

Abstract
LacI and PurR are highly homologous proteins. Their functional units are homodimers, with an N-terminal DNA binding domain that comprises the helix-turn-helix (HTH), N-linker, and hinge regions from both monomers. Hinge structural changes are known to occur upon DNA dissociation but are difficult to monitor experimentally. The initial steps of hinge unfolding were therefore examined using molecular dynamics simulations, utilizing a truncated, chimeric protein comprising the LacI HTH/N-linker and PurR hinge. A terminal Gly-Cys-Gly was added to allow "dimerization" through disulfide bond formation. Simulations indicate that differences in LacI and PurR hinge primary sequence affect the quaternary structure of the hinge•hinge` interface. However, these alternate hinge orientations would be sterically restricted by the core domain. These results prompted detailed comparison of recently available DNA-bound structures for LacI and truncated LacI(1–62) with the PurR structure. Examination revealed that different N-linker and hinge contacts to the core domain of the partner monomer (which binds effector molecule) affect the juxtapositions of the HTH, N-linker, and hinge regions in the DNA binding domain. In addition, the two full-length repressors exhibit significant differences in the interactions between the core and the C-linker connection to the DNA binding domain. Both linkers and the hinge have been implicated in the allosteric response of these repressors. Intriguingly, one functional difference between these two proteins is that they exhibit opposite allosteric response to effector. Simulations and observed structural distinctions are correlated with mutational analysis and sequence information from the LacI/GalR family to formulate a mechanism for fine-tuning individual repressor function.
Keywords: Allostery, hinge domain, repressor, induction, LacI, PurR, molecular dynamics
After decades of investigations of the complex interrelationship between protein structure and function, one truth is clear: No single approach will elucidate the fundamental laws that govern this process. Each avenue of study—biophysical characterization, structure determination, mutational analysis, computational simulation, and bioinformatics comparisons—provides unique but limited information. However, these methods can be combined to generate new insights, even for "well-studied" systems.
The lactose and purine repressor proteins (LacI and PurR, respectively) have ∼35% sequence identity of their monomers and highly homologous secondary and tertiary structures (Rolfes and Zalkin 1988; Weickert and Adhya 1992; Chuprina et al. 1993; Schumacher et al. 1994; Friedman et al. 1995; Nagadoi et al. 1995; Lewis et al. 1996; Spronk et al. 1999a; Bell and Lewis 2000). The dimeric units of these proteins are also similar, even though LacI has an additional C-terminal sequence that mediates tetramer formation. Not surprisingly, these proteins use homologous domains to carry out similar functions. Both bind DNA via their N-terminal domains (thus repressing transcription of downstream genes), bind a small effector ligand in a cleft between core N- and C-subdomains, and transmit an allosteric message between the two types of binding sites that decreases the DNA binding affinity and relieves repression (Fig. 1A ▶; Platt et al. 1973; Files and Weber 1976; Geisler and Weber 1977; Jovin et al. 1977; Ogata and Gilbert 1978; Meng and Nygaard 1990; Rolfes and Zalkin 1990; Khoury et al. 1991; Alberti et al. 1991, 1993; Chakerian et al. 1991; Chen and Matthews 1992; Choi and Zalkin 1992; Schumacher et al. 1993).
Fig. 1.
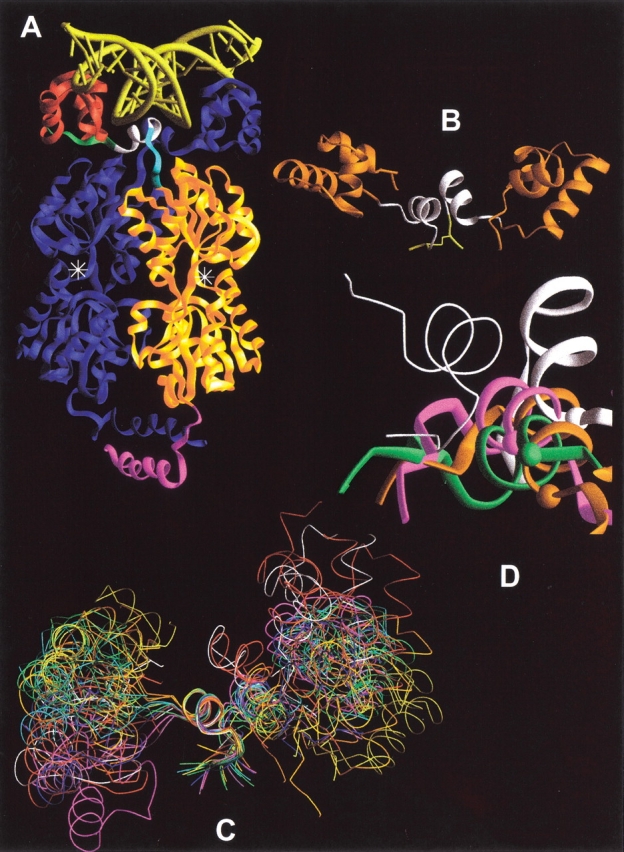
(A) LacI dimer bound to DNA. One half of the LacI tetramer, which is homologous to the PurR dimer, is shown as a Ribbons structure bound to DNA (yellow). One LacI monomer is colored blue. Note that the DNA binding domains "cross over" the core domains of their partners. On the second monomer, the HTH DNA binding region is in red, the N-linker is green, the hinge helix is white, the C-linker is cyan, the core effector binding domain is gold, and the C-terminal helix, which forms a 4-helix bundle in tetrameric LacI, is magenta. PurR does not have a C-terminal helix tetramerization domain. The effector binding site is denoted with the symbol "❉". This structure was created from pdb file 1lbg (Lewis et al. 1996). (B) Starting structure of NlacP2. The HTH domains from LacI are orange (Chuprina et al. 1993), and the hinge helices derived from PurR are white (Schumacher et al. 1994). The C-terminal Gly-Cys-Gly sequence and disulfide bond are in yellow. (C) Structures from the MD simulation of NlacP2. The starting structure of NlacP2 (white), as well as the conformation at 60 ps after "annealing," and representative structures from 100–1600 ps are represented as ribbons aligned on one hinge helix. Starting with red at 60 and 100 ps, the color scheme follows the pattern of colors for visible light (ROYGBIV) to track the time course of the simulation. (D) Hinge•hinge` interfaces sampled during the NlacP2 simulation. The hinges of the starting structure are in white. These are structurally equivalent to the PurR hinges of Fig. 6C ▶–D. The "start" hinge utilized as the alignment reference (and thus essentially identical in all structures) is the wire. The partner hinges at 200 (orange), 900 (green) and 1600 ps (magenta) are shown as wide ribbons. The Cα atoms of A51 are indicated by colored balls to provide a point of reference.
Despite these similarities, the two repressors have some striking functional differences. Most notably, effector binding has opposite allosteric effects in the two proteins: LacI has high affinity for DNA only in the absence of inducer (Riggs et al. 1970b; for review, see Matthews and Nichols 1998), whereas corepressor binding to PurR enhances DNA binding (Meng and Nygaard 1990; Rolfes and Zalkin 1990; Choi and Zalkin 1992). Other functional differences are evident in binding affinities, cooperativities, and kinetics, as well as binding dependences on salt concentration and temperature (Table 1; Riggs et al. 1970a,b; Barkley et al. 1975; Dunaway et al. 1980; O'Gorman et al. 1980; Whitson 1985; Whitson and Matthews 1986; Whitson et al. 1986; Xu et al. 1998; Moraitis et al. 2001). Kinetic differences have been rationalized in terms of the distinct roles these two repressors play in bacterial metabolic regulation (Xu et al. 1998): LacI must be able to quickly respond to changes in nutrient levels (for review, see Matthews and Nichols 1998), whereas PurR regulates a complex set of genes controlling nucleotide synthesis (for review, see Choi and Zalkin 1994). Structurally, the conformational changes in the core domain between the effector-free and effector-bound structures are also very different for the two repressors (Schumacher et al. 1994, 1995; Lewis et al. 1996; Mowbray and Björkman 1999; Bell and Lewis 2000, 2001; Matthews et al. 2000).
Table 1.
Comparison of LacI and purR functions
| LacI | PurR | Reference | ||
| DNA binding | ||||
| Matched conditionsa | Kd (M) | 7.2 × 10−9 | b+G: 1.9 × 10−10 | Xu et al. 1998 |
| c+I: n.d. | no G: 4.0 × 10−8 | |||
| kdiss (s−1) | 3.7 × 10−2 | b+G: 1.2 × 10−3 | ||
| +I: n.d. | no G: 2.8 × 10−2 | |||
| Optimal conditions | kdiss (s−1)d | 6 × 10−4 | Riggs et al. 1970b | |
| e+I: 1 × 10−1 | ||||
| Kd (M)/ | 4.3 × 10−13/ | +G: 3 × 10−12/ | Whitson 1985 | |
| [salt] (M) | 100 mM KClf | 200 mM KClg | Moraitis et al. 2001 | |
| Effector bindingb,d | ||||
| Matched conditionsa | Kd (M)/nh | 0.8 × 10−6/1 | 7.0 × 10−6/1 | Xu et al. 1998 |
| +DNA: 9.8 × 10−7/1 | ||||
| Optimal conditions | Kd (M)/nh | 4 × 10−6/1i | O'Gorman et al. 1980 | |
| +DNA: 8 × 10−5/1.5 | ||||
a Measurements were made in 100 mM HEPES-KOH, pH 7.5, 250 mM potassium glutamate, 150 mM sodium chloride, 10 mM magnesium acetate, and 1 mM EDTA. Operator DNA sequences were 30 bp purF for PurR and 40 bp O1 for LacI. Equilibrium constants for DNA binding are expressed in units of M dimer. Constants for effector binding have the units M monomer.
