

Abstract
Tetrahydrodipicolinate N-succinyltransferase (DapD) catalyzes the succinyl-CoA-dependent acylation of L-2-amino-6-oxopimelate to 2-N-succinyl-6-oxopimelate as part of the succinylase branch of the meso-diaminopimelate/lysine biosynthetic pathway of bacteria, blue-green algae, and plants. This pathway provides meso-diaminopimelate as a building block for cell wall peptidoglycan in most bacteria, and is regarded as a target pathway for antibacterial agents. We have solved the X-ray crystal structures of DapD in ternary complexes with pimelate/succinyl-CoA and L-2-aminopimelate with the nonreactive cofactor analog, succinamide-CoA. These structures define the binding conformation of the cofactor succinyl group and its interactions with the enzyme and place its thioester carbonyl carbon in close proximity to the nucleophilic 2-amino group of the acceptor, in support of a direct attack ternary complex mechanism. The acyl group specificity differences between homologous tetrahydrodipicolinate N-acetyl- and N-succinyltransferases can be rationalized with reference to at least three amino acids that interact with or give accessible active site volume to the cofactor succinyl group. These residues account at least in part for the substrate specificity that commits metabolic intermediates to either the succinylase or acetylase branches of the meso-diaminopimelate/lysine biosynthetic pathway.
Keywords: Succinyltransferase, acetyltransferase, coenzyme A, lysine, protein structure, X-ray crystallography
The biosynthesis of meso-diaminopimelate and lysine in bacteria and plants is accomplished by one of three biosynthetic pathways. The common succinylase branch (Berges et al. 1986) makes use of succinylated intermediates, whereas the more restricted acetylase branch uses acetylated intermediates (Sundaharas and Gilvarg 1967). A distinct dehydrogenase pathway is also observed in several species (Misono et al. 1986; Ishino et al. 1987). The succinylase and acetylase pathways diverge at the acetyl transfer step that is catalyzed by two homologous N-acyltransferases. A tetrahydrodipicolinate N-succinyltransferase (DapD) is present in organisms that use the succinylase pathway, and a tetrahydrodipicolinate N-acetyltransferase, such as YkuQ of Bacillus subtilis, is used by those organisms that possess the acetylase branch. In both cases, the transferred acyl group is used to enforce a linear conformation on ensuing metabolites and to expose the 6-keto group for subsequent transamination (Fig. 1 ▶). The biosynthesis of meso-diaminopimelate provides an essential component of cell wall peptidoglycan in many bacterial species, and enzymes of the meso-diaminopimelate/lysine pathway are regarded as targets for the development of antibacterial agents.
Fig. 1.

(A) The reaction sequence leading from tetrahydrodipicolinate to lysine via the succinylase branch of the meso-diaminopimelate/lysine biosynthetic pathway. (B) The substrates and analogs used in this study.
The crystal structure of DapD, a tetrahydrodipicolinate N-succinyltransferase, has been determined and revealed the presence of three left-handed parallel β-helix (LβH) domains in its trimeric structure (Beaman et al. 1997). The distinctive LβH domain is formed by tandem repeated imperfect copies of a six-residue repeated sequence generally described as [LIV]-[GAED]-X2–[STAV]-X and termed a hexapeptide repeat (Dicker and Seetharam 1992; Vaara 1992). These hexapeptide repeats direct folding of the polypeptide chain into a left-handed coil that takes the form of a triangular prism. Amino acid sequence ranges within the LβH that disobey the hexapeptide repeat sequence rule form loops that are excluded from the coiled LβH fold and frequently participate at the active site of several such hexapeptide acyltransferases (Raetz and Roderick 1995; Beaman et al. 1998a,b; Kostrewa et al. 2001; Olsen and Roderick 2001; Sulzenbacher et al. 2001).
The available crystallographic models of DapD in complex with substrates are limited to its ternary complexes with CoA and either the natural substrate L-2-amino-6-oxopimelate or the close structural analog L-2-aminopimelate (Beaman et al. 1998a). These structures revealed that the enzyme binds the linear conformation of its acceptor substrate and that a dramatic conformational change is required. Each of the three identical and independent active sites are formed at the junction of two adjacent subunits, termed A and B. The residues that contribute to substrate binding are donated by an NH2-terminal domain, a 17-residue C-terminal tail that orders upon substrate binding, and two adjacent LβH domains (Beaman et al. 1998a). However, these crystal structures of DapD in complex with substrates lack the succinyl group present on either the cofactor or the product. We describe here the crystal structure of DapD in complex with pimelate/succinyl-CoA as well as L-2-aminopimelate and the synthetic cofactor, succinamide-CoA.
