

Abstract
A major goal of this paper was to estimate a dynamic range of equilibrium constant for the opening of a single peptide bond in a model protein, bovine pancreatic trypsin inhibitor (BPTI). Ten mutants of BPTI containing a single Xaa→Met substitution introduced in different parts of the molecule were expressed in Escherichia coli. The mutants were folded, purified to homogeneity, and cleaved with cyanogen bromide to respective cleaved forms. Conformation of the intact mutants was similar to the wildtype, as judged from their circular dichroism spectra. Substantial conformational changes were observed on the chemical cleavage of three single peptide bonds—Met46-Ser, Met49-Cys, and Met53-Thr—located within the C-terminal helix. Cleavage of those peptide bonds caused a significant destabilization of the molecule, with a drop of the denaturation temperature by 56.4°C to 68°C at pH 4.3. Opening of the remaining seven peptide bonds was related to a 10.8°C to 39.4°C decrease in Tden. Free energies of the opening of 10 single peptide bonds in native mutants (ΔGop,N) were estimated from the thermodynamic cycle that links denaturation and cleavage free energies. To calculate those values, we assumed that the free energy of opening of a single peptide bond in the denatured state (ΔGop,D) was equal to −2.7 kcal/mole, as reported previously. Calculated ΔGop,N values in BPTI were in the range from 0.2 to 10 kcal/mole, which was equivalent to a >1 million–fold difference in equilibrium constants. The values of ΔGop,N were the largest for peptide bonds located in the C-terminal helix and significantly lower for peptide bonds in the β-structure or loop regions. It appears that opening constants for single peptide bonds in various proteins span across 33 orders of magnitude. Typical equilibrium values for a single peptide bond opening in a protein containing secondary structure elements fall into negligibly low values, from 10−3 to 10−8, and are efficient to ensure stability against proteolysis.
Keywords: Bovine pancreatic trypsin inhibitor, calorimetry, denaturation, hydrolysis, peptide bond, thermodynamic stability
Proteolysis of a single peptide bond is one of the key postranslational modifications of proteins, and it is responsible for maturation of numerous inactive preproproteins and proproteins (Neurath 1991). When uncontrolled, proteolysis can lead to numerous pathological processes, including production of the Alzheimer Aβ-peptide (Selkoe 2001), asthma, other allergic or inflammatory disorders (Seife 1997), and thrombosis (Stubbs and Bode 1994). Specific proteolytic cleavage can also be a useful analytical tool in protein sequencing or protein folding studies (Fontana et al. 1997). However, our current understanding of structural and thermodynamic factors governing peptide bond cleavage reaction is far from complete.
Limited proteolysis occurs in protein regions that contain a proteinase-specific amino acid sequence accessible for the attacking proteinase (Otlewski et al. 1994; Hubbard 1998). Conformational requirements seem to be equally important, as proteinase-sensitive sites show high mobility of a local polypeptide segment and are localized in highly exposed loops or in peptide segments connecting protein domains (Fontana, et al. 1993). Because of high rigidity, secondary structure elements are not susceptible to proteinase action (Krokoszynska and Otlewski 1996). The sensitivity of a denatured protein to proteolysis is dramatically different. In this state, the whole protein molecule shows high flexibility, and peptide bonds are easily accessible to a proteinase active site (Hubbard 1998).
Limited proteolysis of a native protein is usually considered to be an irreversible event, and the equilibrium constant between the hydrolyzed and intact forms, called the hydrolysis constant (Khyd,N), can be determined only occasionally. In fact, the P1-P1' (notation of Schechter and Berger 1967) reactive site in canonical inhibitors of serine proteinases is a unique peptide bond for which Khyd,N is not far from unity and can be known with high accuracy (Ardelt and Laskowski 1991; Otlewski and Zbyryt 1994). After proteolytic cleavage, there are only local conformational changes in the neighborhood of the P1-P1' peptide bond, as shown by structural analysis (Musil et al. 1991; Betzel et al. 1993; Walkenhorst et al. 1994; Cai et al. 1995). In the case of the protein inhibitors, the value of Khyd,N reflects the strength of noncovalent interactions, maintaining the local conformation around the P1-P1' peptide bond as deduced from Khyd,N values determined for numerous single amino acid–substituted variants of ovomucoid third domain (Ardelt and Laskowski 1991).
In our previous paper, we estimated the values of Khyd,N for four peptide bonds in two inhibitors, soybean trypsin inhibitor and bovine pancreatic trypsin inhibitor (BPTI; Krokoszynska and Otlewski 1996). Comparison of stability of intact and cleaved forms allowed us to calculate hydrolysis constants using thermodynamic cycle–linking reactions of hydrolysis and denaturation. We found that Khyd,N values for the Lys15-Ala (P1-P1') and Met52-Arg (central part of α-helix) peptide bonds in BPTI differed ∼1 million times. We also estimated the hydrolysis constant for different single peptide bonds in denatured protein inhibitors (Khyd,D). In the present report, to study the range Khyd,N values can adopt in a single protein, we extended our analysis to 10 peptide bonds, covering all different segments of BPTI structure.
BPTI seems to be a particularly attractive model for such purposes as it contains different secondary structure elements (α-helix, 310 helix, antiparallel β-sheet, and irregular structure) and is stabilized by three disulphide bonds (Cys5-Cys55, Cys30-Cys51, and Cys14-Cys38). BPTI denaturation follows a two-state reversible thermal transition, and its denaturation temperature equals 86°C at pH 2.0 (Moses and Hinz 1983; Makhatadze et al. 1993). Such extraordinary stability provides a useful dynamic range to measure thermal transitions for cleaved forms, which are expected to occur at significantly lower temperatures. Native BPTI wildtype is known to be extremely resistant to proteolysis (Fritz and Wunderer 1983). The only proteinase-sensitive peptide bond is the reactive site: Lys15-Ala, which can be extremely slowly hydrolyzed by trypsin (Siekman et al. 1988).