b G corresponds to the co-repressor guanine. PurR + G is the state with high affinity for DNA.
c I corresponds to inducer IPTG. LacI + I is the state with low affinity for DNA.
d Solution conditions were 10 mM magnesium acetate, 10 mM potassium chloride, 10 mM Tris-HCl, pH 7.4, 10−4 M EDTA, 10−4 M dithiothreitol, 5% dimethylsulfoxide, 50 μg/ml BSA. Operator was λφ80dlac (30 × 106 daltons).
e This experiment measured the DNA off-rate upon the addition of inducer and thus reflects the rate with which LacI responds to its allosteric signal.
f Experimental conditions were 10 mM Tris-HCl, pH 7.4, 10−4 M dithiothreitol, 10−4 M EDTA, 5% dimethylsulfoxide, and 50 μg/ml BSA as well as 100 mM KCl noted in the table. Operator DNA was a 40 bp fragment containing the O1 binding site. The equilibrium DNA binding constant has the units M tetramer.
g Solution conditions included 10 mM Tris-HCl, pH 7.6, 100 μg/ml BSA, and 5% DMSO as well as 200 mM KCl noted in the table. Operator DNA was 30 bp purF. The equilibrium constant for DNA binding was calculated as M dimer.
h Hill coefficient.
i Measurements performed in 10 mM Tris-HCl, 3 × 10−4 M dithiothreitol, 5% glycerol, pH 7.5. Inducer binding is not sensitive to changes in salt concentration. 40 bp operator DNA corresponding to O1 was isolated from pBR345.
Nevertheless, the allosteric conformational change of both repressors appears to involve a helix to coil transition of the hinge region (Spronk et al. 1996, 1999a,Spronk et al. b; Nagadoi et al. 1995; Schumacher et al. 1995; Bell and Lewis 2000). The 10-amino acid hinge region covalently links the DNA binding domain [helix-turn-helix (HTH) plus N-linker] to the larger core domain via the C-linker sequence (Fig. 1A ▶). When bound to operator DNA, the two hinge helices of a dimer are antiparallel, so that the HTH of monomer 1 is positioned over the core domain of monomer 2 (Fig. 1A ▶). NMR studies of the truncated LacI DNA binding domain (amino acids 1–62) show that protein•protein interactions within a dimer are necessary to fold the hinge helices (Spronk et al. 1996, 1999a). However, hinge helix folding for LacI(1–62) also required binding operator DNA (Spronk et al. 1996, 1999a; Kalodimos et al. 2001), and therefore these experiments do not distinguish between protein•protein and protein•DNA contributions to hinge folding. Furthermore, the helix-coil transition of the hinge in intact repressor has been difficult to monitor experimentally. In the absence of DNA, this region exhibits no electron density in X-ray crystallographic studies (Schumacher et al. 1995; Lewis et al. 1996), whereas the small size of the hinge precludes use of other spectroscopic techniques in the context of the complete repressor.
Molecular dynamics (MD) simulations can approximate protein motions that are not experimentally accessible. Here we report the results of exploring contributions to the hinge•hinge` interface using MD simulations of a dimeric, truncated DNA binding domain. These simulations were designed to predict the initial steps in hinge helix unfolding upon dissociation from DNA. By comparison to a previous simulation utilizing the monomeric species (Swint-Kruse et al. 1998), contributions from contacts between specific hinge residues may be assessed apart from protein•DNA interactions. At the time these simulations were begun, no high-resolution coordinates were available for the LacI hinge. Therefore, we designed a chimeric construct (NlacP2) from the NMR structure of the LacI HTH/N-linker (DNA-bound form; Chuprina et al. 1993) and the PurR hinge (DNA-bound form of full-length repressor; Schumacher et al. 1994), plus a terminal Gly-Cys-Gly sequence to create a "dimer" through disulfide bond formation (Fig. 1B ▶). However, the primary sequences of the hinges in the two repressors are actually quite different (Fig. 2 ▶; Beyreuther et al. 1973, 1975; Farabaugh 1978; Rolfes and Zalkin 1988; Weickert and Adhya 1992). Simulations reveal stabilizing interactions between side chains of the PurR hinges that are not possible for analogous positions of the LacI hinges. Additionally, the range of subdomain motions observed in the simulation would be restricted in the full-length protein by the presence of the core domain.
Fig. 2.

(A) LacI and PurR sequence alignments. Residue numbers for LacI are above the one-letter amino acid code; numbers for PurR are below. Structural regions are denoted above the LacI residue numbers. Sites in the N- and C-linkers are denoted with a thin line, and those in the hinge helix are indicated with a bold line. Note that the hinge helix of PurR extends one residue further than that of LacI. The sequences of LacI 141 and PurR 136, which do not align with each other, are also included because these residues make contacts to the N-linker regions. Intervening residues are omitted for clarity. (B) Sequences of LacI and PurR operators. DNA bases are denoted with the one-letter code. LacI binds both the natural operator O1 and an "optimized" sequence Osym with high affinity (Gilbert and Maxam 1973; Sadler et al. 1983; Simons et al. 1984). PurR binds both purF and a palindromic form of that operator (purFsym; Schumacher et al. 1994; Makaroff and Zalkin 1985). LacI crystal structures are in complex with Osym (Bell and Lewis 2000), whereas the PurR structure 1wet was determined in complex with purF (Schumacher et al. 1997). However, this structure must contain some disorder, since the unit cell only contains one purR monomer and one DNA strand even though purF is asymmetric (Schumacher et al. 1997). The NMR structure of LacI(1–62) is also in complex with Osym (1cjg; Spronk et al. 1999a). Numbers at the top of the figure are those of Lac Osym from the pdb file 1efa and are used throughout this report. Numbers at the bottom of this alignment correspond to the base pair numbers used by Schumacher et al. (1994) for PurR structural descriptions.
As information became available from the simulation, new structures with high resolution in the hinge and linker regions were published for intact and truncated LacI bound to DNA (Spronk et al. 1999a; Bell and Lewis 2000), prompting a detailed comparison with the structure available for PurR (Schumacher et al. 1997). The number of cross-subunit/cross-domain and DNA contacts is strikingly different for corresponding regions of LacI and PurR, and the hinge•hinge` juxtapositions also differ significantly. Furthermore, the hinge positions in the NMR structure of the LacI(1–62) are more similar to PurR than to holo-LacI. None of these DNA-bound proteins exhibit the hinge•hinge` interface modeled in the dynamics simulation for DNA-free NlacP2.
To determine which of the observed structural differences are functionally important, we compared these results to the functional properties for the many LacI and PurR mutants in the linker and hinge regions. Additional information was garnered from sequence comparisons within the large LacI/GalR family. We found that cross-domain interactions are most different at positions with very divergent primary sequences, a circumstance we propose is tied to important functional distinctions between LacI and PurR. Therefore, we postulate that the sites identified in this study are locations at which the highly similar structures are "fine-tuned" for their respective, unique functions.
Much effort is currently directed towards utilizing sequence homologies to assign the >30,000 proteins identified by the Human Genome Project to classes of known structures (Venter et al. 2001; Vitkup et al. 2001). However, even this level of structural information will be insufficient to adequately define the exquisitely detailed functions that control intricate life processes. Results from our dynamics simulations and comparison of two closely related members of the LacI/GalR repressor family illustrate the subtle structural divergence that can give rise to functional distinctions critical to cellular life.
Results and Discussion
Molecular dynamics simulations: Contributions to the hinge•hinge` interface
For LacI and PurR, both protein•DNA and protein•protein interactions can contribute to a helix⇌coil conformational change that occurs upon DNA dissociation (Spronk et al. 1996, 1999a,b; Nagadoi et al. 1995; Schumacher et al. 1995; Bell and Lewis 2000). However, protein•protein contributions to this process have been difficult to experimentally isolate. Because previous simulations of a monomeric NlacP are in good agreement with experimental results (Slijper et al. 1997; Swint-Kruse et al. 1998), we have employed MD to isolate the protein•protein contributions to hinge conformational change in a dimeric variant, NlacP2. Dimerization was mediated by a disulfide bond between cysteine residues introduced at position 62, C-terminal to the hinge helix. Cysteine oxidation provides a mechanism for preserving close proximity of the partner hinge helices, which is otherwise facilitated by DNA binding (Spronk et al. 1996, 1999a,b; Kalodimos et al. 2001).
Several results of the molecular dynamics simulations for dimeric NlacP2 parallel those of monomeric NlacP. First, the Cα root mean square deviations for each half of the dimer (HTH plus hinge) are very similar to those of the monomer (data not shown; Swint-Kruse et al. 1998). Second, the HTH and hinge regions of the dimer exhibit motional independence similar to that for the monomer (Fig. 1C ▶); this result can only arise from significant flexibility in the N-linker. However, simulations indicate that the distances between hinge atoms participating in helical hydrogen bonds are ≤2.5 Å for the duration of the trajectory (Fig. 3 ▶, black and gray), in contrast to those observed for the monomer (Fig. 3 ▶, open circles). Therefore, protein•protein interactions can stabilize the hinge helix structure in the absence of DNA.
Fig. 3.

Interatomic distances for atoms involved in hinge helix hydrogen bonds as a function of simulation time. Distances are plotted for the entire 1600 ps simulation; black lines indicate those for one monomer, and gray lines represent the second. Generally, distances are the same for the two monomers and the gray lines obscure the black lines. Small open circles are used to plot the same data from the simulation of NlacP monomer. Arrows at the right of various graphs denote differences in monomer and dimer behavior. The dashed lines at 2.5 Å represent the length of an average hydrogen bond.