Results and Discussion
The absence in the database of structural models that identify the conformation and binding interactions of the acyl group of any tetrahydrodipicolinate N-acyltransferase led us to determine the crystal structure of DapD in ternary complex with pimelate/succinyl-CoA and L-2-aminopimelate/succinamide-CoA (the analog of succinyl-CoA in which the thioester is replaced with an amide bond). These structures have been determined to 2.3 and 2.0 Å resolution and refined to conventional crystallographic R-factors of 17.1% and 17.9%, respectively (Table 1, Figs. 2, 3A ▶ ▶).
Table 1.
Data measurement and structure refinement statistics
| Pimelate/succinyl-CoA | L-2-aminopimelate/ succinamide-CoA | |
| Data measurement | ||
| Resolution (Å) | 2.3 | 2.0 |
| Observed reflections | 47749 | 59293 |
| Unique reflections | 11389 | 16693 |
| Redundancy | 4.3 | 3.5 |
| Rmerge (%)a,b | 7.2 (33.9) | 4.8 (21.7) |
| 〈I/σI〉 | 21.3 (3.6) | 20.8 (3.2) |
| Completeness (%)c | 99.8 (99.9) | 99.5 (97.9) |
| Atoms | ||
| Protein atoms | 2078 | 2079 |
| Substrate atoms | 11/55 | 12/55 |
| Water molecules | 59 | 87 |
| RMS deviation from ideality | ||
| Bond lengths (Å) | 0.024 | 0.022 |
| Bond angles (°) | 2.2 | 2.2 |
| Average thermal factors (Å2) | ||
| Protein atoms | 33.9 | 27.7 |
| Substrate/cofactor atoms | 24.2/40.4 | 13.8/32.8 |
| Solvent atoms | 42.1 | 42.3 |
| Rfree (%)d | 26.1 (35.0) | 25.0 (34.1) |
| Rfactor (%)e | 17.1 (26.0) | 17.9 (27.0) |
a Rmerge (%) = ∑| Ii − |∑| Ii | × 100 where Ii is an individual intensity observation, ≤I> is the mean intensity for that reflection and the summation is over all reflections.