Results
Hydrolysis of a given single peptide bond in a globular protein cannot be easily performed, because proteinase action is limited to very few peptide bonds, and it is dictated by sequence specificity (Fontana 1989). We introduced a single Met residue at 10 positions of BPTI to generate unique cyanogen bromide (CNBr) cleavage sites using an approach similar to that of Ladurner et al. (1997), who studied complementation of peptide fragments of chymotrypsin inhibitor 2. The mutated sites cover different parts of BPTI structure (Fig. 1 ▶). In all mutants, naturally occurring Met52 was replaced by Leu residue to enable chemical cleavage at a single mutated site. The Met52→Leu mutation in BPTI does not lead to significant stability changes (Yu et al. 1995). After the cleavage reaction, all mutants still formed a single chain molecule, as all Met residues were introduced within Cys5-Cys55 disulphide bond.
Fig. 1.

Ribbon representation of the spatial structure of bovine pancreatic trypsin inhibitor (BPTI; Wlodawer et al. 1984). The cleavage sites, including Lys15-Ala and Met52-Arg peptide bonds studied previously (Krokoszynska and Otlewski 1996), are shown.
Intact mutants were purified to homogeneity by reverse-phase C18 high-pressure liquid chromatography (HPLC). The cleaved forms were additionally purified on a C18 column to remove the remaining intact form. All intact mutants were active as proteinase inhibitors, and their concentration could be determined by titration with trypsin. The preparations of intact forms were ∼100% active compared with absorbance-determined concentration. For all cleaved forms, the concentration was determined from calculated molar absorbance coefficients (Pace et al. 1995).
Conformation of the mutants
Introduction of a single Met residue in different locations of BPTI structure did not cause significant conformational changes, as judged from respective circular dichroism (CD) spectra. Spectra of the intact mutants looked very similar both in the far- and near-ultraviolet, indicating preservation of secondary structure and tertiary interactions, respectively (Fig. 2A,B).
Compared with the intact forms, the most severe changes in CD spectra were observed for three mutants that were cleaved within the C-terminal α-helix region at positions 46, 49, and 53. Conformational changes accompanying cleavage reaction could be easily observed on CD differential spectra (Fig. 2C ▶). Conversely, Met54-Cys*, cleaved at the C terminus of this helical region just before cysteine 55, showed spectrum more similar to its intact form (Fig. 2C ▶). In addition, for the remaining six cleaved mutants, spectra were similar in shape and intensity to the spectra of their intact forms both in the near- and far-ultraviolet (Fig. 2 ▶).
Fig. 2.

Circular dichroism (CD) spectra of methionine mutants. CD spectra were determined in 10 mM Gly/HCl (pH 2.0, 2°C). The scans were recorded at 100 nm/min with a step resolution of 1 nm. Each spectrum is the average of seven scans. (A) CD spectra of intact Met mutants recorded in the 190- to 260-nm range at a protein concentration of 2 × 10−5 M in a 1-mm cuvette. (B) Spectra of intact Met mutants recorded in the 240- to 340-nm range at a protein concentration 2 × 10−5 M in a 10-mm pathlength cuvette. (C) Differential CD spectra of intact versus cleaved forms of bovine pancreatic trypsin inhibitor (BPTI) Met mutants. The spectra were obtained by subtraction of those obtained for cleaved from those for intact forms, recorded at the same concentration and expressed in molar ellipticity units to facilitate comparison with direct spectra (A). Spectra could be grouped into four categories: Met6, Met11, Met22, Met29, and Met39 mutants (solid line; Met11 spectrum is shown); Met46, Met49, and Met53 mutants (dashed line; Met49 spectrum is shown); Met54 (line with dots and dashes); and Met32 (dotted line).
Thermal denaturation of intact and cleaved forms of BPTI
Thermal denaturation of intact Met mutants of BPTI and their corresponding cleaved forms was performed at pH 4.3 by differential scanning calorimetry (DSC) measurements. Typical DSC denaturation curves for the Leu29→Met mutant and its cleaved form are shown in Figure 3 ▶. All DSC runs showed the ratio of calorimetric (ΔHcal) to van't Hoff (ΔHvH) enthalpy of denaturation close to unity, indicating a two-state mechanism of denaturation (Table 1). Reversibility of denaturation was checked through repeated heating of the protein sample. For cleaved forms, the reversibility was excellent. It was especially important because it proved that no resynthesis of the peptide bond occurs at pH 4.3. Resynthesis of the peptide bond is known to occur at pH >7 via homoserine lactone attack on the free α-amino group (Dyckes et al. 1978). Denaturation reversibility of the intact forms was similarly verified by two consecutive heatings of the same sample. In DSC-monitored denaturation, the second endotherm was up to 70% smaller if the sample was heated up to 125°C. However, monitoring CD signal at 223-nm full reversibility could be confirmed at a temperature that did not exceed 100°C (data not shown). This results from the irreversibility of hydrolysis of two peptide bonds, Pro2-Asp and Asp3-Phe. The effect is observed at temperatures >120°C and pH >3.5 (Kim et al. 1993; Makhatadze et al. 1993).
Fig. 3.

Temperature dependence of the partial molar heat capacity of the Leu29→Met bovine pancreatic trypsin inhibitor (BPTI) mutant and its cleaved form. Differential scanning calorimetry scans were performed in 10 mM GlyGly/HCl (pH 4.3). The best approximation using a two-state equation (software supplied by CSC Company) is depicted by dark continuous lines.