Despite the persistence of secondary structure, we were surprised to note a change in quaternary structure for NlacP2. The orientations of the hinge helices moved relative to each other over the course of the simulation. They quickly adopt (by 200 ps; Fig. 1D ▶, orange) a conformation quite different from that of the starting structure (Fig. 1D ▶, white). Yet a third hinge•hinge` conformation is populated around 800 ps. During the second half of the simulation, the interface samples a continuum of structures similar to that at 800 ps, with extremes represented by the interfaces at 900 and 1600 ps (Fig. 1D ▶, green and magenta, respectively). To more closely examine the structural origins of the alternate hinge conformations, positional data from the simulation were plotted for a variety of atoms, and maps diagramming hinge•hinge` contacts were created for representative time points (Fig. 4 ▶).
Fig. 4.

Hinge•hinge` interface contact maps for representative simulation times. Hinge residues of the NlacP2 half-sites are indicated with numbers, and residues on the partner monomer are denoted by the prime symbol following the number. Contacts across the interface are indicated with solid straight lines, and contacts with the Gly-Cys-Gly C-terminal sequences are depicted with curved lines. The dotted lines between 62 and 62` represent the disulfide bond.
For the first new interface at 200 ps, we also compared the structure at this timepoint with the full-length repressors. (Remember that NlacP2 was constructed from the pdb coordinates for the DNA binding domain of LacI and the hinge region extracted from the structure of full-length PurR, which included the core domain.) Interestingly, the core domain would sterically preclude this alternate hinge juxtaposition, raising the possibility that hinge•core` interactions force a less stable hinge•hinge` interface in the full-length protein. Similarly, steric conflicts with the core domain would restrict the range of positions sampled by the HTH domains of NlacP2 (Fig. 1C ▶). This unrestricted motion in truncated protein may provide a structural explanation for why the isolated DNA binding domains of LacI and PurR have lower affinities for DNA than the intact repressors (Riggs et al. 1970c; Geisler and Weber 1977; Jovin et al. 1977; Whitson et al. 1986; Nagadoi et al. 1995; Xu et al. 1998). In addition to the loss of cross-domain contacts, the 200 ps hinge interface could also be influenced by formation of additional interactions with the Gly-Cys-Gly C-terminal sequences (Fig. 4 ▶, curved lines). Such interactions may be facilitated by the flexibility of the C-terminal sequence. However, the converse may also be a factor—that is, the disulfide bond engineered for the simulation might put an energetic strain on this system and cause the alternate hinge•hinge` juxtapositions.
Flexibility in the Gly-Cys-Gly terminus is intriguing when considered in the context of LacI mutants that have glycine insertions in the C-linker to increase the distance between the hinge and core regions (Falcon 1999; Falcon and Matthews 1999, 2000). These mutants exhibited decreased affinity for O1 operator DNA that correlated with the number of inserted glycines. The current simulations suggest a mechanism by which affinity for DNA might be diminished. Additional length in the C-linker may release the steric constraint of the core domain, thus allowing these proteins to adopt a hinge•hinge` interface similar to that seen at 200 ps, which in turn may be less functional for binding DNA.
Whereas formation of the 200 ps hinge•hinge` interface appears to be driven by interactions outside of the hinge region, the conformations in the second half of the simulation seem to be dictated by interactions between the side chains of residues 51–59. One difference between these conformations is that the two helices slide past each other (see Scheme 1 ▶).
Figure 11.

Scheme 1.
Another change between the 200 and 800–1600 ps structures is a rotation of the C terminus of the partner helix (Fig. 1D ▶). Contact maps of the interface reveal that the interaction between L56 and L56`, which have side chains that make critical contacts to the DNA minor groove (Schumacher et al. 1994; Spronk et al. 1999a; Bell and Lewis 2000), is disrupted. Instead, a new hydrogen bond is formed around 900 ps between the side chains of the serines at position 55 (Figs. 4 and 5 ▶ ▶).
Fig. 5.

Motions of NlacP2 side chains as a function of simulation time. The side chains of S55 and S55` form a hydrogen bond around 900 ps. Note that the interaction between S55 O and S55` OH is symmetrically equivalent to that between S55 OH and S55` O. The dashed lines at 2.5 Å represent the length of an average hydrogen bond. Also around 900 ps, the side chains of V52 and V52` are stabilized in close proximity to each other.
This observation highlights an intriguing difference in primary sequences of LacI and PurR hinge regions. The hinge of NlacP2 comes from PurR; S55 of NlacP2 differs significantly from the equivalent position in full-length LacI, which has glutamine and cannot make the same side chain hydrogen bonds. Furthermore, in the continuum of structures from 800–1600 ps, additional interactions form between A51 and N59 of the partner helix (Fig. 4 ▶). The wild-type LacI sequences for these homologous positions are R and K, respectively. We find it unlikely that two positively charged residues would adopt a conformation similar to that of the neutral A and N residues. Therefore, the simulation suggests that primary sequence of the hinge region can have profound effects on the quaternary structure of the hinge•hinge` interface.
One final residue of interest is V52. In LacI and PurR, the hinge helices are antiparallel, and the side chains of 52 and 52` provide the point of closest approach for any single position in the hinge primary sequence. This feature was exploited to design the V52C mutation of LacI, which when oxidized covalently links the hinge helices near their N-termini (Falcon et al. 1997). This mutation was recently incorporated into a truncated version of LacI, LacI(1–62)V52C (Kalodimos et al. 2001). In both cases, disulfide formation greatly increased affinity for DNA. For NlacP2, the proximity of V52 side chains is apparent in the contact maps of the starting and 60 ps structures but disappears in the 200 ps conformation. Intriguingly, this close juxtaposition is restored during the second half of the simulation (Figs. 4 and 5 ▶ ▶).
Structural comparisons of LacI and PurR: Alternate hinge•hinge` arrangements
The fact that hinge•hinge` conformations for the later time points in the simulation are mediated through sequences highly divergent between LacI and PurR prompted a detailed comparison of the two repressors. This study utilized the recently available high-resolution structures for full-length and truncated LacI bound to DNA (Spronk et al. 1999a; Bell and Lewis 2000) and the structure of full-length PurR (Schumacher et al. 1997). Most comparisons of the DNA binding domains of LacI and PurR are accomplished by aligning the repressors on one or both HTH DNA binding domains. Such an alignment is presented in Fig. 6A ▶, in which the structures of full-length LacI and PurR and truncated LacI(1–62) were aligned using amino acids 1–59 of one monomer, encompassing both the HTH and hinge helix. From this perspective, the three structures appear to have almost identical placements of all four subdomains: HTH, hinge, hinge`, and HTH`. However, the default setting for backbone width in many structure viewing programs hides a systematic deviation in the position of hinge` for the three structures. The significance of these differences only becomes apparent when alignments utilize the 11 residues of one hinge helix (Fig. 6B–D ▶).
Fig. 6.
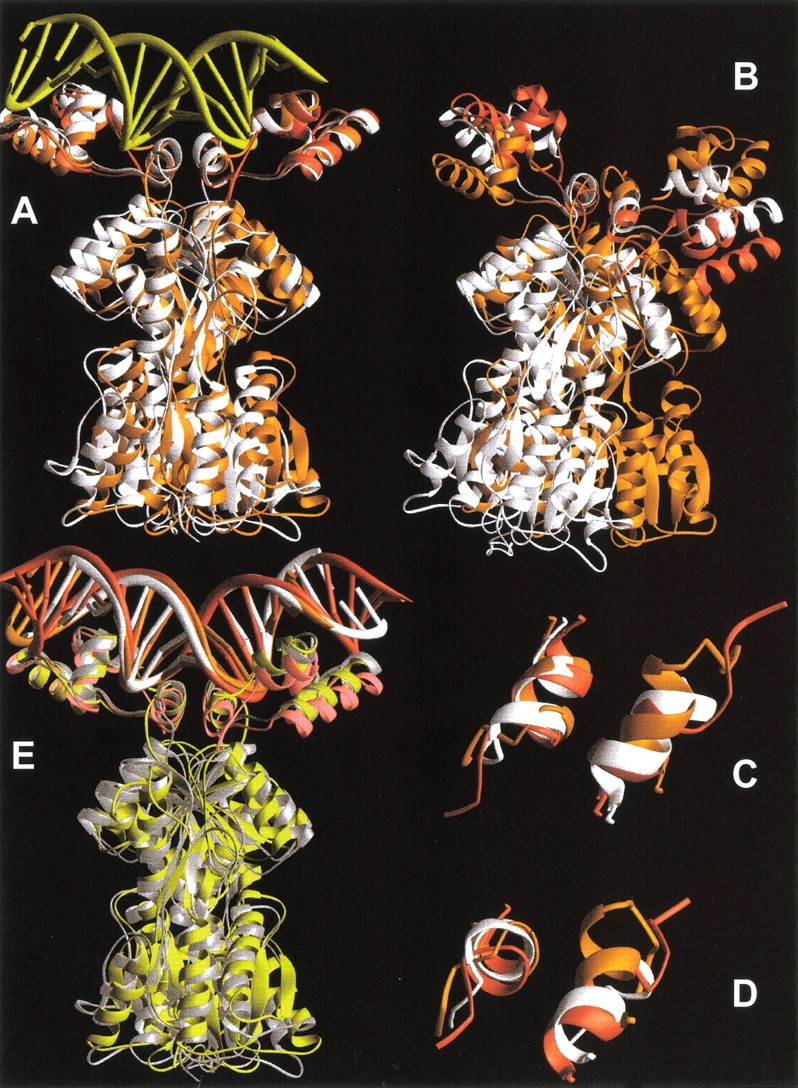
Various alignments of LacI, LacI(1–62), and PurR. The structures of the DNA bound forms of these proteins are plotted as orange, red, and white ribbons, respectively. Structures were created from the pdb files 1efa, 1cjg, and 1wet (Spronk et al. 1999a; Bell and Lewis 2000; Schumacher et al. 1997). (A) Alignment of LacI, LacI(1–62), and PurR on the right HTH/hinge. DNA (yellow) is from the structure of full-length LacI. (B) Alignment of LacI, LacI(1–62), and PurR on only the left hinge helix. (C–D) Two views of aligned hinge helices for LacI, LacI(1–62), and PurR that demonstrate their alternate hinge juxtapositions. (E) Alignment of DNA bound proteins on DNA backbone atoms. Lac Osym bound to LacI is colored orange, Osym bound to LacI(1–62) is red, and purF bound to PurR is white. The respective proteins are shaded yellow, pink, and silver.
