | −8.82 | −9.15 | −0.33 | 6 | ±1.13 | + |
| A11A13R15A17A18A19A34A37A39 | −7.81 | −8.13 | −0.32 | 8 | ±1.52 | + |
a ΔGa,calc was calculated from data for single alanine mutants (Castro and Anderson 1996).
bΔΔGa,calc = ΔGa,calc − ΔGa.
ck is a number of introduced alanine mutations.
d (+) Additivity; (−) non-additivity.
Estimation of additive behavior requires a careful error analysis. The error in ΔGa values for 15 single alanine mutants was similar to that in our work: ±0.2 kcal mole−1 at the 2σ level (Castro and Anderson, 1996). For ΔΔGa,calc, the global error depends on the number of introduced alanines, according to the equation (Lu et al. 2001):
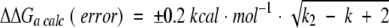 |
(1) |
where k is the number of introduced alanine mutations. This error analysis is valid for k > 2. If k equals 1 or 2, the error is calculated as a sum of individual errors (Lu et al. 2001).
Inspection of Table 4 shows that seven out of 10 alanine mutants are fully additive and three are nonadditive. Interestingly, these three mutants R15A34A37A39, A13R15A17A18A19, and A13R15A17A18A19A34contain three, four, and five alanine substitutions—less than the two most extensive variants (six and eight alanines), which are additive. Upon addition of the Thr11Ala mutation to the latter two variants, additivity is recovered, as observed in the A11A13R15A17A18A19 and A11A13R15A17A18A19A34 variants (Table 4). Respective ΔGa values are decreased by 0.53 and 1.70 kcal mole−1. On the contrary, introduction of the single Thr11Ala substitution increases ΔGa by 0.2 kcal mole−1 (Castro and Anderson 1996). It should be stressed that the most extensive mutant containing eight alanines binds chymotrypsin exactly as predicted from single mutant effects.
Conformation and stability of alanine mutants
Although all our variants inhibited both proteinases, we were aware that some of them might be structurally altered, as we introduced multiple alanine substitutions into a small protein. We tested the structural properties of all the variants using CD spectroscopy and thermodynamic stability analysis.
CD spectra recorded in a near and far UV did not differ significantly from the pseudo wild-type protein spectra, indicating a properly folded structure of the mutants (Fig. 3 ▶). Only the spectrum of the most extensive mutant, A11A13R15A17A18A19A34A37A39 BPTI, showed significant changes in the far UV. Further, transitions of thermal denaturation curves of all mutants, monitored both with CD and DSC, were highly cooperative. We analyzed denaturation of mutants at three pH values: 2.0, 2.5, and 2.7. Denaturation temperatures for all mutants were 6.4°C to 34.8°C lower compared with the pseudo wild type. Generally, introduction of a larger number of alanines led to a more severe destabilization of the molecule. In this respect, mutant A11A13R15A17A18A19A34A37A39 behaves unusually, as it is more stable than several less extensive mutants, which contain four to six alanines (Table 5). Additionally, we determined stability parameters for all the mutants using the DSC method. Tden values were on average 1.5°C higher than those determined from CD-monitored curves, and the ratio of calorimetric to van't Hoff enthalpy was close to unity, suggesting the two-state transition.
Fig. 3.


Circular dichroism spectra of alanine mutants of BPTI in far (top) and near (bottom) UV range. Spectra were recorded in 10 mM Gly/HCl, (pH 2.0 at 25°C). Details are described in the Materials and Methods.
Table 5.
CD spectroscopy determined thermal denaturation parameters for BPTI variants
| BPTI mutant | pH | Tden (°C) | ΔTden (°C) | ΔHden (kcal mole−1) | Δ Cp,dena (kcal mole−1 deg−1) | Δ Gden(25°C) b (kcal mol−1) | ΔΔGden(25°C) (kcal mol−1) |
| R15 | 2.0 | 81.9 | — | 63.0 | 8.27 | — | |
| 2.5 | 83.4 | — | 64.5 | ||||
| 2.7 | 89.3 | — | 66.1 | 0.38 (±0.12) | |||
| A13R15 | 2.0 | 69.9 | 12.0 | 53.7 | 5.95 | 2.32 | |
| 2.5 | 71.9 | 11.5 | 53.9 | ||||
| 2.7 | 74.7 | 14.6 | 55.3 | 0.35 (±0.1) | |||
| A11A13R15 | 2.0 | 69.7 | 12.2 | 54.4 | 6.06 | 2.21 | |
| 2.5 | 72.1 | 11.3 | 55.9 | ||||
| 2.7 | 75.1 | 14.2 | 56.3 | 0.34 (±0.14) | |||
| R15A17 | 2.0 | 75.5 | 6.4 | 69.4 | 8.67 | −0.4 | |
| 2.5 | 77.3 | 6.1 | 70.1 | ||||
| 2.7 | 83.3 | 6.0 | 72.2 | 0.36 (±0.004) | |||