Table 1.
Thermal denaturation parameters for intact and cyanogen bromide–cleaved forms of bovine pancreatic trypsin inhibitor. The denaturation experiments were performed on a Nano differential scanning calorimeter in 10 mM acetate/Na (pH 4.3)
| Protein | ΔHcal (kcal/mole) | ΔHvH (kcal/mole) | ΔHcal/ΔHvH | ΔScal (cal • mol−1 • deg−1) | ΔCp,den (kcal • mol−1 • deg−1) | Tden (°C) |
| Leu6→Met | 77.3 | 78.1 | 0.99 | 210 | 0.38 | 99.3 |
| Met6-Glu* | 37.9 | 38.2 | 0.99 | 117 | 0.29 | 61.7 |
| Thr11→Met | 76.1 | 79.3 | 0.96 | 205 | 0.32 | 97.9 |
| Met11-Gly* | 40.1 | 39.2 | 1.02 | 111 | 0.41 | 87.1 |
| Phe22→Met | 68.4 | 68.6 | 1.00 | 192 | 0.50 | 84.0 |
| Met22-Tyr* | 24.1 | 24.2 | 1.00 | 75 | 0.40 | 54.7 |
| Leu29→Met | 78.9 | 80.0 | 0.99 | 211 | 0.36 | 100.8 |
| Met29-Cys* | 49.4 | 49.6 | 1.00 | 148 | 0.28 | 61.4 |
| Thr32→Met | 64.8 | 67.5 | 0.96 | 185 | 0.46 | 91.0 |
| Met32-Phe* | 33.8 | 34.3 | 0.99 | 101 | 0.47 | 66.6 |
| Arg39→Met | 75.0 | 74.8 | 1.00 | 200 | 0.34 | 100.8 |
| Met39-Ala* | 49.6 | 49.2 | 1.01 | 139 | 0.27 | 83.8 |
| Lys46→Met | 80.2 | 82.2 | 0.98 | 215 | 0.34 | 100.0 |
| Met46-Ser* | 24.0 | 22.6 | 1.06 | 77 | – | 39.7 |
| Glu49→Met | 77.8 | 76.0 | 1.02 | 210 | 0.31 | 98.0 |
| Met49-Asp* | 23.5 | 22.7 | 1.03 | 75 | – | 41.6 |
| Arg53→Met | 75.1 | 77.8 | 0.97 | 210 | 0.32 | 97.3 |
| Met53-Thr* | 31.5 | 31.2 | 1.01 | 104 | – | 29.3 |
| Thr54→Met | 78.2 | 76.8 | 1.02 | 210 | 0.41 | 98.6 |
| Met54-Cys* | 38.5 | 39.1 | 0.98 | 117 | 0.22 | 61.9 |
Table 1 shows DSC-determined thermodynamic parameters for thermal denaturation of both intact and cleaved forms of BPTI Met mutants. Cleavage of the peptide bond in each of 10 variants caused significant destabilization of the protein molecule, manifested in lowering of the Tden value by 10.8°C to 68°C at pH 4.3 (Table 2). In all cleaved forms, both ΔHcal and ΔScal values were also lowered compared with values for their respective intact forms (Table 1). Values of ΔCp,den did not change substantially on cleavage of peptide bonds, with the exception of Met54-Cys*.
Table 2.
Thermodynamic parameters for the thermal denaturation of intact and cleaved forms of bovine pancreatic trypsin inhibitor variants at 25°C and pH 4.3
| Protein | ΔGden (kcal/mole) | Δ(ΔGden)a (kcal/mole) | ΔTdenb (°C) | ΔGop,N (kcal/mole) | Kop,Nc |
| Leu6→Met | 12.4 | 8.8 | 37.6 | 6.1 | 3.2 • 10−5 |
| Met6-Glu* | 3.6 | ||||
| Thr11→Met | 13.1 | 8.7 | 10.8 | 6.0 | 3.8 • 10−5 |
| Met11-Gly* | 4.4 | ||||
| Lys15-Alad | 0.2 | 0.72 | |||
| Phe22→Met | 8.8 | 7.2 | 29.3 | 4.5 | 4.9 • 10−4 |
| Met22-Tyr* | 1.6 | ||||
| Leu29→Met | 13.3 | 8.5 | 39.4 | 5.8 | 5.4 • 10−5 |
| Met29-Cys* | 4.8 | ||||
| Thr32→Met | 9.3 | 6.3 | 24.4 | 3.6 | 2.2 • 10−3 |
| Met32-Phe* | 3.0 | ||||
| Arg39→Met | 12.4 | 5.7 | 17.0 | 3.0 | 6.2 • 10−3 |
| Met39-Ala* | 6.7 | ||||
| Lys46→Met | 13.8 | 12.7 | 60.3 | 10.0 | 4.4 • 10−8 |
| Met46-Ser* | 1.1e | ||||
| Glu49→Met | 12.6 | 11.4 | 56.4 | 8.7 | 5.6 • 10−7 |
| Met49-Asp* | 1.2e | ||||
| Met52-Argd | 7.3 | 4.2 • 10−6 | |||
| Arg53→Met | 12.8 | 12.4 | 68 | 9.7 | 7.2 • 10−8 |
| Met53-Thr* | 0.4e | ||||
| Thr54→Met | 12.0 | 8.1 | 36.7 | 5.4 | 1.1 • 10−4 |
| Met54-Cys* | 3.9 |
a Δ(ΔGden) = ΔGden − ΔG*den, changes in the free energy of denaturation on single peptide bond cleavage.
b ΔTden = ΔTden − ΔTden*, changes in denaturation temperature on single peptide bond cleavage.
cKop,N = e− ΔGop,N/RT.
d From Krokoszynska and Otlewski (1996).
e The value was calculated directly from the denaturation curve.