b Numbers in parentheses refer to the highest resolution shell: pimelate/succinyl-CoA, 2.50 − 2.30 Å; L-2-aminopimelate/succinamide-CoA, 2.10 − 2.00 Å.
c Completeness is the ratio of the number of observed unique reflections to the total number of theoretically possible reflections × 100.
d Rfree (%) = ∑ | Fo − Fc | /∑ | Fo | × 100 for a 5% subset of X-ray diffraction data omitted from refinement calculations.
e Rfactor (%) = ∑ | Fo − Fc | /∑∼ | Fo | × 100 for all available data.
Fig. 2.
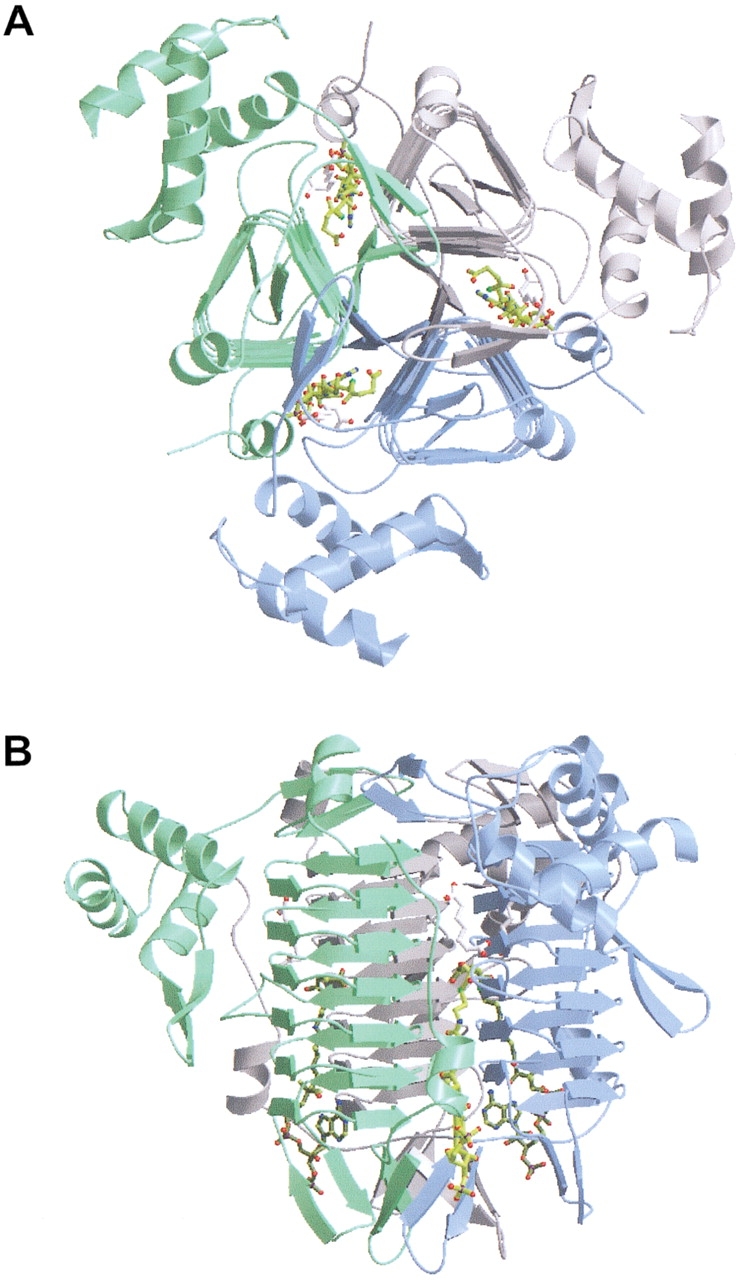
Overall structure of trimeric DapD in complex with pimelate (white) and succinyl-CoA (yellow). Each trimer contains three active sites. (A) View parallel to the molecular threefold axis. (B) View perpendicular to the threefold axis. The proximal active site is composed of two subunits termed A (green) and B (blue).
Fig. 3.
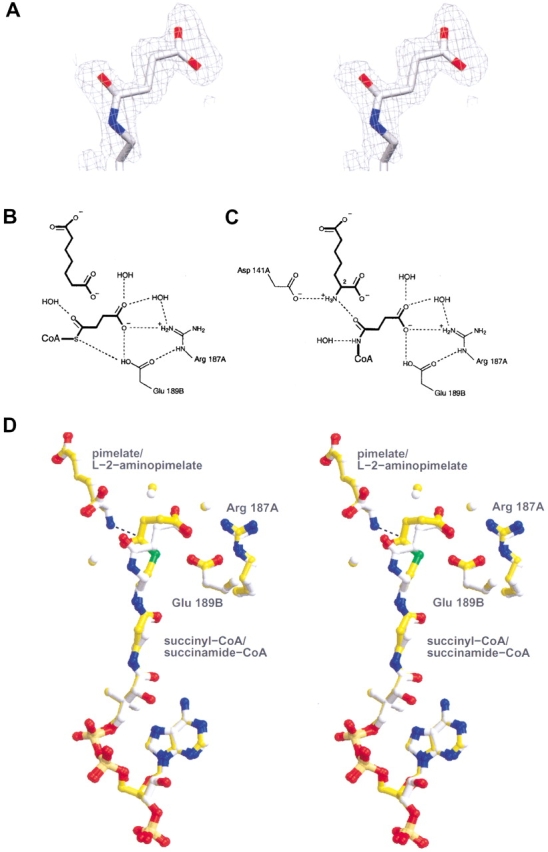
Succinyl group binding pocket of DapD. (A) Stereoview of the unbiased Fo-Fo electron density map surrounding the refined coordinates corresponding to the succinamide moiety of the succinamide-CoA cofactor analog. (B) Hydrophilic interactions at the active site of the pimelate/succinyl-CoA complex. (C) Hydrophilic interactions at the active site of the L-2-aminopimelate/succinamide-CoA complex. (D) Superimposed stereoview of the two complexes, depicting the acceptor and cofactor (or analogs), surrounding water molecules, and the two residues that interact with the succinyl carboxyl group (Arg 187A, Glu 189B). The nucleophilic amino group of L-2-aminopimelate and the carbonyl carbon of the succinyl-CoA thioester are joined by a dotted line (distance 2.9 Å).