| R15A34 | 2.0 | 74.8 | 7.1 | 68.1 | 8.44 | −0.17 | |
| 2.5 | 76.6 | 6.8 | 68.9 | ||||
| 2.7 | 82.1 | 7.2 | 70.7 | 0.35 (±0.01) | |||
| R15A34A37A39 | 2.0 | 64.5 | 17.4 | 35.3 | 3.37 | 4.9 | |
| 2.5 | 68.9 | 14.5 | 37.7 | ||||
| 2.7 | 75.4 | 13.9 | 39.0 | 0.33 (±0.087) | |||
| A13R15A17A18A19 | 2.0 | 47.1 | 34.8 | 30.6 | 1.87 | 6.4 | |
| 2.5 | 49.1 | 34.3 | 31.7 | ||||
| 2.7 | 53.0 | 36.3 | 32.5 | 0.31 (±0.09) | |||
| A11A13R15A17A18A19 | 2.0 | 47.5 | 34.4 | 30.6 | 1.90 | 6.37 | |
| 2.5 | 48.1 | 35.3 | 31.5 | ||||
| 2.7 | 53.1 | 36.2 | 32.6 | 0.31 (±0.11) | |||
| A13R15A17A18A19A34 | 2.0 | 47.3 | 34.6 | 34.1 | 2.18 | 6.09 | |
| 2.5 | 50.9 | 32.5 | 34.9 | ||||
| 2.7 | 51.9 | 37.4 | 35.5 | 0.25 (±0.003) | |||
| A11A13R15A17A18A19A34 | 2.0 | 48.6 | 33.3 | 35.1 | 2.35 | 5.92 | |
| 2.5 | 49.2 | 34.2 | 35.9 | ||||
| 2.7 | 52.0 | 37.3 | 36.5 | 0.25 (±0.067) | |||
| A11A13R15A17A18A19A34A37A39 | 2.0 | 60.9 | 21.0 | 41.7 | — | — | |
| 2.5 | N.D. | — | N.D. | ||||
| 2.7 | N.D. | — | N.D. | — |
The unfolding experiments were performed in 10 mM Gly/HCl, pH 2.0, 2.5, and 2.7 with final protein concentration of 20 μg ml−1; ellipticity was followed at 223 nm, with temperature scan rate of 1 deg min−1 (Jasco model J-715 spectropolarimeter).
a ΔCp,den calculated from ΔHden dependence on Tden (Fig. 4 ▶).
b Denaturation free energy changes were calculated at 25°C at pH 2.0 using Gibbs-Helmholz equation; (N.D.) Not determined.
ΔHden versus Tden data points for individual mutants do not fall on a single line, rather they provide a set of (roughly) parallel lines (Fig. 4 ▶). Therefore, to compare the thermodynamic stability of the mutants, we calculated ΔGden values from the Gibbs-Helmholtz equation at a common temperature (25 °C). The mutants showed decreases in ΔGden values up to >6.4 kcal mole−1, strongly manifested in the case of those containing three or more alanines. Thus, up to 77% of the stabilization energy was lost upon introduction of alanine mutations. Two single mutants: R15A17 and R15A34, despite lower Tden values, were marginally more stable than the pseudo wild type because of larger ΔHden values (Table 5 and Fig. 4 ▶).
Fig. 4.


Denaturation temperature (Tden) dependence of van't Hoff enthalpy (ΔHvH) for 10 alanine mutants of BPTI determined in 10 mM Gly/HCl buffer at pHs 2.0, 2.5, and 2.7.
We checked whether the stability effects were additive through thermodynamic cycle analysis (Fig. 5 ▶). To analyze additivity, we first estimated errors in Tden (±0.1°C), ΔHden (±1 kcal mole−1) and ΔCp,den (±20% of the experimental value). Hence, the error in ΔGden from the Gibbs-Helmholtz equation equals ±0.34 kcal mole−1, and the limit on additivity is │ΔΔΔGden│ = 4 × 0.34 kcal mole−1 = 1.36 kcal mole−1.
Fig. 5.

Thermodynamic stability additivity cycles for alanine substitutions in BPTI. All ΔGden values were calculated at common a temperature of 25°C using the Gibbs-Helmholtz equation. ΔΔGden is equal to ΔGden, mutant with less Ala − ΔGden, mutant with more Ala and describes the stability effect caused by introduction of alanine(s). Similarly as defined for protein–protein additivity, ΔΔΔGden is a difference of two ΔΔGden values and determines whether the effect of the substitution is additive.
│ΔΔΔGden│ values reported for five thermodynamic cycles in Figure 5 ▶ show that introduction of alanine(s) is fully additive, despite very large effects on the stability of individual mutants. This observation suggests that alanine substitutions are independent of each other in terms of free energy changes. However, we noticed unusual behavior of A11A13R15A17A18A19A34A37A39 BPTI. Compared to the A11A13R15A17A18A19A34 variant (Tden = 48.6°C at pH 2.0), the previous one contains two additional mutations, Gly37Ala and Arg39Ala. Yu et al. (1995) showed that the former substitution reduced Tden by 17.2°C, and the latter had no effect on BPTI stability. Although these two effects were observed in the context of the single Cys5–Cys55 disulfide BPTI variant and at pH 7.0, we feel that severe nonadditivity occurs for A11A13R15A17A18A19A34A37A39 BPTI. The two mutations should decrease its Tden to about 31.4°C (48.6°C–17.2°C), and the observed Tden at pH 2.0 is 60.9°C. The determination of the NMR solution structure for this variant is currently underway in our laboratory.