Changes in the free energy of denaturation on single peptide bond cleavage (Δ[ΔGden] = ΔGden − ΔG*den) are shown in Table 2. The most extreme effect is observed for the Met46-Ser peptide bond preceding the C-terminal α-helix. The drop of ΔGden is equal to 12.7 kcal/mole. It is related to a decrease in denaturation temperature from 100°C to 39.7°C at pH 4.3. Cleavage of two peptide bonds within the α-helix, Met49-Asp and Met53-Thr, also gave large, albeit smaller, effects on Δ(ΔGden), equal to 11.4 and 12.4 kcal/mole, respectively. However, opening of the Met54-Cys peptide bond at the C terminus of the helix reduced ΔGden much less, by 8.1 kcal/mole. A very similar decrease in ΔGden (8.8 kcal/mole) was observed for the cleavage of the Met6-Glu peptide bond in the N-terminal 310 helix. For the remaining five sites, values of ΔΔGden were in the range of 5.7 to 8.7 kcal/mole.
Peptide bond opening constants in native BPTI
In the previous paper, we estimated ΔGhyd,D values for P1-P1' peptide bonds in five protein inhibitors of serine proteinases, including the Lys15-Ala reactive site of BPTI, using the following thermodynamic cycle (Krokoszynska and Otlewski 1996):
 |
and
 |
(1) |
where N and D are native and denatured proteins, respectively; N* and D* are cleaved forms of native and denatured protein, respectively; ΔGhyd,N and ΔGhyd,D are free energy changes for hydrolysis of a single peptide bond in native and denatured protein, respectively; and ΔGden and ΔG*den are respective free energy changes for denaturation of intact and cleaved protein.
Interestingly, calculated ΔGhyd,D values were similar for all considered peptide bonds and equaled −2.7 (±0.7) kcal/mole at 25°C, equivalent to Khyd,D = 100 (Krokoszynska and Otlewski 1996). Similar values of ΔGhyd,D for different peptide bonds indicated that denatured states of studied inhibitors were conformationally equivalent. In the present study, we used exclusively chemical cleavage reaction with CNBr rather than enzymatic hydrolysis of a peptide bond, so the hydrolysis parameters will be called the peptide bond opening constant (Kop,N and Kop,D) or free energy change (ΔGhyd,N and ΔGhyd,D).
We estimated the free energy changes of the opening of 10 different peptide bonds in the native state of BPTI according to Equation 2, using denaturation data from pH 4.3 and a ΔGop,D value equal to −2.7 kcal/mole:
 |
(2) |
There is some source of error in the cycle, resulting from the chemical difference of species on the left and right side: N and D contain methionine; N* and D*, homoserine residue. We think, however, that this chemical difference can be neglected, as we introduced Met mutations only at solvent-exposed sites. Yu et al. (1995) showed that introduction of alanine at those sites lowered Tden by <5°C with a single exception: mutation of Phe22 to alanine decreased Tden by 14.7°C. We found that influence on stability of Phe22→Ala (Yu et al. 1995) and Phe22→Met mutations was similar (decrease by 14.7°C and 16.8°C, respectively). Therefore, we expected that Met22→Hse conversion should not cause significant changes in stability.
Table 2 shows ΔGop,N and Kop,N values calculated for all 10 peptide bonds together with two other (Lys15-Ala and Met52-Arg) reported previously (Krokoszynska and Otlewski 1996). ΔGop,N values cover the range from 0.2 to 10.0 kcal/mole, which is equivalent to Kop,N values from 0.72 to 4.4 × 10−8. Hydrolyzed peptide bonds can be divided in three groups: Lys15-Ala shows ΔGop,N close to zero; Met39-Ala, Met32-Phe, Met22-Tyr, Met54-Cys, Met29-Cys, Met11-Gly, and Met6-Glu peptide bonds form the second group with the values in the range from 3.0 to 6.1 kcal/mole; and Met52-Arg, Met49-Asp, Met53-Thr, and Met46-Ser are in the third group with ΔGop,N values changing from 7.3 to 10.0 kcal/mole.
Discussion
It is predicted from the classical theory of polymers that covalent crosslink(s) introduced into a protein molecule stabilize it by decreasing the configurational entropy of the denatured state (Flory 1956; Poland and Scheraga 1965). Indeed, there are many reports showing that introduction of single or multiple disulphide bonds into different proteins significantly improved their stability (Matsumura et al. 1989). Opposite effects were detected after elimination of disulphide bond(s) (Pace et al. 1988).
Surprisingly, although proteins are polymers primarily linked by peptide bonds, very little is known about the energetic role of these covalent bonds. Very recent study on an SH3 domain showed that introduction of an additional peptide bond using intein-based strategy to form a circular form of SH3 did not improve protein stability (Camarero et al. 2001). For the purely entropic reason, there should not be a difference between selective hydrolysis of a peptide bond and reduction (or elimination) of a disulphide bond. In this paper, we studied the thermodynamic effects accompanying elimination of a single peptide bond and the integrity of a protein native structure on opening of a peptide bond.
Cleavage of single peptide bonds led to substantial conformational changes when Met residue was introduced into the central part of C-terminal α-helix: Met49 and Met53 (Fig. 2C ▶). Also opening of the Met46-Ser peptide bond immediately before the helix changed the CD spectrum to a large extent (Fig. 2C ▶). The changes were observed both in the far- and near-ultraviolet range of CD spectrum, indicating that opening of those three peptide bonds strongly perturbed not only the local helix conformation but also tertiary interactions. Changes in the CD spectrum were smaller when the peptide bond in the C-terminal part of the helix (Met54-Cys) was opened. Cleavage of peptide bonds in all other parts of BPTI (310 helix, extended loop, different parts of β-sheet, or irregular structure) produced negligible changes in protein conformation (Fig. 2C ▶).