The structure of DapD in complex with pimelate/succinyl-CoA was determined in order to view the cofactor succinyl group in the presence of an acceptor substrate analog that lacks the attacking 2-amino group (Fig. 1 ▶). Pimelate binds to the active site in a very similar extended conformation to L-2-aminopimelate in the previously reported L-2-aminopimelate/CoA complex (Beaman et al. 1998a). The carboxylate group of pimelate nearest the cofactor interacts with Ser 146B, a water molecule, and the main chain amide groups of Leu 168B and Glu 169B, two residues of a loop excluded from the LβH domain. The proximity of one of the succinyl carboxyl oxygen atoms to a side chain oxygen atom of Glu 189B (2.5 Å) suggests that one of these atoms is protonated, perhaps the carboxyl group of the glutamate, since the succinyl oxygen atom is positioned 3.5 Å from the positively charged guanidinium group of Arg 187A (Fig. 3 ▶). The terminal carboxylate group of succinyl-CoA interacts with Arg 187A, Glu 189B, and two water molecules. In addition, a side chain oxygen atom of Glu 189B appears to interact with the cofactor sulfur atom (distance 3.5 Å).
The structure of DapD in ternary complex with L-2-aminopimelate and succinamide-CoA, an unreactive cofactor amide analog, was determined in order to view the succinyl group in the presence of a catalytically competent acceptor. This structure places the acceptor very nearly superimposed on the position of pimelate in the structure described above (Fig. 3C,D). The succinamide carboxylate group shares the same interactions with the enzyme as succinyl-CoA, but the conformation of the planar cis amide linkage differs from the natural thioester linkage of succinyl-CoA. Since N-methylacetamide exists as only 1.5% cis isomer in water (Radzicka et al. 1988), it is apparent that the enzyme is selectively binding the less stable isomer. The amide oxygen of the succinamide group hydrogen bonds to the 2-amino group of the acceptor.
Superposition of the two complex structures places the thioester and amide carbonyl oxygen atoms in very nearly the same position and demonstrates that the succinyl carboxylate groups form the same set of interactions with the enzyme (Fig. 3D ▶), despite the constraints placed on the planar amide group of succinamide-CoA and the presence or absence of the attacking 2-amino group of the acceptor. The distance between the 2-amino group of the acceptor and the carbonyl carbon of the thioester group determined from the superimposed structures is 2.9 Å, suggesting that a direct attack of the 2-amino group on the thioester is indeed possible. This is in agreement with the ternary complex mechanism proposed on the basis of steady-state kinetic data (Berges et al. 1986). The structures determined here also suggest that Asp 141A may be positioned to function as a general base capable of abstracting a proton from the nucleophilic 2-amino group of the acceptor. A negatively charged tetrahedral intermediate could then be stabilized by the peptide nitrogen of Gly 166B. The interaction of the succinyl carboxylate group with Glu 189B suggests that this residue or Asp 141A could serve to protonate the cofactor sulfur atom to form the thiol group of the product CoA.
In an effort to rationalize the acyl group specificity differences between tetrahydrodipicolinate N-acetyl- and N-succinyltransferases, we compared the identity of several active site residues for these enzymes. The tetrahydrodipicolinate acyltransferase from Bacillus pumilus (YkuQ) is known to catalyze acetyl transfer but not succinyltransfer (Gilvarg and Weinberger 1970). We observed the same cofactor specificity for the purified enzyme from Bacillus subtilis (T. Beaman and S. Roderick, unpubl.). The amino acid sequence alignment of DapD with these acetyltransferases suggests that at least three key residues are required for succinyl group recognition. Arg 187A and Glu 189B contact the succinyl group directly but are replaced by Asn and Val, respectively, in the acetyltransferases. In addition, the absence of a side chain for Gly 163B gives accessible volume to the succinyl binding pocket which would not be provided by Val of the acetyltransferases. These three substitutions would clearly alter the interactions of an acetyltransferase with the acyl group and prevent succinyl-CoA from binding, using the pattern of interactions observed here. It is interesting that the same three substitutions are present in the tetrahydrodipicolinate N-acyltransferase from Staphylococcus aureus and Streptococcus pneumoniae, indicating that some pathogenic cocci may also catalyze reactions of the acetylase branch.