Discussion
The main aim of this study was to analyze the influence of surrounding residues on the energy of interaction of a functional epitope formed by the single P1 residue of a protein inhibitor, BPTI, with trypsin and chymotrypsin. To accomplish that, we applied additivity cycle analysis using 10 single or multiple BPTI mutants, in which different sets of contact residues were converted to alanine(s). Serine proteinase–protein inhibitor interaction seems to be well suited for such analysis. In all known complexes of this type, the shape of the contact area and the geometry of the interaction, including the network of hydrogen bonds, are well preserved, despite differences in the sequences of proteinases and inhibitors (Bode and Huber 1992). For example, geometry of trypsin recognition by 10 P1 mutants of BPTI that differ in strength of association with trypsin by nine orders of magnitude is identical, even at the level of the length of individual intermolecular hydrogen bonds (Helland et al. 1999). It has also been shown that the same mutations at the P1 position of inhibitors from families of totally different scaffoldings led to similar energetic effects (Qasim et al. 1997; Krowarsch et al. 1999). An analogous intrascaffolding additivity rule works when contact residues of one interacting component are mutated and analyzed through a thermodynamic cycle (Wells, 1990; Lu et al. 2001). Finally, Kuroda and Kim (2000) showed that BPTI scaffolding is tolerant of extensive mutagenesis as it is able to accept up to 50% of alanines without major conformational changes thus justifying extensive alanine mutagenesis of the inhibitor.
The following analysis of protein–protein association data is based on the assumption that, for all mutants, no conformational adjustments occur upon alanine(s) introduction when compared with the pseudo wild-type BPTI, which was verified by CD spectra measurements. Additionally, the atomic resolution structure for A11A13R15 BPTI showed a similar conformation of its primary binding loop to that of the wild-type BPTI with an rmsd of 0.4 Å for backbone atoms (Czapinska et al. 2000; Addlagatta et al. 2001). We are aware that the conformation of A11A13R15A17A18A19A34A37A39 BPTI may differ from all others, as its CD spectrum was changed in the far UV range. Also, the thermodynamic stability of this mutant showed significant deviation from additivity.
We feel that the additivity tests performed here are rigorous as the mutations compared lead to severe changes in the complex contact area. Figure 6 ▶ shows estimated changes in the size of the contact area accompanying complex formation calculated for all alanine mutants interacting with trypsin and chymotrypsin. To illustrate the extent of the changes, a comparison of structural epitopes of the pseudo wild type and of the most shaved A11A13R15A17A18A19A34A37A39 mutant is shown (Fig. 1 ▶). In this mutant, the total contact area with both trypsin and chymotrypsin is reduced by about 22% compared with the respective pseudo wild-type complexes (Fig. 6 ▶). In all modeled trypsin complexes Arg 15 remains 100% buried, as it is in the pseudo wild-type complex. Arg 15 in chymotrypsin complexes, including the pseudo wild type, is 92% buried, and exposes about 15 Å2 to the solvent. Thus, in all trypsin and chymotrypsin complexes, interactions involving Arg 15 should not be, in principle, affected by alanine shaving. Also, the relative contribution of Arg 15 is larger in more extensively mutated variants. For example, it provides 16% of the total contact area in the wild-type complex with trypsin and 20% of the total area in the A11A13R15A17A18A19A34A37A39–trypsin complex.
Fig. 6.
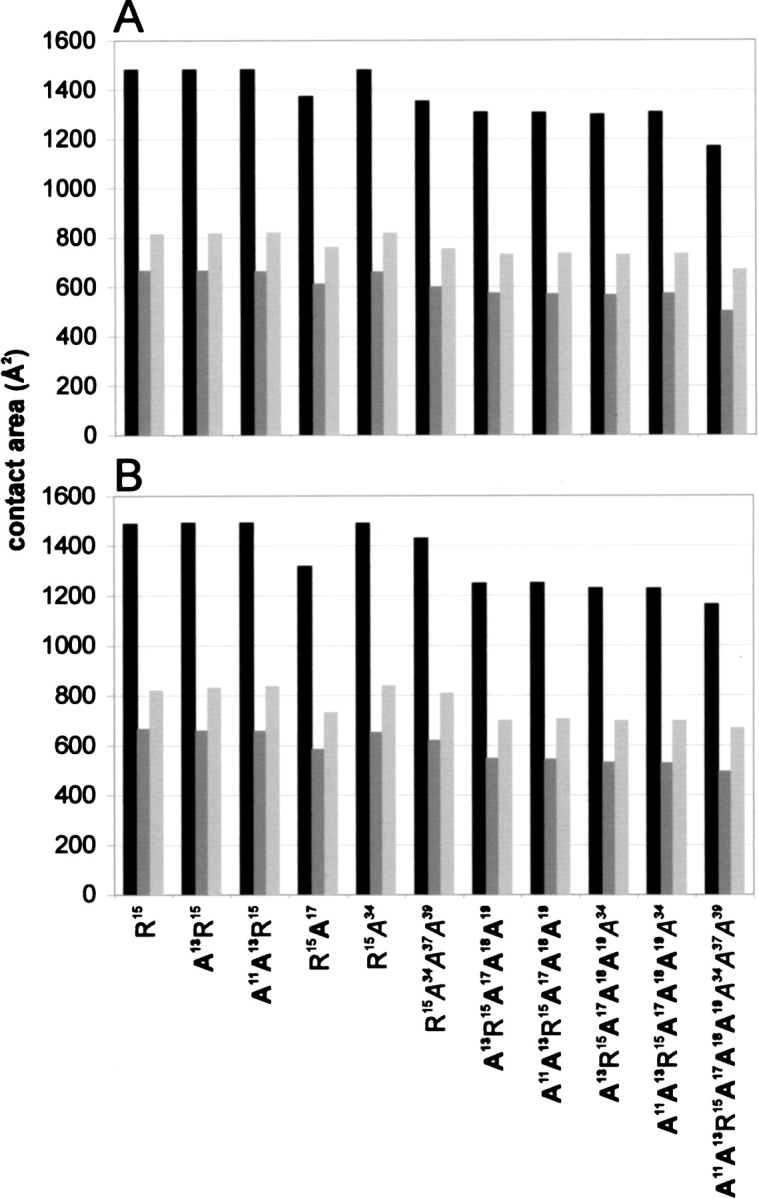
Contact area values for the complexes between individual alanine mutants of BPTI and trypsin (A) or chymotrypsin (B). Total contact area (black bar) and contact area of enzyme (gray) and inhibitor (light gray) components are shown.