The extent of conformational changes resulting from a single peptide bond cleavage is correlated with degree of destabilization, as opening of peptide bonds at residues 46, 49, and 53 causes drop of Tden by 56.4°C to 68°C, at least 17°C larger than that of any other bond. Because of a peptide bond cleavage, those three mutants lost from 89% to 97% of the total stabilization free energy at 25°C (Table 2). The remaining seven mutants lost from 46% to 82% of the total stabilization free energy.
To calculate ΔGop,N from the thermodynamic cycle, we had to estimate ΔGop,D. In our previous paper, we calculated the value of ΔGhyd,D, taking advantage from the fact that ΔGhyd,N for the P1-P1' peptide bond in proteinase inhibitors is close to zero and, therefore, could be accurately determined in a direct equilibrium experiment using proteolytic cleavage. Essentially in all other proteins, proteolysis is considered to be an irreversible event (i.e., very low and negative value of ΔGhyd,N), it can not be detected (i.e., very large and positive value of ΔGhyd,N), or it occurs at multiple sites. Comparison of the thermodynamic stability of intact and cleaved forms of BPTI mutants together with ΔGop,D = −2.7 kcal/mole allowed us to estimate free energy of opening of 10 single peptide bonds in native BPTI.
The major result of this paper is a very large range of ΔGop,N values reported for a single protein from 0.2 to 10.0 kcal/mole. We did not find a peptide bond with ΔGop,N value smaller than that for the reactive site Lys15-Ala peptide bond, which is located in the extended and exposed loop. Neither solvent accessibility nor temperature factors analysis explains why this particular peptide bond is more favorable to opening than the remaining studied. The low values of ΔGop,N have also been observed for reactive sites of many other protein proteinase inhibitors (Ardelt and Laskowski 1991; Otlewski and Zbyryt 1994). Because all these inhibitors show similar conformation of the P3-P3' loop, it seems plausible that this canonical conformation is a major reason of particularly low values of ΔGop,N. Simultaneously this conformation is highly complementary to the proteinase active site and can be recognized as a typical protein substrate.
For all remaining peptide bonds, ΔGop,N is larger by at least 2.8 kcal/mole. Cleavage of the Met11-Gly peptide bond, which is also located in this loop, has ΔGop,N = 6.0 kcal/mole. The only peptide bond located in irregular loop is Met39-Ala. Notably, it shows the second smallest value of ΔGop,N = 3.0 kcal/mole. Opening of three peptide bonds located in the helix region (Met46-Ser, Met49-Asp, and Met53-Thr) produced large structural changes comprising not only this helix but also tertiary interactions in the hydrophobic core. In agreement, cleavage of these peptide bonds in native mutants is thermodynamically very unfavorable, and their ΔGop,N values are >7 kcal/mole. We conclude that breaking of peptide bonds located in the helical region destroys a highly cooperative network of hydrogen bonds. It should be noted that the Met54-Cys peptide bond, which is next to Met53-Thr, behaved differently. Its cleavage did not produce extensive conformational changes, and ΔGop,N was >4 kcal/mole smaller than that for Met53-Thr; that is, from the thermodynamic point of view, its cleavage can be much easier performed. We suppose that opening of this peptide bond does not affect the local helix conformation and tertiary interactions within BPTI, as it is the last C-terminal bond of the helix and Cys55, which is at the N terminus of newly formed peptide and is disulphide bridged to Cys5. Met6-Glu peptide bond in the 310 helix shows similar characteristics.
For seven peptide bonds located outside the C-terminal helix, we did not observe major conformational changes after CNBr cleavage. ΔGop,N values for three peptide bonds—Met22-Tyr, Met29-Cys, and Met32-Phe—located in the β-sheet are from 1.5 to 6.4 kcal/mole smaller than those in the helical region (Table 2). This, together with negligible structural changes observed in CD difference spectra for the three β-sheet mutants, indicates that cleavage of peptide bonds in the β-sheet is not as disadvantageous as in the α-helix.
From the above analysis, it seems that peptide bonds in the central part of the α-helix are most difficult to open, followed by those in the β-sheet and loop regions. In this respect, our results are qualitatively similar to observations that proteinase sensitive sites are located in flexible and exposed loops (Fontana et al. 1986; Fontana 1989; Polverino et al. 1999). However, we did not observe correlation between main-chain temperature B factors and ΔGop,N (data not shown).
We expect that our data provide upper limit of ΔGop,N values in proteins, which means that peptide bonds located in helices are most difficult to cleave. Regarding the lower limit, there is a genuine example of proteins containing a peptide bond that can be virtually irreversibly broken. Im et al. (2000) reported that cleavage of a single peptide bond within a 30-residue loop in α1-antitrypsin is coupled with enormous increase in ΔGden, by 32.2 kcal/mole. Thus, according to the thermodynamic cycle, ΔGop,N for this unusual peptide bond equals −34.9 kcal/mole, equivalent to Kop,N = 4.6 × 1025. The extremely low value of ΔGop,N results from huge gain in stability on peptide bond cleavage, what drives hydrolysis reaction to completion. Thus, dynamic range of equilibrium constant for opening of a single peptide bond in proteins covers 33 orders of magnitude. However, to ensure stability against proteolysis, typical equilibrium values for a single peptide bond opening in a protein containing secondary structure elements fall into negligibly low values from 10−3 to 10−8.