This work has demonstrated the proximity of the thioester group of succinyl-CoA in the pimelate/succinyl-CoA complex to the acceptor 2-amino group of L-2-aminopimelate in the L-2-aminopimelate/succinamide-CoA complex, in support of a direct attack ternary complex mechanism. In addition, the pattern of substitution for three key amino acid residues at the active site of DapD may explain the substrate specificity of the tetrahydrodipicolinate N-acyltransferases and the means by which these enzymes commit metabolic flux to either the succinylase or acetylase branches of the meso-diaminopimelate/lysine biosynthetic pathway. The structural basis for cofactor acyl group specificity reported here should be of interest in the development of antibiotics that disrupt this pathway, thus reducing the levels of meso-diaminopimelate, an essential component of bacterial cell wall peptidoglycan.
Materials and methods
DapD was prepared and crystallized as described (Binder et al. 1996). The succinamide-CoA cofactor analog was prepared from N-(2-aminoethyl)-succinamic acid (Grinberg et al. 1975) by reaction with an enzymatically prepared CoA analog synthon, as described (Martin et al. 1994). Crystals of DapD in complex with pimelate and succinyl-CoA were prepared by cocrystallization using the hanging drop vapor diffusion method. The drops were formed by mixing 5 μL 27 mg/mL DapD with an equal volume of precipitant solution containing 17% (w/v) polyethyleneglycol 4000, 188 mM ammonium sulfate, 94 mM MES, pH 6.4, 4.7% (v/v) 2-propanol, 20 mM pimelate, and 5 mM succinyl-CoA and inverted over a reservoir containing 0.5 mL of the precipitant solution (Beaman et al. 1998a). Crystals of the complex with L-2-aminopimelate and succinamide-CoA were prepared by mixing 30 mg/mL DapD with a solution containing 16% (w/v) polyethyleneglycol 4000, 188 mM ammonium sulfate, 94 mM MES, pH 6.4, 4.7% (v/v) 2-propanol, 16 mM (D,L)-2-aminopimelate, and 5 mM succinamide-CoA.
All crystals obtained from these cocrystallizations belong to space group R3 with unit cell parameters a = 95.8 and c = 72.6 Å (hexagonal setting) and contain a single subunit of the trimeric molecule in the asymmetric unit. X-ray diffraction data sets were measured at 22°C using a Siemens X1000 area detector mounted on a Rigaku RU200 rotating anode generator operating with fine focus at 4 kW. The data were reduced with XDS and XSCALE (Kabsch 1988) (Table 1).
The structure of these complexes were determined by molecular replacement by comparison to the isomorphous structure of the L-2-aminopimelate/CoA complex (Beaman et al. 1998a). Iterative cycles of model building and refinement were carried out with O (Jones et al. 1991) and TNT (Tronrud et al. 1987), reserving 5% of the X-ray diffraction data for cross-validation (Brunger 1992; Kleywegt and Brunger 1996) (Table 1).
Acknowledgments
This work was supported by NIH grants AI-33696 (to J.S.B.) and GM-45831 (to D.G.D.). The atomic coordinates for the DapD complexes have been deposited in the Protein Data Bank with PDB accession codes 1KGQ and 1KGT.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.4310102.
References
- Beaman, T.W., Binder, D.A., Blanchard, J.S., and Roderick, S.L. 1997. Three-dimensional structure of tetrahydrodipicolinate N-succinyltransferase. Biochemistry 36 489–494. [DOI] [PubMed] [Google Scholar]
- Beaman, T.W., Blanchard, J.S., and Roderick, S.L. 1998a. The conformational change and active site structure of tetrahydrodipicolinate N-succinyltransferase. Biochemistry 37 10363–10369. [DOI] [PubMed] [Google Scholar]
- Beaman, T.W., Sugantino, M., and Roderick, S.L. 1998b. Structure of the hexapeptide xenobiotic acetyltransferase from Pseudomonas aeruginosa. Biochemistry 37 6689–6696. [DOI] [PubMed] [Google Scholar]
- Berges, D.A., DeWolf Jr., W.E., Dunn, G.L., Newman, D.J., Schmidt, S.J., Taggart, J.J., and Gilvarg, C. 1986. Studies on the active site of succinyl-CoA: Tetrahydrodipicolinate N-succinyltransferase. J. Biol. Chem. 261 6160–6167. [PubMed] [Google Scholar]