Thermal stability curves recorded at three pH values provided thermodynamic parameters, ΔHden, ΔCp,den, and Tden for all mutants. The ΔHden versus Tden plots for individual mutants did not provide a single but a series of (roughly) parallel lines, in a marked difference to previously analyzed P1 mutants of BPTI (Krowarsch and Otlewski 2001). Thus, to compare their stability, we turned to ΔGden values. Arrangement of ΔGden values in five thermodynamic additivity cycles showed that all the effects of substitutions are fully independent of each other and, therefore, additive. In the free inhibitor, there is a hydrogen bond Thr 11 Oγ–Val 34 O that involves a side chain of a mutated residue and may cause nonadditive stability behavior (Wells 1990). However, elimination of this bond as a result of the Thr11Ala substitution had a marginal effect on stability of the respective variants and was fully additive (cycles B and C).
We also applied a simpler method of additivity estimation, suggested by Kuroda and Kim (2000), through comparison of respective ΔTden values at a common pH, rather than through ΔGden (data not shown). This method did not allow us to close cycles D and E.
Data obtained on interaction of 10 alanine mutants with chymotrypsin enable more extensive calculations of additivity effects, also including data for the single alanine mutants described by Castro and Anderson (1996). It should be stressed that our data for all three single mutants (A13R15, R15A17, and R15A34) agree very well with alanine scanning data justifying joint analysis of two data sets. Table 4 shows that ΔGa,calc values estimated from single mutation effects agree very well with experimental ΔGas for seven mutants. However, for three mutants: A13R15A17A18A19A34, R15A34A37A39, and A13R15A17A18A19, the discrepancies from additivity excess allowed error by −1.25, −0.62, and −0.3 kcal mole−1, respectively. It is intriguing that these deviations occur just for these three mutants and not for the more extensive ones. Lack of respective structures for inhibitor mutants and their complexes with chymotrypsin does not allow more detailed interpretation of this phenomenon.
The association constant for the A11A13R15A17A18A19 A34A37A39 BPTI–chymotrypsin interaction is 6.6 × 105 M−1 and agrees extremely well with the value calculated from single mutation effects. The same agreement is observed for mutant containing six alanines A11A13R15A17A18A19A34. We conclude that these two extensively mutated variants fulfill a very strong additivity test.
It could be assumed that the Ka values for A11A13R15A17A18A19A34A37A39 BPTI interacting with both enzymes should be equal to the contribution of Arg 15 (the P1 residue) to the total energy of association. In the case of chymotrypsin, this contribution was calculated to be 4.7 kcal mole−1 from the effect of the Arg15Gly substitution on the binding energy (Krowarsch et al. 1999). Thus, we observed a large gap between the two values, equivalent to 3 kcal mole−1. This means that the association energy for the pseudo wild-type BPTI cannot be reproduced from a sum of single residue mutational effects. There are two possible reasons for this discrepancy. First, we suppose that the alanine background surrounding the P1 site might not be fully neutral and might still provide some binding energy as a result of the presence of numerous methyl groups of alanines. For example, we observe that the Ala15Gly mutation removes an additional1.9 kcal mole−1 of binding energy with chymotrypsin (Krowarsch et al. 1999), and the analogous mutation at P1` in BPTI removes 1.5 kcal mole−1 (Grzesiak et al. 2000a). This residual energy could easily be removed if we performed glycine shaving analysis. However, it appears to be a formidable task because of the much higher susceptibility to proteolysis of glycine mutants. Also, introduction of glycines would significantly increase the conformational entropy of the contact loops complicating the interpretation of the binding data.