The hinge-only alignment produced a number of striking observations (Fig. 6B–D ▶). First, hinge•hinge` quaternary structures are different in full-length LacI (Fig. 6 ▶, orange) and PurR (Fig. 6 ▶, white). Second, the structure of truncated LacI(1–62) (Fig. 6 ▶, red) better aligns with PurR. Interestingly, the hinge•hinge` juxtapositions of the three DNA-bound proteins are not the same as those observed during the simulation of NlacP2 dissociation from DNA. Third, within a monomer, the HTH•hinge orientation is different for the three proteins (Fig. 6B ▶). For LacI and LacI(1–62), this arrangement can only result from flexibility of the N-linker, consistent with behavior observed in the simulation. Additional alignments show that the structures of all the HTH domains are essentially identical (not shown), as are the three DNA structures (Fig. 6E ▶). Although comparison of the positions of HTH relative to HTH` for each structure in the DNA alignment does show some discrepancies (Fig. 6E ▶), these can probably be attributed to the high mobility of these regions (Swint-Kruse et al. 1998; Bell and Lewis 2000). Additional structural differences for the three proteins are observed in the N-linker region connecting the HTH to the hinge and the C-linker that connects the hinge to the core domain (see below).
We have examined multiple other structures of LacI and PurR and verified that these differences are consistently present. Additional structures include: (1) tetrameric LacI bound to DNA, which contains only the positions of Cα atoms (Lewis et al. 1996), (2) the 11 alternate NMR structures reported for LacI(1–62) (Spronk et al. 1999a), (3) wild-type PurR bound to different corepressors and DNA sequences (Schumacher et al. 1994; Glasfeld et al. 1999), and (4) PurR with mutations in the hinge at positions 54 and 55 (Glasfeld et al. 1996, 1999; Arvidson et al. 1998). With the exception of PurR L55M, all other structures have hinge•hinge` orientations that are essentially identical to those of their respective structures shown in Figure 6B–C ▶. Therefore, we attempted to identify which structural features give rise to these differences and whether mutational analysis at identified sites correlates with changed repressor function.
To address the question of structural origin, we investigated several possibilities. Because LacI(1–62) aligns better with PurR than with full-length LacI, the observed disparities in hinge quaternary structure cannot arise simply from changes in hinge primary sequence. Differences between truncated and full-length LacI might arise from the fact that the latter was crystallized bound to antiinducer o-nitrophenyl-β-D-fucopyranoside (ONPF; Bell and Lewis 2000). The LacI•ONPF complex has increased affinity for DNA relative to LacI alone (Riggs et al. 1970b), which may arise from either of two possibilities: (1) LacI•ONPF adopts an alternate protein structure, or (2) binding ONPF produces a population shift between normally accessible states. However, comparison of the previously published low-resolution structure, LacI•DNA, to LacI•ONPF•DNA and LacI(1–62) demonstrates that the full-length structures are very similar (not shown).
Another potential origin of the structural differences in LacI, LacI(1–62), and PurR comes from cross-domain contacts to the rest of the repressor. To more easily visualize these interactions, diagrams of the interfaces are presented in Figures 7, 8, and 9 ▶ ▶ ▶. We first looked for differences in the hinge•hinge` interface. Whereas full-length LacI has more of these interactions than does PurR (Fig. 7A ▶, dashed lines), the LacI(1–62) hinge•hinge` interface involves the same residues as its parent repressor. However, the lengths of these hydrophobic interactions do change (Fig. 7B ▶, bold dashed lines).
Fig. 7.

Hinge•hinge` interface contact maps for LacI, PurR, and LacI(1–62). Hinge residues of the protein monomers are shown as large black numbers, sites in the partner core domain as large gray numbers, and DNA positions as small black numbers. The prime symbol denotes a residue of the partner monomer or a base of the cognate strand. Contacts across the hinge•hinge` interface are indicated with dashed lines, solid lines represent interactions with the partner core domain, and dotted lines symbolize contacts to DNA. For clarity, DNA positions 12 and 13 occur in two places in this figure. Bold lines (solid, dash, or dot) indicate a difference between two structures represented on each panel—either an increased bond length or additional interaction (see text). (A) LacI versus PurR. Homologous sites align horizontally. For example, LacI 50 is homologous to PurR 48. (B) LacI versus LacI(1–62).
Fig. 8.

Maps of N-linker contacts for LacI, PurR, and LacI(1–62). N-linker residues of the protein monomers are indicated by large black numbers, as are sites on the HTH of the same monomer. Sites in the core domains are shown as large gray numbers, and DNA positions as small black numbers. The prime symbol indicates a residue of the partner monomer or a base of the cognate strand. Solid lines depict interactions to the partner core domain, dashed lines represent intrasubunit HTH contacts, and dotted lines are interactions with DNA. Bold lines (solid, dash, or dot) indicate an additional interaction in one of a pair of proteins. (A) LacI versus PurR. Homologous sites align horizontally. For example, LacI 5 is homologous to PurR site 3. (B) LacI versus LacI(1–62). Additional N-linker contacts to the HTH of the same monomer are indicated on the right of the diagram for LacI(1–62); these occur in place of full-length LacI N-linker interactions with the partner core domain.
Fig. 9.

C-linker contact maps for LacI and PurR. C-linker residues of the protein monomers are shown as large black numbers, and sites in the core domains as large gray numbers. The prime symbol indicates a residue of the partner monomer. Solid lines depict interactions within a subunit or to the partner core domain. Solid bold lines denote interactions that differ between the two repressors. Homologous sites align horizontally. For example, LacI 117` is structurally equivalent to PurR 114`.
More striking are the differences in hinge•core` interactions (Fig. 7A ▶, solid lines). Full-length LacI has more interactions with its partner core domain than does PurR. A critical difference appears to occur at LacI Q55, which interacts with Q117` and R118`. LacI(1–62) is of necessity missing these partner core contacts, and the side chain of Q55 occupies a different location in this structure. Interestingly, the side chain of the PurR homolog S53 is not long enough to make analogous contacts to K114` and R115`. An additional consequence of alternate hinge•hinge` juxtapositions is a change in the hinge•DNA interactions: full-length LacI makes more contacts than either PurR or LacI(1–62) (Fig. 7A ▶, bold dotted lines).
The region of the partner core that interacts with the hinge also contacts a linker region just N-terminal to the hinge (Fig. 8 ▶). Again, the N-linker of full-length LacI has a more extensive interface with the core` than does that for PurR (Fig. 8A ▶). As before, these distinctions appear to derive from differences in primary sequence (Fig. 2 ▶). In this interface, LacI I48 appears to be a key residue and is a much larger side chain than PurR S46. LacI partner core residues H112` and L115` are also larger than PurR S109` and A112`. LacI I48 is again identified as important by comparing LacI and LacI(1–62). For the truncated protein, missing partner core interactions are compensated by additional HTH contacts to residue 48 (Fig. 8B ▶).
A third region that differs significantly between LacI and PurR is the linker between the C-terminus of the hinge and the core domain (Figs. 9 and 10 ▶ ▶). The key difference results from the length of the hinge helices: the PurR helix is one amino acid longer (V56) than that of LacI, which has a helix breaker at G58. In hindsight, this structural deviation adds unanticipated complexity to the design of chimeric NlacP2 and may contribute to the complex behavior observed in the simulation. As a consequence of different helix lengths in the two full-length proteins, the two C-linkers of a PurR dimer are much closer to the center of the repressor than in LacI (Fig. 10 ▶). PurR residues 58–61 make extensive contacts with both the intrasubunit core and that of the partner monomer. The LacI C-linker forms only intrasubunit interactions (Fig. 9 ▶). Interestingly, whereas the absolute number of interactions is similar for the two repressors, the PurR C-linker makes more contacts to the region of the core N-subdomain (residues 114–117) that interacts with the hinge and N-linker.
Fig. 10.

Structures of the C-linkers in LacI and PurR. The ribbon for LacI is colored black; that of PurR is gray. Arrows point to the C-linker structures. The figure was derived from pdb files 1efa and 1wet (Bell and Lewis 2000; Schumacher et al. 1997).
Sequence analysis of LacI and PurR: Correlation with function
Despite their high overall sequence identity (35%), LacI and PurR display significant functional and structural properties. Functionally, LacI and PurR have opposite allosteric modes and very different kinetics of allosteric response (Table 1). Observations from simulations and structural comparisons suggest that intriguing differences in tertiary and quaternary structure arise from discrepancies between the primary sequences of LacI and PurR. To test hypotheses that specific sites are functionally important, mutagenesis experiments are required. Fortuitously, many mutations of LacI and PurR have already been either phenotypically or biochemically characterized. However, mutations of a single protein cannot distinguish whether the site is required for the general function of that fold family or is a specific feature of a unique function. This issue may be resolved by comparing sequence and function across a large family of homologous proteins.