Materials and methods
Materials
Guanidinium chloride, urea, and acetonitrile were purchased from Merck. Trifluoroacetic acid, formic acid (88%), and CNBr were obtained from Fluka. Tris, sodium acetate, DTT, chloramphenicol, ampicillin, and reduced and oxidized glutathione were supplied by Sigma. Sephadex G-25 was obtained from Amersham Pharmacia Biotech.
The basic components of culture media were purchased from Merck. DNA-modifying enzymes (phage T4 polymerase, phage T4 ligase, and phage T4 polynucleotide kinase) were supplied by Boehringer Mannheim. The DNA sequencing kit was from Amersham Pharmacia Biotech, and the DNA purification kit was from Qiagen. Oligonucleotides were chemically synthesized by Ransom Hill. IPTG was obtained from Bachem.
BPTI (Traskolan) was a generous gift from Jelfa Pharmaceutical Company S.A. (Poland). Before use, protein was desalted on the Sephadex G-25 and lyophilized from 50 mM acetic acid.
Plasmid construction
The pAED4 plasmid bearing a coding sequence for BPTI and a portion of the Escherichia coli trp operon, which serves as a hydrophobic leader sequence and facilitates formation of inclusion bodies, was a generous gift from Professor P.S. Kim (Whitehead Institute, MIT) (Staley and Kim 1994). To enable the selective peptide bond cleavage at the single Met residue by CNBr, the hydrophobic leader sequence was removed from the plasmid after the introduction of NdeI site at the 3` end of the leader sequence.
Single Met substitutions were introduced individually into the gene coding BPTI Met52→Leu by oligonucleotide-directed mutagenesis (Kunkel 1985), and the introduced mutations were confirmed by DNA sequencing.
Expression and purification of BPTI mutants
All Met mutants of BPTI were overexpressed in the E. coli strain BL 21 (DE3) pLysS, using the T7 promotor system (Studier et al. 1990). Purification of BPTI mutants was performed as described (Krokoszynska et al. 1998). Despite the lack of the hydrophobic leader sequence, all the mutants formed inclusion bodies. Because of the absence of the leader sequence, the CNBr cleavage step was omitted in the current purification protocol. To ensure high degree of purity, mutant proteins were purified in both the reduced and the oxidized state on Vydac C18 semi-preparative HPLC columns. A typical yield of oxidative refolding was 50% to 60% (1 to 5 mg of pure protein per liter of culture medium).
Mass spectrometry
Electrospray mass spectrometry of all recombinant mutants used in this study was performed with a Finningan MAT TSQ-700 equipped with ESI source. Samples were dissolved in methanol/water (1:1, v/v) containing 1% acetic acid. Molecular masses of all mutants were within 1.0 atomic mass unit from those expected from amino acid sequences.
CNBr cleavage of BPTI Met mutants
CNBr cleavage of single peptide bond in BPTI mutants was performed as described (Dyckes et al. 1978). Each mutant was dissolved in CNBr solution (30 mg/mL in 70% formic acid) to give a CNBr/BPTI weight ratio of 1.5:1.0. After 12 h of incubation in the dark at room temperature, the solution was diluted 10-fold with water and lyophilized. The cleaved form of the mutant was dissolved (2 mg/mL) in 0.02 M Tris (pH 9.1) and 6 M guanidinium chloride to enable hydrolysis of homoserine lactone to homoserine. The presence of denaturant prevented resynthesis of the Hse-Xaa' peptide bond. After 6 h, the material was desalted on Sephadex G-25 column equilibrated in 50 mM acetic acid. The cleaved form was separated from the intact one on HPLC column (Vydac C18). Solvents A (0.1% trifluoroacetic acid) and B (90% acetonitrile and 0.1% trifluoroacetic acid) were mixed to form a linear gradient of 10% to 60% solvent B (1.5 mL/min flow). A typical yield of cyanogen bromide cleavage was 50% to 70%.
Calorimetry
Denaturation experiments were performed on a NANO II differential scanning microcalorimeter (CSC Corp.). The temperature range was 0°C to 125°C; the heating rate, 1°C/min; and protein concentration, 1.5 × 10−5 to 3 × 10−5 M. BPTI concentration was determined spectrophotometrically using the molar absorbance coefficient at 280 nm, ɛ = 5377 M−1/cm (Pace et al. 1995). Denaturations were performed in 10 mM GlyGly/HCl (pH 4.3). Before loading the protein solution into the calorimetric cell, samples were dialyzed against the experimental buffer using 3-kD cut-off dialysis tubing, centrifugated, and degassed under lowered pressure. All the procedures were performed at 4 °C. Baseline was determined by running the calorimeter with both cells filled with the dialysis buffer.
The values of Tden, ΔHcal, ΔHvH, and ΔCp,den were calculated directly from thermograms using Cpcalc software provided by CSC Corp. The partial specific volume of BPTI was estimated to be 0.71 cm3/g for intact forms and 0.73 cm3/g for cleaved forms of BPTI (Makhatadze et al. 1993). The temperature dependence of ΔH, ΔS, and ΔG was calculated from the following equations:
 |
(3) |
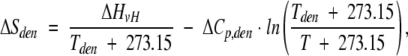 |
(4) |
 |
(5) |
where ΔHden, ΔSden, and ΔGden are the enthalpy, entropy, and free energy of denaturation at the temperature (T) of 25°C; ΔHvH is the van't Hoff enthalpy of denaturation; ΔCp,den is the temperature-independent change of the denaturational heat capacity; and Tden is the midpoint temperature.