The second possible reason for the observed discrepancy is related to the presence of the main chain–main chain intermolecular hydrogen bond network, which is well conserved among different proteinase–inhibitor complexes (Jackson 1999). In our opinion, preservation of the binding mode is the main reason for the high nonadditivity described above. The total energetic contribution of these hydrogen bonds remains unknown, but should be substantial. A study on the chemical variant of the turkey ovomucoid third domain in which the P2–P1 amide bond has been replaced with an ester shows that a single intermolecular hydrogen bond provides 0.8–2.0 kcal mole−1 of free energy, depending on a proteinase (Lu et al. 1997). Thus, the retained 3 kcal mole−1 can come from binding energy provided by the methyls of alanines and/or by intermolecular backbone hydrogen bonds.
Because of the extremely strong electrostatic interaction, analysis of additivity and hot spot energetics is more complicated in the case of the trypsin interaction. A11A13R15A17A18A19A34A37A39 BPTI binds trypsin with a Ka of 3.4 × 106 M−1, and the contribution of Arg 15 estimated from the effect of the Arg15Gly substitution on binding energy with this proteinase approaches −12.7 kcal mole−1 (Krowarsch et al. 1999; Helland et al. 1999). Thus, this fully shaved variant binds to trypsin 3.95 kcal mole−1 weaker than expected from the contribution of Arg 15 to the association energy. Interestingly, as the effect is opposite to the discrepancy described above for chymotrypsin, we suppose that, when corrected for the presence of alanine methyls rather than glycines and main chain hydrogen bonds, the total disagreement can be about 3 kcal mole−1 larger, that is, up to about 7 kcal mole−1. This indicates that in A11A13R15A17A18A19A34A37A39 BPTI even 45% of the Arg 15 contribution can be destroyed. Clearly, this points to large nonadditivity comprising the P1 side chain.
This nonadditivity can also be shown through the following reasoning. Castro and Anderson (1996) reported one single alanine substitution effect: The Ile18Ala mutation decreases ΔGa by 4.94 kcal mole−1. We measured the effects of four single alanine substitutions: Thr11Ala (0 kcal mole−1), Pro13Ala (2.7 kcal mole−1), Arg17Ala (0.2 kcal mole−1) and Val34Ala (0 kcal mole−1). The former effect is calculated on the basis of the additivity assumption, which works in all three cycles (A, B, and C) including the single Thr11Ala substitution. The sum of all these effects (−7.84 kcal mole−1) subtracted from ΔGa for the pseudo wild type (−18.3 kcal mole−1) equals −10.86 kcal mole−1. This value is already 2 kcal mole−1 higher than expected from Arg15Gly (−12.7 kcal mole−1) and still does not cover effects from all contact residues.
We suppose that the nonadditivity observed in the F and G cycles and particularly large nonadditivity observed in the D and E cycles results from weakening of the electrostatic P1–S1 interaction. It can be speculated that the nonadditivity observed in these four cycles may result from different perturbations of the electrostatic interaction caused by various numbers of alanine substitutions introduced in the individual mutants. On the other hand, the Thr11Ala substitution does not seem to influence the strength of this interaction, as the A, B, and C cycles are fully additive.
Interestingly, lack of additivity connected with the P1 electrostatic interaction has been also observed for the ecotin–trypsin system (Yang et al. 1998). In this case, the interaction is extremely weak, as the effect of the substitutions at this position on Ka is only several fold, compared with 106-fold in BPTI–trypsin system (Krowarsch et al. 1999). In ecotin, the P1–S1 interaction appears to be highly coupled and nonadditive with interactions formed by two remote secondary binding loops that take over the majority of the association energy and relax the loop comprising the P1 site.
Materials and methods
Materials
Guanidinium chloride (GdmCl), urea, dimethylsulfoxide (DMSO), N,N-dimethylformamide (DMF), methanol, and acetonitrile were purchased from Merck. Trifluoroacetic acid (TFA), formic acid (88%) and cyanogen bromide (CNBr) were obtained from Fluka. Tris, sodium acetate, DTT, chloramphenicol, ampicillin, reduced and oxidized glutathione (GSH and GSSG), and 2–[[N-morpholino]]ethanesulfonic acid (MES) were supplied by Sigma. Sephadex G-25 was obtained from Amersham Pharmacia Biotech.
The components of the culture media were purchased from Merck. DNA-modifying enzymes (T4 DNA polymerase, T4 DNA ligase, and T4 polynucleotide kinase) and the DNA sequencing kit was from Amersham Pharmacia Biotech, and the DNA purification kit was from Qiagen. Oligonucleotides were synthesized by Ransom Hill. IPTG was obtained from Bachem.
Bovine trypsin and bovine chymotrypsin were purchased from Worthington. 4-Nitrophenyl 4`-guanidinobenzoate (NPGB) was from Merck and N-Bz-L-Arg-4-nitroanilide (Bz-Arg-pNA) was from Sigma. N-suc-Gly-Gly-L-Phe-4-nitroanilide and N-suc-Ala-Ala-Pro-L-Phe-4-nitroanilide were purchased from Bachem. N-suc-L-Phe-4-nitroanilide, N-p-tosyl-Gly-Pro-L-Arg-4-nitroanilide and N-Bz-Pro-Phe-L-Arg-4-nitroanilide were from Sigma.