LacI and PurR are members of the LacI/GalR family of bacterial metabolic transcription repressors, which at this writing includes 50 proteins. We surveyed the N-linker, hinge, and C-linker regions of these family members (Table 2). Cross-family analysis of partner sites on the core N-subdomains was not performed; these regions of the repressors have less than 30% sequence identity, which decreases confidence in sequence alignment (Vitkup et al. 2001). Residues corresponding to LacI positions 46, 48, 51, 52, 55, 57, 58, 59, 60, 62, and 63 are highly variable across the family. Variability in protein sequence could derive from either of two very different evolutionary pressures: (1) the site might be structurally and functionally unimportant, and therefore experiences no evolutionary pressure to resist change, or (2) the site serves as a point for functional "fine-tuning" in this extended protein family. We propose that some of the LacI/PurR hinge and linker sites fall into the latter category.
Table 2.
Amino acid frequencies in the N-linker, hinge, and C-linker regions of the LacI/GalR transcription repressor familya
| Amino acid frequency | |||||||||||||||||||
| Hydrophobic | Polar | Charged | |||||||||||||||||
| LacI residue number | A | V | I | L | M | P | F | Y | S | T | N | Q | H | D | E | K | R | G | gap |
| 45 | 3 | 3 | 1 | 30 | 1 | 1 | 2 | 1 | 2 | 2 | 3 | 1 | |||||||
| 46 | 2 | 2 | 1 | 11 | 1 | 6 | 4 | 2 | 2 | 4 | 15 | ||||||||
| 47 | 1 | 2 | 3 | 37 | 1 | 1 | 4 | 1 | |||||||||||
| 48 | 2 | 4 | 5 | 1 | 3 | 5 | 3 | 8 | 5 | 4 | 1 | 8 | 1 | ||||||
| 49 | 1 | 36 | 3 | 2 | 2 | 5 | 1 | ||||||||||||
| 50 | 5 | 5 | 7 | 1 | 28 | 1 | 1 | 2 | |||||||||||
| 51 | 15 | 3 | 1 | 3 | 2 | 1 | 3 | 4 | 3 | 2 | 1 | 2 | 10 | ||||||
| 52 | 5 | 12 | 1 | 4 | 2 | 4 | 1 | 1 | 6 | 1 | 4 | 2 | 1 | 1 | 1 | 4 | |||
| 53 | 34 | 1 | 3 | 1 | 2 | 3 | 3 | 1 | 1 | 1 | |||||||||
| 54 | 6 | 3 | 1 | 1 | 6 | 1 | 31 | 1 | |||||||||||
| 55 | 8 | 2 | 3 | 1 | 1 | 12 | 2 | 2 | 4 | 2 | 1 | 2 | 5 | 5 | |||||
| 56 | 5 | 1 | 33 | 3 | 1 | 1 | 1 | 4 | 1 | ||||||||||
| 57 | 13 | 3 | 1 | 4 | 1 | 6 | 2 | 1 | 3 | 9 | 7 | ||||||||
| 58 | 5 | 5 | 1 | 1 | 10 | 5 | 2 | 3 | 1 | 3 | 2 | 11 | 1 | ||||||
| 59 | 2 | 1 | 5 | 1 | 3 | 1 | 6 | 8 | 13 | 4 | 6 | ||||||||
| 60 | 1 | 1 | 1 | 5 | 3 | 3 | 6 | 2 | 3 | 8 | 11 | 3 | 3 | ||||||
| 61 | 5 | 1 | 1 | 9 | 23 | 1 | 3 | 3 | 1 | 3 | |||||||||
| 62 | 1 | 1 | 1 | 1 | 1 | 7 | 5 | 2 | 2 | 5 | 2 | 12 | 4 | 3 | 3 | ||||
| 63 | 3 | 5 | 5 | 2 | 1 | 1 | 4 | 17 | 2 | 3 | 4 | 3 | |||||||
a A BLAST search against SwissProt (Altschul et al. 1990; Bairoch and Apweiler 2000) identified 81 proteins with reasonable homology to LacI. Of these, 50 proteins contain both a DNA binding domain and full-length core and are included in this table.
b Bold numbers correspond to the amino acid residues of LacI.
c Underlined numbers correspond to the amino acid residues of PurR.
To differentiate between these two possibilities, we combined the sequence analysis with comparison of the LacI and PurR structural data, MD simulations, and phenotypic and biochemical analyses of mutations in this region. Compiled results are presented in Table 3. Sites with highly variable amino acid residues are noted (Table 3, "V"). Any variable site "V" must also demonstrate mutational sensitivity and structural difference in order to be a candidate for providing structural fine-tuning of the LacI and PurR functions. The five sites that meet these three criteria are discussed below. (Note that variation in the absence mutational sensitivity or structural distinction between LacI and PurR may be functionally important for other family members.)
Table 3.
Summary of results for hinge and linker residues

a Y, yes. N, no.
b V, variable. C, conserved.
c R, required by most family members for common structural/functional roles. FT, Site utilized by specific repressors to fine-tune individual functions.
d Suckow et al. 1996. Phenotypic analysis of LacI mutants.
e Lu et al. 1998. Phenotypic and biophysical analysis of PurR mutants.
f Choi and Zalkin 1994. Biochemical and spectral analysis of PurR mutants.
g Falcon 1999. Biophysical analysis of LacI mutants.
h Glasfeld et al. 1996, 1999. Structural determination and biochemical analysis of PurR mutants.
i Falcon and Matthews 1999, 2000. Biophysical analysis of LacI mutants.
j Betz 1987. Biophysical analysis of LacI mutants.
Finally, the residues of the linkers and hinge with little variability across the LacI/GalR family (Table 3, "C") are very sensitive to mutations, consistent with typical analyses of multiple protein alignments. These residues make very similar intraresidue contacts in LacI and PurR (Figs. 7A and 8A ▶ ▶), consistent with their conserved functional roles and illustrating that conserved residues do not participate in functional fine-tuning.
N-linker residues that fine-tune function
Structural analyses indicate that the difference in primary sequence at homologous positions LacI I48/PurR S46 may be critical for functional distinction between the two repressors. Mutational analysis gives a similar result: phenotypic characterization of LacI I48S and many other mutations at this position demonstrate a loss in repressor function (Suckow et al. 1996). Perhaps altering this key residue in the N-linker•core` network of interactions effectively locks the protein in its "induced" state, with low affinity for DNA. (Since both the HTH DNA binding domain and the core domain maintain folded structures as isolated domains (Friedman et al,1995; Chuprina et al. 1993), we find unlikely the possibility that mutations at 48 disrupt the structure of the entire protein.) Interestingly, substitutions of LacI I48 partners—112`, 115`, 116`, and 118`—have little or no effect on repression. Mutations in the cluster of interactions around PurR 46 show an opposite pattern. S46A has no effect on PurR repression, whereas S46G diminishes DNA binding less than 100-fold. However, PurR R115A exhibits no measurable binding in vitro and little repression in vivo (Lu et al. 1998), reflecting the importance of this residue in maintaining functional structure. These results suggest an important transposition: I48 is a pivotal residue in LacI allosteric communication, whereas R115 is key in PurR.
C-linker residues that fine-tune function
A similar line of reasoning indicates that the N- and C-linkers may be differentially important to allosteric communication in the two repressors, with the N-linker more critical for LacI and the C-linker for PurR. First, the C-linker in PurR makes more contacts to the core 115 region than it does in LacI (Fig. 9 ▶). In fact, the C-linker of LacI makes no cross-monomer contacts. This hypothesis can be tested by mutagenesis; one might expect the PurR C-linker to be more sensitive to substitution than that of LacI. Although this region has not yet been targeted for mutagenesis in PurR, data for LacI are consistent with this proposal (Table 3).
An alternate hypothesis for how the different C-linker structures contribute to differences in LacI and PurR allostery is that the C-linkers of the two proteins receive allosteric information from different regions of the core domain. The C-linker (in the DNA-bound structures) has many more contacts to LacI core residues 91–95 than are observed for PurR 89–93 (Fig. 9 ▶). In LacI, these core residues form part of the monomer–monomer interface of the inducer-bound structure, but not the DNA-bound structure (Lewis et al. 1996; Bell and Lewis 2000; Swint-Kruse et al. 2001). In contrast, for PurR, these residues participate in the interface of both conformations (Schumacher et al. 1995, 1997; Swint-Kruse et al. 2001). Phenotypic analysis indicates that mutations at LacI 92, 94, and 95 render the repressor insensitive to inducer (Suckow et al. 1996), and Pace et al. (1997) postulate that this region is critical to allosteric response. However, parsing disruption of allosteric communication from a structural effect that precludes requisite conformational changes is difficult. Nonetheless, in support of this hypothesis, the contributions from the LacI C-linker are suggested by mutational sensitivity of S61 (Table 3).
Hinge residues that fine-tune function
Likewise, data for residues in the hinge regions were correlated to identify any sites that might differentiate the functions of LacI and PurR. The residue implicated as structurally important by both molecular dynamics simulations and contact maps, LacI Q55, is at the edge of the web of cross-domain interactions (see Fig. 4 ▶ in Bell and Lewis 2000). The PurR residue at this position is S53, and LacI Q55S does diminish (but not abrogate) repression (Suckow et al. 1996). In fact, LacI position 55 shows little sensitivity to a variety of mutations (Suckow et al. 1996; Falcon 1999). Conversely, PurR mutations at position 53 to I, R, or V abolish the ability of this protein to repress, but C, A, or G substitutions have lesser effects (Choi and Zalkin 1994). Since PurR S53 does not participate in any long-range intraresidue interactions (Fig. 7A ▶), one possible conclusion from these observations is that PurR requires a small, unbranched side chain at position 53. In contrast, LacI can accommodate a range of larger residues, and the additional Q55•core` interactions are a gratuitous result.