CD measurements
CD spectra of BPTI variants were recorded in 10 mM Gly/HCl (pH 2.0) with a Jasco J-715 spectropolarimeter at 2°C. Measurements were made at a protein concentration of 2 × 10−5 M using a 10-mm cuvette (range, 240 to 340 nm ) or at 2 × 10−5 M protein in a 1-mm pathlength cuvette (range, 190 to 260 nm). Spectra were averaged from seven individual scans.
Acknowledgments
We thank Professor Zbigniew Szewczuk for mass spectra analysis. We are grateful to Honorata Czapinska and Katarzyna Koscielska-Kasprzak for reviewing this manuscript. The research of Jacek Otlewski was partially supported by an International Scholar's award from the Howard Hughes Medical Institute. Daniel Krowarsch is a recipient of the Young Scholar Award from the Foundation for Polish Science.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
BPTI, bovine pancreatic trypsin inhibitor
*, denotes a cyanogen bromide
cleaved form of BPTI Met mutant
CD, circular dichroism
DSC, differential scanning calorimetry
Tden, temperature of denaturation
ΔCp,den, denaturational heat capacity change
ΔHvH, van't Hoff enthalpy of denaturation
ΔHcal, calorimetric enthalpy of denaturation
ΔScal, calorimetric entropy of denaturation
ΔGden and ΔG*den, free energy change for denaturation of an intact or cleaved protein, respectively
Kop,N and Kop,D, equilibrium constant for opening of a single peptide bond in a native or denatured protein, respectively
ΔGop,N and ΔGop,D, free energy change for opening of single peptide bond in a native or denatured protein, respectively
Δ(ΔGden), difference between free energy of denaturation for an intact and cleaved forms of BPTI
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.4460102.
References
- Ardelt, W. and Laskowski, Jr., M. 1991. Effect of single amino acid replacements on the thermodynamics of the reactive site peptide bond hydrolysis in ovomucoid third domain. J. Mol. Biol. 220 1041–1053. [DOI] [PubMed] [Google Scholar]
- Betzel, C., Dauter, Z., Genov, N., Lamzin, V., Navaza, J., Schnebli, H.P., Visanji, M., and Wilson, K.P. 1993. Structure of the proteinase inhibitor eglin c with hydrolyzed reactive center at 2.0 Å resolution. FEBS Lett. 317 185–188. [DOI] [PubMed] [Google Scholar]
- Cai, M., Gong, Y., Prakash, O., and Krishnamoorthi, R. 1995. Reactive-site hydrolyzed Cucurbita maxima trypsin inhibitor-C: Function, thermodynamic stability, and NMR solution structure. Biochemistry 34 12088–12094. [DOI] [PubMed] [Google Scholar]
- Camarero, J.A., Fushman, D., Sato, S., Giriat, I., Cowburn, D., Raleigh, D.P., and Muir, T.W. 2001. Rescuing a destabilized protein fold through backbone cyclization. J. Mol. Biol. 308 1045–1062. [DOI] [PubMed] [Google Scholar]
- Dyckes, D.E., Creighton, T.E., and Shephard, R.C. 1978. [52-homoserine]-basic pancreatic trypsin inhibitor: Preparation and properties of a protein analog. Int. J. Peptide Protein Res. 11 258–268. [PubMed] [Google Scholar]
- Flory, P. 1956. Theory of elastic mechanisms in fibrous proteins. J. Am. Chem. Soc. 78 5222–5235. [Google Scholar]
- Fontana, A. 1989. Limited proteolysis of globular proteins occur at exposed and flexible loops. In Highlights of modern biochemistry (eds. A. Kotyk et al.), pp. 1711–1726. VSP International Publications, Amsterdam.
- Fontana, A., Fassina, G., Vita, C., Dalzoppo, D., Zamai, M., and Zambonin, M. 1986. Correlation between sites of limited proteolysis and segmental mobility in thermolysin. Biochemistry 25 1847–1851. [DOI] [PubMed] [Google Scholar]
- Fontana, A., Polverino de Laureto, P., and De Filippis, V. 1993. Molecular aspects of proteolysis of globular proteins. In Protein stability and stabilization (eds. W. Van den Tweel et al.), pp. 101–110. Elsevier, Amsterdam.