Plasmid construction
Single or multiple alanine substitutions were introduced individually into the gene coding for Lys15Arg, Met52Leu BPTI by oligonucleotide-directed mutagenesis (Kunkel 1985), and the introduced mutations were confirmed by DNA sequencing.
Expression and purification of BPTI mutants
All 10 alanine mutants of BPTI and the pseudo wild type (Lys15Arg, Met52Leu) were overexpressed in Escherichia coli strain BL 21 (DE3) pLysS by use of the T7 polymerase promoter system (Studier et al. 1990). Purification of BPTI mutants was performed as described (Krokoszynska et al. 1998). Protein samples were >96% pure, as judged from C18 reverse-phase HPLC runs. A typical yield of in vitro oxidative refolding was 30%–80% (1–3 mg of pure protein per liter of culture medium). Electrospray mass spectrometry of all mutants used in this study was performed with a Finningan MAT TSQ-700 equipped with an ESI source. Samples were dissolved in methanol/water (1:1, vol/vol) containing 1% acetic acid. Molecular masses of all mutants were within 1.0 atomic mass unit from those expected from amino acid sequences.
Association constant determination
The association constant (Ka) values were determined as described by Empie and Laskowski (1982) and Otlewski and Zbyryt (1994). All measurements were performed in 100 mM Tris-HCl, 20 mM CaCl2, 0.005% Triton X-100 (pH 8.2) or in 100 mM MES, 20 mM CaCl2, 0.005% Triton X-100 (pH 5.5) at 22°C. To determine an association constant increasing amounts of the inhibitor were added to a constant concentration of a given proteinase. The initial enzyme concentration (E0) was chosen so that the condition 1 < [E0] · Ka < 50 was fulfilled. For the determination of low Ka values (103–107 M−1) inhibitors were used at much higher concentrations than enzymes to force complex formation. After 8–10t1/2 incubation time, the residual enzyme activity was measured by monitoring linear release of p-nitroaniline for the period of 300 sec using a diode array spectrophotometer HP 8452A. The average value of absorbance in the 380–410 nm range was recorded and corrected for the background average absorbance in the 480–510 nm range. The value of Ka was calculated by a three-parameter algorithm (E) = f(E0, Ka, F) using the nonlinear regression analysis program GraFit, according to the equation
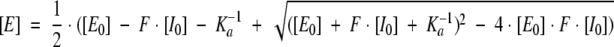 |
(2) |
where [E0] and [I0] are the total enzyme and inhibitor concentrations, respectively, [E] is the residual enzyme concentration, and F is the enzyme:inhibitor equimolarity factor. In the case of weak associations (Ka<107 M−1), the default value of F = 1 was applied.
Calorimetry
Differential scanning calorimetry (DSC) experiments were performed with a Nano II calorimeter (CSC Corp.). The experiments were carried out at protein concentrations of 100–200 μg mL−1 (total cell volume 323 μL), at a scan rate of 1.0 deg min−1, under a pressure excess of 2.5 atm. Before a DSC run, the protein solution was extensively dialyzed against 10 mM Gly/HCl (pH 2.0). Repeated measurements of the same sample demonstrated ∼70% reproducibility of ΔHcal values. The baseline was determined by running the calorimeter with both cells filled with the dialysis buffer. Experiments were performed in triplicate. The partial specific volume of BPTI was assumed to be 0.71 cm3 g−1 (Makhatadze et al. 1993). Tden, ΔHcal and ΔHvH values were calculated from thermogram analysis using Cpcalc software provided by CSC Corp.
CD measurements
Spectra
CD spectra of BPTI variants were recorded in 10 mM Gly/HCl buffer (pH 2.0), at 25°C on a Jasco J-715 spectropolarimeter. Measurements were performed at a protein concentration of 2×10−5 M using a 10-mm cuvette (240–360 nm range) or at 2×10−4 M protein in a 1-mm pathlength cuvette (190–260 nm range). Spectra were averaged from seven separate scans.
Thermal transitions
Thermal denaturation was monitored following the ellipticity at 223 nm using a band slit of 2 nm and a 4-sec response time. An automatic Peltier accessory PFD 350S allowed continuous monitoring of the thermal transition at a constant rate of 1 deg min−1. The temperature of the sample was monitored directly using a probe immersed in the cuvette and controlled with a PFD-350S/350L Peltier type FDCD attachment. Protein was dissolved in 10 mM Gly/HCl buffer, (pH 2.0, 2.5, and 2.7). Before measurement, a protein solution was passed through a 0.22–μm Millipore filter. Each denaturation experiment was performed in duplicate. The data were analyzed assuming a two-state reversible equilibrium transition:
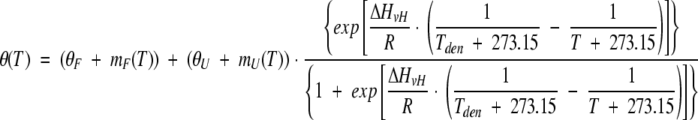 |
(3) |
where T is the temperature, R is the gas constant, θ(T) is the ellipticity signal at 223 nm; ΔHvH is the van't Hoff enthalpy, θF is the value of the folded signal extrapolated to −273.15 °C, mF is the slope of the temperature dependence of the CD signal for the folded protein, θU is the value of the unfolded signal extrapolated to −273.15 °C, mU is the slope of the temperature dependence of the CD signal for the unfolded protein, and Tden is denaturation temperature.