- Binder, D.A., Blanchard, J.S., and Roderick, S.L. 1996. Crystallization and preliminary crystallographic analysis of tetrahydrodipicolinate-N-succinyltransferase. Proteins 26 115–117. [DOI] [PubMed] [Google Scholar]
- Brunger, A.T. 1992. Free R-value: A novel statistical quantity for assessing the accuracy of crystal structures. Nature 355 472–475. [DOI] [PubMed] [Google Scholar]
- Dicker, I.B. and Seetharam, S. 1992. What is known about the structure and function of the Escherichia coli protein FirA ? Mol. Microbiol. 6 817–823. [DOI] [PubMed] [Google Scholar]
- Gilvarg, C. and Weinberger, S. 1970. Bacterial distribution of the use of succinyl and aectyl blocking groups in diaminopimelic acid biosynthesis. J. Bacteriol. 101 323–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinberg, H., Lamdan, S., and Gaozza, C.H. 1975. Heterocycles derived from condensation of aliphatic diamines with succinic and glutaric acid-derivatives. J Heterocyclic Chem 12 763–766. [Google Scholar]
- Ishino, S., Mizukami, T., Yamaguchi, K., Katsumata, R., and Araki, K. 1987. Nucleotide sequence of the meso-diaminopimelate D-dehydrogenase gene from Corynebacterium glutamicum. Nucleic Acids Res. 15 3917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, T.A., Zou, J.-Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47 110–119. [DOI] [PubMed] [Google Scholar]
- Kabsch, W. 1988. Evaluation of single crystal X-ray diffraction data from a position sensitive detector. J Appl Crystallogr 21 916–924. [Google Scholar]
- Kleywegt, G.J. and Brunger, A.T. 1996. Checking your imagination: Applications of the free R value. Structure 4 897–904. [DOI] [PubMed] [Google Scholar]
- Kostrewa, D., D'Arcy, A., Takacs, B., and Kamber, M. 2001. Crystal structure of Streptococcus pneumoniae N-acetylglucosamine-1-phosphate uridyltransferase, GlmU, in apo form at 2.33 Å resolution and in complex with UDP-N-acetylglucosamine and Mg2+ at 1.96 Å. J. Mol. Biol. 305 279–289. [DOI] [PubMed] [Google Scholar]
- Martin, D.H., Bibart, R.T., and Drueckhammer, D.G. 1994. Synthesis of novel analogs of acetyl coenzyme A: Mimics of enzyme reaction intermediates. J. Am. Chem. Soc. 116 4660–4666. [Google Scholar]
- Misono, H., Ogaswara, M., and Nagasaki, S. 1986. Properties of meso-α,ɛ-diaminopimelate D-dehydrogenase from Bacillus sphaericus. Agric. Biol. Chem. 50 2729–2734. [Google Scholar]
- Olsen, L.R. and Roderick, S.L. 2001. Structure of the Escherichia coli GlmU pyrophosphorylase and acetyltransferase active sites. Biochemistry 40 1913–1921. [DOI] [PubMed] [Google Scholar]
- Radzicka, A., Pedersen, L., and Wolfenden, R. 1988. Influences of solvent water on protein folding: Free energies of solvation of cis and trans peptides are nearly identical. Biochemistry 27 4538–4541. [DOI] [PubMed] [Google Scholar]
- Raetz, C.R.H., Roderick, S.L. 1995. A left-handed parallel β helix in the structure of UDP-N-acetylglucosamine acyltransferase. Science 270 997–1000. [DOI] [PubMed] [Google Scholar]
- Sulzenbacher, G., Gal, L., Peneff, C., Fassy, F., and Bourne, Y. 2001. Crystal structure of Streptococcus pneumoniae N-acetylglucosamine-1-phosphate uridyltransferase bound to acetyl-coenzyme A reveals a novel active site architecture. J. Biol. Chem. 276 11844–11851. [DOI] [PubMed] [Google Scholar]
- Sundaharas, G. and Gilvarg, C. 1967. Biosynthesis of α,ɛ-diaminopimelic acid in Bacillus megaterium. J. Biol. Chem. 242 3983–3988. [PubMed] [Google Scholar]
- Tronrud, D.E., Ten Eyck, L.F., and Matthews, B.W. 1987. An efficient general-purpose least-squares refinement program for macromolecular structures. Acta Crystallogr. A 43 489–501. [Google Scholar]
- Vaara, M. 1992. Eight bacterial proteins, including UDP-N-acetylglucosamine acyltransferase (LpxA) and three other transferases of Escherichia coli, consist of a six-residue periodicity theme. FEMS Microbiol. Lett. 97 249–254. [DOI] [PubMed] [Google Scholar]