- Fontana, A., Polverino de Laureto, P., De Fillips, V., Scaramella, E., and Zambonin, M. 1997. Probing the partly folded states of proteins by limited proteolysis. Fold. Des. 2 17–26. [DOI] [PubMed] [Google Scholar]
- Fritz, H. and Wunderer, G. 1983. Biochemistry and applications of aprotinin, the kallikrein inhibitor from bovine organs. Arzneimittelforschung 33 479–494. [PubMed] [Google Scholar]
- Hubbard, S.J. 1998. The structural aspects of limited proteolysis of native proteins. Biochim. Biophys. Acta 1382 191–206. [DOI] [PubMed] [Google Scholar]
- Im, H., Ahn, H.-Y., and Yu, M.-H. 2000. Bypassing the kinetic trap of serpin protein folding by loop extension. Protein Sci. 9 1497–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, K.-S., Tao, F., Fuchs, J., Danishefsky, A.T., Housset, D., Wlodawer, A., and Woodward, C. 1993. Crevice-forming mutants in the rigid core of bovine pancreatic trypsin inhibitor: Stability changes and new hydrophobic surface. Protein Sci. 2 588–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krokoszynska, I. and Otlewski, J. 1996. Thermodynamic stability effects of single peptide bond hydrolysis in protein inhibitors of serine proteinases. J. Mol. Biol. 256 793–802. [DOI] [PubMed] [Google Scholar]
- Krokoszynska, I., Dadlez, M., and Otlewski, J. 1998. Structure of single-disulphide variants of bovine pancreatic trypsin inhibitor (BPTI) as probed by their binding to bovine β-trypsin. J. Mol. Biol. 275 503–513. [DOI] [PubMed] [Google Scholar]
- Kunkel, T.A. 1985. Rapid and efficient site-specific mutagenesis without phenotopic selection. Proc. Natl. Acad. Sci. 82 488–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladurner, A.G., Itzhaki, L.S., and Prat Gay, G.D. 1997. Complementation of peptide fragments of the single domain protein chymotrypsin inhibitor 2. J. Mol. Biol. 273 317–329. [DOI] [PubMed] [Google Scholar]
- Makhatadze, G.I., Kim, K., Woodward, C., and Privalov, P.L. 1993. Thermodynamics of BPTI folding. Protein Sci. 2 2028–2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura, M., Becktel, W.J., Levitt, M., and Matthews, B.W. 1989. Stabilization of phage T4 lysozyme by engineered disulfide bonds. Proc. Natl. Acad. Sci. 86 6562–6566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moses, E. and Hinz, H.J. 1983. Basic pancreatic trypsin inhibitor has unusual thermodynamic stability parameters. J. Mol. Biol. 170 765–776. [DOI] [PubMed] [Google Scholar]
- Musil, D., Bode, W., Huber, R., Laskowski, Jr., M., Lin, T.Y.L., and Ardelt, W. 1991. Refined X-ray crystal structures of the reactive site modified ovomucoid third domains from silver pheasant and from Japanese quail. J. Mol. Biol. 220 739–755. [DOI] [PubMed] [Google Scholar]
- Neurath, H. 1991. Proteolytic processing and regulation. Enzyme 45 239–243. [DOI] [PubMed] [Google Scholar]
- Otlewski, J. and Zbyryt, T. 1994. Single peptide bond hydrolysis/resynthesis in squash inhibitors of serine proteinases, I: Kinetics and thermodynamics of the interaction between squash inhibitors and bovine β-trypsin. Biochemistry 33 200–207. [DOI] [PubMed] [Google Scholar]
- Otlewski, J., Zbyryt, T., Dryjanski, M., Bulaj, G., and Wilusz, T. 1994. Single peptide bond hydrolysis/resynthesis in squash inhibitors of serine proteinases, II: Limited proteolysis of Cucurbita maxima trypsin inhibitor I (CMTI I) by pepsin. Biochemistry 33 208–213. [DOI] [PubMed] [Google Scholar]
- Pace, C.N., Grimsley, G.R., Thomson, J.A., and Barnett, B.J. 1988. Conformational stability and activity of ribonuclease T1 with zero, one, and two intact disulfide bonds. J. Biol. Chem. 263 11820–11825. [PubMed] [Google Scholar]
- Pace, C.N., Vajdos, F., Fee, L., Grimsley, G., and Gray, T. 1995. How to measure and predict the molar absorption coefficients of a protein? Protein Sci. 4 2411–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poland, D.C. and Scheraga, H.A. 1965. Statistical mechanics of noncovalent bonds in polyamino acids. Biopolymers 3 379–399. [Google Scholar]
- Polverino de Laureto, P., Scaramella, E., Frigo, M., Wondrich, F.G., De Filippis, V., Zambonin, M., and Fontana, A. 1999. Limited proteolysis of bovine α-lactalbumin: isolation and characterization of protein domains. Protein Sci. 8 2290–2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schechter, P. and Berger, A. 1967. On the size of the active site in proteases, I: Papain. Biochem. Biophys. Res. Commun. 27 157–162. [DOI] [PubMed] [Google Scholar]
- Seife, C. 1997. Blunting nature's Swiss army knife. Science 277 1602–1603. [DOI] [PubMed] [Google Scholar]
- Selkoe, D.J. 2001. Alzeimer's disease: Genes, proteins, and therapy. Physiol. Rev. 81 741–766. [DOI] [PubMed] [Google Scholar]
- Siekman, J., Wenzel, H.R., Matuszak, E., Goldammer, E., and Tschesche, H. 1988. The pH dependence of the equilibrium constant Khyd for hydrolysis of the Lys15-Ala16 reactive-site peptide bond in bovine pancreatic trypsin inhibitor (aprotinin). J. Protein Chem. 7 633–640. [DOI] [PubMed] [Google Scholar]
- Staley, J.P. and Kim, P.S. 1994. Formation of a native-like subdomain in a partially folded intermediate of bovine pancreatic trypsin inhibitor. Protein Sci. 3 1822–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubbs, M.T. and Bode, W. 1994. Coagulation factors and their inhibitors. Curr. Opin. Struct. Biol. 4 823–832. [DOI] [PubMed] [Google Scholar]
- Studier, F.W., Rosenberg, A.H., Dunn, J.J., and Dubendorff, J.W. 1990. Use of T7 polymerase to direct expression of cloned genes. Methods Enzymol. 185 60–89. [DOI] [PubMed] [Google Scholar]
- Walkenhorst, W.F., Krezel, A.M., Rhyu, G.I., and Markley, J.L. 1994. Solution structure of reactive site hydrolyzed turkey ovomucoid third domain by nuclear magnetic resonance and distance geometry methods. J. Mol. Biol. 242 215–230. [DOI] [PubMed] [Google Scholar]
- Wlodawer, A., Walter, J., Huber, R., and Sjolin, L. 1984. Structure of bovine pancreatic trypsin inhibitor: Results of joint neutron and X-ray refinement of crystal form II. J. Mol. Biol. 180 301–329. [DOI] [PubMed] [Google Scholar]
- Yu, M.H., Weissman, J.S., and Kim, P.S. 1995. Contribution of individual side chains to the stability of BPTI examined by alanine-scanning mutagenesis. J. Mol. Biol. 249 388–397. [DOI] [PubMed] [Google Scholar]