Acknowledgments
This work was supported by grant 6 PO4B 014 17 from the Polish Committee for Scientific Research. We thank Professor Zbigniew Szewczuk for mass spectra analysis and Marta Gladysz for help with mutant refolding. Daniel Krowarsch is a recipient of the Young Scholar Award from the Foundation for Polish Science.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.3510102.
References
- Addlagatta, A., Krzywda, S., Czapinska, H., Otlewski, J., and Jaskolski, M. 2001. Ultra high resolution structure of BPTI. Acta Cryst. D57 649–663. [DOI] [PubMed] [Google Scholar]
- Bode, W. and Huber, R. 1992. Natural protein proteinase inhibitors and their interaction with proteinases. Eur. J. Biochem. 204 433–451. [DOI] [PubMed] [Google Scholar]
- Bogan, A.A. and Thorn, K.S. 1998. Anatomy of hot spots in protein interfaces. J. Mol. Biol. 280 1–9. [DOI] [PubMed] [Google Scholar]
- Capasso, C., Rizzi, M., Menegatti, E., Ascenzi, P., and Bolognesi, M. 1997. Multiple protein–protein recognition sites in the crystal structure of α-chymotrypsin in complex with basic pancreatic trypsin inhibitor. J. Mol. Recogn. 10 26–35. [DOI] [PubMed] [Google Scholar]
- Castro, M.J. and Anderson, S. 1996. Alanine point-mutations in the reactive region of bovine pancreatic trypsin inhibitor: Effects on kinetics and thermodynamics of binding to β-trypsin and α-chymotrypsin. Biochemistry 35 11435–11446. [DOI] [PubMed] [Google Scholar]
- Czapinska, H., Otlewski, J., Krzywda, S., Sheldrick, G.M., and Jaskólski, M. 2000. High resolution structure of bovine pancreatic trypsin inhibitor with altered binding loop sequence. J. Mol. Biol. 295 1237–1249. [DOI] [PubMed] [Google Scholar]
- Empie, M.W. and Laskowski, Jr., M. 1982. Thermodynamics and kinetics of single residue replacements in avian ovomucoid third domains: Effect on inhibitor interactions with serine proteinases. Biochemistry 21 2274–2284. [DOI] [PubMed] [Google Scholar]
- Grzesiak, A., Helland, R., Smalås, A.O., Krowarsch, D., Dadlez, M., and Otlewski, J. 2000a. The P1` position in BPTI is a major determinant of the association energy with serine proteinases. J. Mol. Biol. 301 207–218. [DOI] [PubMed] [Google Scholar]
- Grzesiak, A., Krokoszynska, I., Krowarsch, D., Buczek, O., Dadlez, M., and Otlewski, J. 2000b. Inhibition of six serine proteinases of human coagulation system by mutants of bovine pancreatic trypsin inhibitor. J. Biol. Chem. 275 33346–33352. [DOI] [PubMed] [Google Scholar]
- Helland, R., Otlewski, J., Sundheim, O., Dadlez, M., and Smalas, A.O. 1999. The crystal structures of the complexes between bovine β-trypsin and ten P1 variants of BPTI. J. Mol. Biol. 287 923–942. [DOI] [PubMed] [Google Scholar]
- Hubbard, S.J. and Thornton, J.M. 1993. A computer algorithm to calculate surface accessibility. Department of Biochemistry and Molecular Biology, University College, London.