Two other highly variable sites in the LacI/GalR family are at LacI positions 57 and 58 (Table 2), which make different hinge•DNA contacts in LacI and PurR. The PurR mutation K55A (homologous to LacI A57) has been the subject of much crystallographic and functional analysis that demonstrates that this residue contributes to specificity of DNA binding (Glasfeld et al. 1996, 1999). Consistent with this result, and with the fact that it makes many DNA contacts, substitutions at LacI A57 and G58 abolish repression (Suckow et al. 1996). Given the variability at this position across the LacI/GalR family, these may be additional points for structurally fine-tuning the DNA binding function.
Conclusions
Flexibility in repressor structure: Cross-domain and cross-DNA interactions dictate structure and allosteric function
Several structure-function models have been proposed for the DNA binding domain of LacI. Spronk et al. (1999b) postulated a model that correlates LacI DNA binding with the spacing of operator half-sites. Hinge helix formation, and thus high-affinity binding, is precluded if half-sites are too far to allow hinge•hinge` interactions, or, conversely, overlapping half-sites can sterically preclude binding by both hinge regions (Spronk et al. 1999b). More recently, Bell and Lewis (2000) proposed that contacts between the N-linker and its partner core domain are important for LacI allosteric response. Finally, Falcon and Matthews (1999, 2000, 2001) demonstrated that the allosteric response of various LacI hinge mutants is dependent upon operator DNA sequence.
Results presented herein for the molecular dynamics simulations of NlacP2 and structural comparisons of LacI, LacI(1–62), and PurR indicate that flexibility of the N-linker allows different cross-domain contacts in the context of different full-length proteins. (This observation should be taken as a cautionary note when examining the structures of isolated protein domains.) These observations can be integrated with previous models to formulate one theory of LacI allosteric communication. In this model, the core domain and DNA serve as templates while flexibility between the HTH/N-linker/hinge/C-linker regions of the repressor dimer allows optimization of both protein•protein and protein•DNA contacts. When these contacts include the N- or C-linker to core interactions that appear to transmit allosteric response, effector binding can elicit changes in DNA affinity.
Mutational studies of a single protein cannot distinguish between a contribution to conserved function and involvement with a unique function. Differentiating between these possibilities requires the context of family members with similar structures and functions. The current structural comparisons, in combination with mutational studies of LacI and PurR and sequence analysis of the LacI/GalR family, suggest that the family utilizes sequence variability to fine-tune a unique allosteric response for each member. Interestingly, whereas the molecular dynamics and structural studies appear to indicate that hinge•hinge` interactions contribute to allosteric response, mutagenesis indicates that the N- and C-linkers probably play a central role.
The observations presented here provide a means for testing the proposed models for allostery in this family. The disrupted allosteric communication for various LacI hinge/linker mutants when bound to particular operator DNA sequences should be restored by second site mutations in the linkers and/or core domains. Exploration of these issues will be of significant future interest.
Materials and methods
Molecular dynamics
These simulations are part of an ongoing effort to match dynamics simulations with biochemical experiments. To complete simulations on a reasonable timescale, only the small DNA binding domains of dimeric LacI were used. Two domains are required to encompass a complete DNA binding site. The design of the hybrid protein NlacP2 was reported in Swint-Kruse et al. (1998). The sequence of each monomer is: MKPVT5 LYDVA10 EYAGV15 SYQTV20 SRVVN25 QASHV30 SAKTR35 EKVEA40 AMAEL45 NYIPS50 AVARS55 LKVNH60 GCG63. The side chain of Cys62 was modeled as a disulfide bond with Cys62`, in order to covalently connect the two polypeptides.
Molecular dynamics simulations parallel those of monomeric NlacP (Swint-Kruse et al. 1998). These simulations used an Ewald sum to model long-range electrostatic interactions (de Leeuw et al. 1980; Smith and Pettitt 1995). For efficient calculations, a balance between box volume and shape must be determined. By placing the protein on the diagonal, box size could be reduced and its shape better approximate a cube. This goal was accomplished by rotating the protein 37° around both the Y and Z axes. The final box size was 53 nm × 47 nm × 49 nm. Although a noncubic box can limit the time available before the simulation is disturbed by spontaneous reorientation, this was not a factor for a system of this size. The protein was solvated with TIP3P waters (Jorgensen et al. 1983), and replicas of the NlacP2 images were buffered by at least three water molecules in all directions. No water molecules had oxygen atoms initially within 2.3 Å of a heavy atom in the protein. During the following minimization and trial MD simulations, additional water molecules were randomly added as needed to adjust the pressure of the system near 1 atm. The final number of waters was 3510, and the total number of atoms in the simulation was 12,442.
We modeled an ionic atmosphere with a neutralizing background with equal and opposite sign to the total charge on the monomer (Tosi 1964). The energy of the system was minimized with approximately 50 steps of steepest descent. The first 60 ps of simulations included alternate rounds of "annealing": first, the protein was fixed and 5 ps of dynamics simulations performed at 300 K. Next, NlacP2 dynamics were simulated at 100 K for 5 ps whereas the solvent was fixed. These iterations were repeated twice more at 5 ps intervals while the simulated temperature of the protein dynamics was increased 100 K per cycle with random velocity assignments, to a final temperature of 300 K. To accommodate temperature changes, a velocity scaling factor "H" was employed at the beginning of each annealing interval (see Equation 1 in Swint-Kruse et al. 1998). The simulation was performed in a microcanonical NVE ensemble with interactions described by the all-atom force field of Charmm23 (MacKerell et al., 1992). A 2 fs time step was used to integrate equations. The Ewald electrostatic convergence parameter, α, was 1.9 nm-1 using all lattice vectors with n2 less than or equal to 64. The Lennard-Jones and real-space electrostatic cutoff distance was 1.5 nm. The SHAKE algorithm (Ryckaert et al. 1977) was used to constrain bonds with a tolerance of 1×10−6 nm. The complete simulation encompassed 1600 ps, and configurations were sampled every 0.1 ps (50 steps). Before calculating root mean square deviations from the simulations, global translational and rotational motions were removed with a least squares fit of the Cα atoms (Smith et al. 1995). These used either the first three helices of the HTH (residues 1–48) or the hinge helix (residues 49–63).
Structural analysis
Structural alignments of LacI and PurR that were based on more than 16 amino acids were accomplished using the web-based program Combinatorial Extension (CE, http://cl.sdsc.edu/ce.html, Shindyalov and Bourne 1998). For alignments on shorter protein segments, the program Molmol was utilized (http://www.mol.biol.ethz.ch/wuthrich/software/molmol/, Koradi et al. 1996). To preserve the correct quaternary structure, the protein pdb files were modified in order to rotate the oligomeric proteins as one unit instead of as individual subunits. For both programs, this was accomplished by setting the "Chain ID" parameter of the pdb file to the same value for all subunits. Alignment with CE also required modifying "SEQRES" lines to indicate that all residues of all subunits were assigned the same chain ID. These changes were restored to original values in the resulting, aligned pdb files as needed. Protein•protein and protein•DNA contacts were identified by a combination of the web-based program Contacts of Structural Units (http://bioinfo.weizmann.ac.il:8500/oca-bin/lpccsu, Sobolev et al. 1999), visual inspection of the structures (Rasmol, http://www.umass.edu/microbio/rasmol/getras.htm), and comparison to the reports describing the structures (Schumacher et al. 1997; Spronk et al. 1999a; Bell and Lewis 2000). Generally, contacts between hydrophilic residues were defined as ≤3.5 Å and contacts between hydrophobic groups as ≤4.5 Å. Protein structures presented in the figures were created with Ribbons (http://sgce.cbse.uab.edu/ribbons/, Carson 1997). A BLAST search (SIB; http://ca.expasy.org/cgi-bin/BLASTEMBnet-CH.pl, Altschul et al. 1990) against SwissProt (http://ca.expasy.org/sprot/, Bairoch and Apweiler 2000) utilizing BLOSUB62 with a 100 protein limit was used to identify current members of the LacI/GalR family and to perform a sequence alignment for comparing the N-linker, hinge, and C-linker primary sequences.
Acknowledgments
This work was supported by grants to K.S.M. from the National Institutes of Health (NIH) (GM22441) and the Robert A. Welch Foundation (C-576) and by grants to B.M.P. from the NIH and the Robert A. Welch Foundation. L.S.K. was supported by a postdoctoral training fellowship from the W.M. Keck Center for Computational Biology (National Library of Medicine LM07093). We thank Dr. Paul E. Smith (Kansas State University) for assistance implementing the dynamics simulation and Dr. Gillian Lynch (The University of Houston) for assistance with data manipulation. Drs. Sarah Bondos and Xin-Xing Tan (Rice University) provided valuable comments on the manuscript.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
LacI, lactose repressor protein
PurR, purine repressor protein
HTH, helix-turn-helix structure of LacI/PurR DNA binding domains
ONPF, o-nitrophenyl-β-D-fucopyranoside
IPTG, isopropyl-β-D-thiogalactopyranoside
MD, molecular dynamics
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.4050102.