- Huber, R., Kukla, D., Bode, W., Schwager, P., Bartels, K., Deisenhofer, J., and Steigemann, W. 1974. Structure of the complex formed by bovine trypsin and bovine pancreatic trypsin inhibitor. J. Mol. Biol. 89 73–101. [DOI] [PubMed] [Google Scholar]
- Jackson, R.M. 1999. Comparison of protein–protein interaction in serine proteinase–inhibitor and antibody–antigen complexes: Implications for the protein docking problem. Protein Sci. 8 603–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, L. and Wells, J.A. 1994. Dissecting the energetics of an antibody-antigen interface by alanine shaving and molecular grafting. Protein Sci. 3 2351–2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, S. and Thorton, J.M. 1996. Principles of protein–protein interactions. Proc. Natl. Acad. Sci. 93 13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krokoszynska, I., Dadlez, M., and Otlewski, J. 1998. Structure of single-disulphide variants of bovine pancreatic trypsin inhibitor (BPTI) as probed by their binding to bovine β-trypsin. J. Mol. Biol. 275 503–513. [DOI] [PubMed] [Google Scholar]
- Krowarsch, D. and Otlewski, J. 2001. Amino-acid substitutions at the fully exposed P1 site of bovine pancreatic trypsin inhibitor affect its stability. Protein Sci. 10 715–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krowarsch, D., Dadlez, M., Buczek, O., Krokoszynska, I., Smalas, A.O., and Otlewski, J. 1999. Interscaffolding additivity: Binding of P1 variants of bovine pancreatic trypsin inhibitor to four serine proteases. J. Mol. Biol. 289 175–186. [DOI] [PubMed] [Google Scholar]
- Kunkel, T.A. 1985. Rapid and efficient site-specific mutagenesis without phenotopic selection. Proc. Natl. Acad. Sci. 82 488–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda, Y. and Kim, P.S. 2000. Folding of bovine pancreatic trypsin inhibitor (BPTI) variants in which almost half the residues are alanine. J. Mol. Biol. 298 493–501. [DOI] [PubMed] [Google Scholar]
- LiCata, V.J. and Ackers, G.K. 1995. Long-range, small magnitude non-additivity of mutational effects in proteins. Biochemistry 34 3133–3139. [DOI] [PubMed] [Google Scholar]
- Lo Conte, L., Chothia, C., and Janin, J. 1999. The atomic structure of protein–protein recognition sites. J. Mol. Biol. 285 2177–2198. [DOI] [PubMed] [Google Scholar]
- Lu, W., Qasim, M.A., Laskowski, Jr., M., and Kent, S.B.H. 1997. Probing intermolecular main chain hydrogen bonding in serine proteinases-protein inhibitor complexes: chemical synthesis of backbone-engineered turkey ovomucoid third domain. Biochemistry 36 673–679. [DOI] [PubMed] [Google Scholar]
- Lu, S.M., Lu, W., Qasim, M.A., Anderson, S., Apostol, I., Ardelt, W., Bigler, T., Chiang, Y.W., Cook, J., James, M.N.G., et al. 2001. Predicting the reactivity of proteins from their sequence alone. Kazal family of protein inhibitors of serine proteinases. Proc. Natl. Acad. Sci. 98 1410–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makhatadze, G.I., Kim, K., Woodward, C. and Privalov, P.L. 1993. Thermodynamics of BPTI folding. Protein Sci. 2 2028–2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald, I. K. and Thornton, J. M. 1994. Satisfying hydrogen bonding potential in proteins. J. Mol. Biol. 238 777–793. [DOI] [PubMed] [Google Scholar]
- Otlewski, J. and Apostoluk, W. 1997. Structural and energetic aspects of protein-protein recognition. Acta Biochim. Polon. 44 367–388. [PubMed] [Google Scholar]
- Otlewski, J. and Zbyryt, T. 1994. Single peptide bond hydrolysis/resynthesis in squash inhibitors of serine proteinases. I. Kinetics and thermodynamics of the interaction between squash inhibitors and bovine β-trypsin. Biochemistry 33 200–207. [DOI] [PubMed] [Google Scholar]
- Qasim, M.A., Ganz, P.J., Saunders, C.W., Bateman, K.S., James, M.N.G., and Laskowski, M.Jr 1997. Interscaffolding additivity. Association of P1 variants of eglin c and ovomucoid third domain with serine proteinases. Biochemistry 36 1598–1607. [DOI] [PubMed] [Google Scholar]
- Schechter, P. and Berger, A. 1967. On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun.. 27 157–162. [DOI] [PubMed] [Google Scholar]
- Scheidig, A.J., Hynes, T.R., Pelletier, L.A., Wells, J.A., and Kossiakoff, A.A. 1997. Crystal structures of bovine chymotrypsin and trypsin complexed to the inhibitor domain of Alzhaimer's amyloid β-precursor (APPI) and basic pancreatic trypsin inhibitor (BPTI): Engineering of inhibitors with altered specificities. Protein Sci. 6 1806–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber, G. and Fersht, A.R. 1995. Energetics of protein–protein interactions: Analysis of the barnase-barstar interface by single mutations and double mutant cycles. J. Mol. Biol. 248 478–486. [DOI] [PubMed] [Google Scholar]
- Studier, F.W., Rosenberg, A.H., Dunn, J.J., and Dubendorff, J.W. 1990. Use of T7 polymerase to direct expression of cloned genes. Methods Enzymol. 185 60–89. [DOI] [PubMed] [Google Scholar]
- Wells, J.A. 1990. Additivity of mutational effects in proteins. Biochemistry 29 8509–8517. [DOI] [PubMed] [Google Scholar]
- ———. 1991. Systematic mutational analyses of protein–protein interfaces. Methods Enzymol. 202 390–411. [DOI] [PubMed] [Google Scholar]
- Yang, S.Q., Wang, C.-I., Gillmor, S.A., Fletterick, R.J., and Craik, C.S. 1998. Ecotin: A serine protease inhibitor with two distinct and interesting binding sites. J. Mol. Biol. 279 945–957. [DOI] [PubMed] [Google Scholar]
- Yu, M.-H., Weissman, J.S., and Kim, P.S. 1995. Contribution of individual side-chains to the stability of BPTI examined by alanine-scanning mutagenesis. J. Mol. Biol. 249 388–397. [DOI] [PubMed] [Google Scholar]