References
- Alberti, S., Oehler, S., v. Wilcken-Bergmann, B., Krämer, H., and Müller-Hill, B. 1991. Dimer-to-tetramer assembly of Lac repressor involves a leucine heptad repeat. The New Biologist 3 57–62. [PubMed] [Google Scholar]
- Alberti, S., Oehler, S., von Wilcken-Bergmann, B., and Müller-Hill, B. 1993. Genetic analysis of the leucine heptad repeats of Lac repressor: Evidence for a 4-helical bundle. EMBO J. 12 3227–3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul, S.F., Gish, W., Miller, W., Myers, E.W., and Lipman, D.J. 1990. Basic local alignment search tool. J. Mol. Biol. 215 403–410. [DOI] [PubMed] [Google Scholar]
- Arvidson, D.N., Lu, F., Faber, C., Zalkin, H., and Brennan, R.G. 1998. The structure of PurR mutant L54M shows an alternative route to DNA kinking. Nat. Struct. Biol. 5 436–441. [DOI] [PubMed] [Google Scholar]
- Bairoch, A. and Apweiler, R. 2000. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 28 45–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkley, M.D., Riggs, A.D., Jobe, A., and Bourgeois, S. 1975. Interaction of effecting ligands with lac repressor and repressor-operator complex. Biochemistry 14 1700–1712. [DOI] [PubMed] [Google Scholar]
- Bell, C.E. and Lewis, M. 2000. A closer view of the conformation of the Lac repressor bound to operator. Nat. Struct. Biol. 7 209–214. [DOI] [PubMed] [Google Scholar]
- Bell, C.E. and Lewis, M. 2001. The Lac repressor. A second generation of structural and functional studies. Curr. Opin. Struct. Biol. 11 19–25. [DOI] [PubMed] [Google Scholar]
- Betz, J.L. 1987. Affinities of tight-binding lactose repressors for wild-type and pseudo-operators. J. Mol. Biol. 195 495–504. [DOI] [PubMed] [Google Scholar]
- Beyreuther, K., Adler, K., Fanning, E., Murry, C., Klemm, A., and Geisler, N. 1975. Amino-acid sequence of lac repressor from Escherichia coli. Isolation, sequence analysis and sequence assembly of tryptic peptides and cyanogen bromide fragments. Eur. J. Biochem. 59 491–509. [DOI] [PubMed] [Google Scholar]
- Beyreuther, K., Adler, K., Geisler, N., and Klemm, A. 1973. The amino-acid sequence of lac repressor. Proc. Nat. Acad. Sci. 70 3576–3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson, M. 1997. Ribbons. Methods Enzymol. 277 493–505. [PubMed] [Google Scholar]
- Chakerian, A.E., Tesmer, V.M., Manly, S.P., Brackett, J.K., Lynch, M.J., Hoh, J.T., and Matthews, K.S. 1991. Evidence for leucine zipper motif in lactose repressor protein. J. Biol. Chem. 266 1371–1374. [PubMed] [Google Scholar]
- Chen, J. and Matthews, K.S. 1992. Deletion of lactose repressor carboxyl-terminal domain affects tetramer formation. J. Biol. Chem. 267 13843–13850. [PubMed] [Google Scholar]
- Choi, K.Y. and Zalkin, H. 1992. Structural characterization and corepressor binding of the Escherichia coli purine repressor. J. Bacteriol. 174 6207–6214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, K.Y. and Zalkin, H. 1994. Role of the purine repressor hinge sequence in repressor function. J. Bacteriol. 176 1767–1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuprina, V.P., Rullmann, J.A.C., Lamerichs, R.M.J.N., van Boom, J.H., Boelens, R., and Kaptein, R. 1993. Structure of the complex of lac repressor headpiece and an 11 base-pair half-operator determined by nuclear magnetic resonance spectroscopy and restrained molecular dynamics. J. Mol. Biol. 234 446–462. [DOI] [PubMed] [Google Scholar]
- de Leeuw, S.W., Perram, J.W., and Smith, E.R. 1980. Simulation of electrostatic systems in periodic boundary conditions. I. Lattice sums and dielectric constants. Proc. R. Soc. Lond. A. 373 27–56. [Google Scholar]
- Dunaway, M., Olson, J.S., Rosenberg, J.M., Kallai, O.B., Dickerson, R.E., and Matthews, K.S. 1980. Kinetic studies of inducer binding to lac repressor–operator complex. J. Biol. Chem. 255 10115–10119 [PubMed] [Google Scholar]
- Falcon, C.M. 1999. Role of lac repressor hinge region and operator DNA sequence in complex formation. Ph. D. Thesis, Rice University, Houston, Texas.
- Falcon, C.M. and Matthews, K.S. 1999. Glycine insertion in the hinge region of lactose repressor protein alters DNA binding. J. Biol. Chem. 274 30849–30857. [DOI] [PubMed] [Google Scholar]
- Falcon, C.M. and Matthews, K.S. 2000. Operator DNA sequence variation enhances high affinity binding by hinge helix mutants of lactose repressor protein. Biochemistry 39 11074–11083. [DOI] [PubMed] [Google Scholar]
- Falcon, C.M. and Matthews, K.S. 2001. Engineered disulfide linking the hinge regions within lactose repressor dimer increases operator affinity, decreases sequence selectivity, and alters allostery. Biochemistry, 40 15650–15659. [DOI] [PubMed] [Google Scholar]
- Falcon, C.M., Swint-Kruse, L., and Matthews, K.S. 1997. Designed disulfide between N-terminal domains of lactose repressor disrupts allosteric linkage. J. Biol. Chem. 272 26818–26821. [DOI] [PubMed] [Google Scholar]
- Farabaugh, P.J. 1978. Sequence of the lacI gene. Nature 274 765–769. [DOI] [PubMed] [Google Scholar]
- Files, J.G. and Weber, K. 1976. Limited proteolytic digestion of lac repressor by trypsin. Chemical nature of the resulting trypsin-resistant core. J. Biol. Chem. 251 3386–3391. [PubMed] [Google Scholar]
- Friedman, A.M., Fischmann, T.O., and Steitz, T.A. 1995. Crystal structure of lac repressor core tetramer and its implications for DNA looping. Science 268 1721–1727. [DOI] [PubMed] [Google Scholar]
- Geisler, N. and Weber, K. 1977. Isolation of amino-terminal fragment of lactose repressor necessary for DNA binding. Biochemistry 16 938–943. [DOI] [PubMed] [Google Scholar]
- Gilbert, W. and Maxam, A. 1973. The nucleotide sequence of the lac operator. Proc. Natl. Acad. Sci. 70 3581–3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasfeld, A., Koehler, A.N., Schumacher, M.A., and Brennan, R.G. 1999. The role of lysine 55 in determining the specificity of the purine repressor for its operators through minor groove interactions. J. Mol. Biol. 291 347–361. [DOI] [PubMed] [Google Scholar]
- Glasfeld, A., Schumacher, M.A., Choi, K. -Y., Zalkin, H., and Brennan, R.G. 1996. A positively charged residue bound in the minor groove does not alter the bending of a DNA duplex. J. Am. Chem. Soc. 118 13073–13074. [Google Scholar]
- Jorgensen, W.L., Chandrasekhar, J., Madura, J.D., Impey, R.W., and Klein, M.L. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79 926–935. [Google Scholar]
- Jovin T.M., Geisler N., and Weber K. 1977. Amino-terminal fragments of Escherichia coli lac repressor bind to DNA. Nature 269 668–672. [DOI] [PubMed] [Google Scholar]
- Kalodimos, C.G., Folkers, G.E., Boelens, R., and Kaptein, R. 2001. Strong DNA binding by covalently linked dimeric Lac headpiece: Evidence for the crucial role of the hinge helices. Proc. Natl. Acad. Sci. 98 6039–6044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury A.M., Nick, H.S., and Lu, P. 1991. In vivo interaction of Escherichia coli lac repressor N-terminal fragments with the lac operator. J. Mol. Biol. 219 623–634. [DOI] [PubMed] [Google Scholar]
- Koradi, R., Billeter, M., and Wüthrich, K. 1996. MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graph. 14 51–55. [DOI] [PubMed] [Google Scholar]
- Lewis, M., Chang, G., Horton, N.C., Kercher, M.A., Pace, H.C., Schumacher, M.A., Brennan, R.G., and Lu, P. 1996. Crystal structure of the lactose operon repressor and its complexes with DNA and inducer. Science 271 1247–1254. [DOI] [PubMed] [Google Scholar]
- Lu, F., Brennan, R.G., and Zalkin, H. 1998. Escherichia coli purine repressor: Key residues for the allosteric transition between active and inactive conformations and for interdomain signaling. Biochemistry 37 15680 – 15690. [DOI] [PubMed] [Google Scholar]
- MacKerell, A.D., Jr., Bashford, D., Bellott, M., Dunbrack, R.L., Jr., Field, M.J., Fischer, S., Gao, J., Guo, H., Ha, S., Joseph, D., et al. 1992. Self-consistent parameterization of biomolecules for molecular modeling and condensed phase simulations. FASEB J. 6 A143. [Google Scholar]
- Makaroff, C.A. and Zalkin, H. 1985. Regulation of Escherichia coli purF. Analysis of the control regions of a pur regulon gene. J. Biol. Chem. 260 10378–10387. [PubMed] [Google Scholar]
- Matthews, K.S., Falcon, C.M., and Swint-Kruse, L. 2000. Relieving repression. Nat. Struct. Biol. 7 184–187. [DOI] [PubMed] [Google Scholar]
- Matthews, K.S. and Nichols, J.C. 1998. Lactose repressor protein: Functional properties and structure. Prog. Nucleic Acid Res. Mol. Biol. 58 127–164. [DOI] [PubMed] [Google Scholar]
- Meng, L.M. and Nygaard, P. 1990. Identification of hypoxanthine and guanine as the co-repressors for the purine regulon genes of Escherichia coli. Mol. Microbiol. 4 2187–2192. [DOI] [PubMed] [Google Scholar]
- Moraitis, M.I., Xu, H., and Matthews, K.S. 2001. Ion concentration and temperature dependence of DNA binding: Comparison of PurR and LacI repressor proteins. Biochemistry 40 8109–8117. [DOI] [PubMed] [Google Scholar]
- Mowbray, S.L. and Björkman, A.J. 1999. Conformational changes of ribose-binding protein and two related repressors are tailored to fit the functional need. J. Mol. Biol. 294 487–499. [DOI] [PubMed] [Google Scholar]
- Nagadoi, A., Morikawa, S., Nakamura, H., Enari, M., Kobayashi, K., Yamamoto, H., Sampei, G., Mizobuchi, K., Schumacher, M.A., Brennan, R.G., and Nishimura, Y. 1995. Structural comparison of the free and DNA-bound forms of the purine repressor DNA-binding domain. Structure 3 1217–1224. [DOI] [PubMed] [Google Scholar]
- Ogata, R.T. and Gilbert, W. 1978. Contacts between the lac repressor and the thymines in the lac operator. Proc. Natl. Acad. Sci. 75 5851–5854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Gorman, R.B., Rosenberg, J.M., Kallai, O.B., Dickerson, R.E., Itakura, K., Riggs, A.D., and Matthews, K.S. 1980. Equilibrium binding of inducer to lac repressor-operator DNA complex. J. Biol. Chem. 255 10107–10114. [PubMed] [Google Scholar]
- Pace, H.C., Kercher, M.A., Lu, P., Markiewicz, P., Miller, J.H., Chang, G., and Lewis, M. 1997. Lac repressor genetic map in real space. Trends Biochem. Sci. 22 334–339. [DOI] [PubMed] [Google Scholar]
- Platt, T., Files, J.G., and Weber, K. 1973. Lac repressor. Specific proteolytic destruction of the NH2-terminal region and loss of the deoxyribonucleic acid-binding activity. J. Biol. Chem. 248 110–121. [PubMed] [Google Scholar]
- Riggs, A.D., Bourgeois, S., and Cohn, M. 1970a. The lac repressor-operator interaction. 3. Kinetic studies. J. Mol. Biol. 53 401–417. [DOI] [PubMed] [Google Scholar]
- Riggs, A.D., Newby, R.F., and Bourgeois, S. 1970b. lac repressor-operator interaction II. Effect of galactosides and other ligands. J. Mol. Biol. 51 303–314. [DOI] [PubMed] [Google Scholar]
- Riggs, A.D., Suzuki, H., and Bourgeois, S. 1970c. Lac repressor-operator interaction. I. Equilibrium studies. J. Mol. Biol. 48 67–83. [DOI] [PubMed] [Google Scholar]
- Rolfes, R.J. and Zalkin, H. 1988. Escherichia coli gene purR encoding a repressor protein for purine nucleotide synthesis. Cloning, nucleotide sequence, and interaction with purF operator. J. Biol. Chem. 263 19653–19661. [PubMed] [Google Scholar]
- Rolfes, R.J. and Zalkin, H. 1990. Purification of the Escherichia coli purine regulon repressor and identification of corepressors. J. Bacteriol. 172 5637–5642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryckaert, J.-P., Ciccotti, G., and Berendsen, H.J.C. 1977. Numerical integration of the Cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 23 327–341. [Google Scholar]
- Sadler, J.R., Sasmor, H., and Betz, J.L. 1983. A perfectly symmetric lac operator binds the lac repressor very tightly. Proc. Natl. Acad. Sci. 80 6785–6789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher, M.A., Choi, K.Y., Zalkin, H., and Brennan, R.G. 1994. Crystal structure of LacI member, PurR, bound to operator DNA: Minor groove binding by α helices. Science 266 763–770. [DOI] [PubMed] [Google Scholar]
- Schumacher, M.A., Choi, K.Y., Lu, F., Zalkin, H., and Brennan, R.G. 1995. Mechanism of corepressor-mediated specific DNA binding by the purine repressor. Cell 83 147–155. [DOI] [PubMed] [Google Scholar]
- Schumacher, M.A., Glasfeld, A., Zalkin, H., and Brennan, R.G. 1997. The X-ray structure of the PurR-guanine-purF operator complex reveals the contributions of complementary electrostatic surfaces and a water-mediated hydrogen bond to corepressor specificity and binding affinity. J. Biol. Chem. 272 22648–22653. [DOI] [PubMed] [Google Scholar]
- Schumacher, M.A., Macdonald, J.R., Björkman, J., Mowbray, S.L., and Brennan, R.G. 1993. Structural analysis of the purine repressor, an Escherichia coli DNA binding protein. J. Biol. Chem. 268 12282–12288. [PubMed] [Google Scholar]
- Shindyalov, I.N. and Bourne, P.E. 1998. Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng. 11 739–747. [DOI] [PubMed] [Google Scholar]
- Simons, A., Tils, D., von Wilcken-Bergmann, B., and Müller-Hill, B. 1984. Possible ideal lac operator: Escherichia coli lac operator-like sequences from eukaryotic genomes lack the central G:C pair. Proc. Natl. Acad. Sci. 81 1624–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slijper, M., Boelens, R., Davis, A.L., Konings, R.N.H., van der Marel, G.A., van Boom, J.H., and Kaptein, R. 1997. Backbone and side chain dynamics of lac repressor headpiece(1–56) and its complex with DNA. Biochemistry 36 249–254. [DOI] [PubMed] [Google Scholar]
- Smith, P.E. and Pettitt, B.M. 1995. Efficient Ewald electrostatic calculations for large systems. Comput. Phys. Commun. 91 339–344. [Google Scholar]
- Smith, P.E., van Schaik, R.C., Szyperski, T., Wüthrich, K., and van Gunsteren, W.F. 1995. Internal mobility of the basic pancreatic trypsin inhibitor in solution: A comparison of NMR spin relaxation measurements and molecular dynamics simulations. J. Mol. Biol. 246 356–365. [DOI] [PubMed] [Google Scholar]
- Sobolev, V., Sorokine, A., Prilusky, J., Abola, E.E. and Edelman, M. 1999. Automated analysis of interatomic contacts in proteins. Bioinformatics 15 327–332. [DOI] [PubMed] [Google Scholar]
- Spronk, C.A.E.M., Slijper, M., van Boom, J.H., Kaptein, R. and Boelens, R. 1996. Formation of the hinge helix in the lac repressor is inducer upon binding to the lac operator. Nat. Struct. Biol. 3 916–919. [DOI] [PubMed] [Google Scholar]
- Spronk, C.A.E.M., Bonvin, A.M.J.J., Radha, P.K., Melacini, G., Boelens, R., and Kaptein, R. 1999a. The solution structure of Lac repressor headpiece complexed to a symmetrical lac operator. Structure 7 1483–1492. [DOI] [PubMed] [Google Scholar]
- Spronk, C.A.E.M., Folkers, G.E., Noordman, A.-M.G.W., Wechselberger, R., van den Brink, N., Boelens, R., and Kaptein, R. 1999b. Hinge-helix formation and DNA bending in various lac repressor-operator complex. EMBO J. 18 6472–6480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suckow, J., Markiewicz, P., Kleina, L.G., Miller, J., Kisters-Woike, B., and Müller-Hill, B. 1996. Genetic studies of the Lac repressor. XV. 4000 single amino acid substitutions and analysis of the resulting phenotypes on the basis of the protein structure. J. Mol. Biol. 261 509–523. [DOI] [PubMed] [Google Scholar]
- Swint-Kruse, L., Elam, C.R., Lin, J.W., Wycuff, D.R., and Matthews, K.S. 2001. Plasticity of quaternary structure: Twenty-two ways to form a LacI dimer. Protein Sci. 10 262–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swint-Kruse, L., Matthews, K.S., Smith, P.E., and Pettitt, B.M. 1998. Comparison of simulated and experimentally determined dynamics for a variant of the LacI DNA binding domain, NlacP. Biophys. J. 74 413–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosi, M.P. 1964. Cohesion of ionic solids in the Born model. In Solid State Physics (eds F. Seitz and D. Turnbull), Vol. 16, pp. 1–12. Academic Press, New York.
- Venter, J.C., Adams, M.D., Myers, E.W., Li, P.W., Mural, R.J., Sutton, G.G., Smith, H.O., Yandell, M., Evans, C.A., Holt, R.A., et al. 2001. The sequence of the human genome. Science 291 1304–1351. [DOI] [PubMed] [Google Scholar]
- Vitkup, D., Melamud, E., Moult, J. and Sander, C. 2001. Completeness in structural genomics. Nat. Struct. Biol. 8 559–566. [DOI] [PubMed] [Google Scholar]
- Weickert, M.J. and Adhya, S. 1992. A family of bacterial regulators homologous to Gal and Lac repressors. J. Biol. Chem. 267 15869–15874. [PubMed] [Google Scholar]
- Whitson, P.A. 1985. The lactose repressor-operator DNA interaction: Chemical and physical studies of the complex. Ph. D. Thesis, Rice University, Houston, Tx.
- Whitson, P.A. and Matthews, K.S. 1986. Dissociation of the lactose repressor-operator DNA complex: Effects of size and sequence context of operator-containing DNA. Biochemistry 25 3845–3852. [DOI] [PubMed] [Google Scholar]
- Whitson, P.A., Olson, J.S., and Matthews, K.S. 1986. Thermodynamic analysis of the lactose repressor-operator interaction. Biochemistry 25 3852–3858. [DOI] [PubMed] [Google Scholar]
- Xu, H., Moraitis, M., Reedstrom, R.J., and Matthews, K.S. 1998. Kinetic and thermodynamic studies of purine repressor binding to corepressor and operator DNA. J. Biol. Chem. 273 8958–8964. [DOI] [PubMed] [Google Scholar]