

Abstract
The HasASM hemophore, secreted by Serratia marcescens, binds free or hemoprotein bound heme with high affinity and delivers it to a specific outer membrane receptor, HasR. In HasASM, heme is held by two loops and coordinated to iron by two residues, His 32 and Tyr 75. A third residue His 83 was shown recently to play a crucial role in heme ligation. To address the mechanistic issues of the heme capture and release processes, the histidine protonation states were studied in both apo- and holo-forms of HasASM in solution. Holo-HasASM was formed with gallium-protoporphyrin IX (GaPPIX), giving rise to a diamagnetic protein. By use of heteronuclear correlation NMR spectroscopy, the imidazole side-chain 15N and 1H resonances of the six HasASM histidines were assigned and their pKa values and predominant tautomeric states according to pH were determined. We show that protonation states of the heme pocket histidines can modulate the nucleophilic character of the two axial ligands and, consequently, control the heme binding. In particular, the essential role of the His 83 is emphasized according to its direct interaction with Tyr 75.
Keywords: Histidine, tautomer, pKa, NMR, hemophore, gallium, protoporphyrin
Bacteria have developed various mechanisms for scavenging iron, thus allowing them to survive in iron-poor environments (Braun and Killmann 1999). One general mechanism involves the excretion of small inorganic iron chelators, termed siderophores, which display very high affinities for the metallic ion. The iron-loaded siderophores are recognized by specific outer membrane receptors that act in an energy-dependent way and allow the delivery of iron to the bacteria via a tonB-dependent process. Heme iron utilization is also widespread among bacterial pathogens. Various heme-containing compounds such as hemopexin, hemoglobin, haptoglobin–hemoglobin complex, or heme-loaded albumin are available within the host organism. One of these heme uptake systems is dependent on hemophores, which have a similar function to siderophores.
Hemophores (HasA) are small extracellular proteins secreted by an ABC transporter via their carboxy-terminal signal (Létoffé et al 1994a). They form an independent family of heme-binding proteins that are not homologous to any known proteins. The role of the hemophores is to bind free or hemoprotein-bound heme and to deliver it to a specific outer membrane receptor, HasR (Ghigo et al. 1997). The Serratia marcescens HasASM hemophore is a monomer (19 kD) that binds b heme with a stoichiometry of one and a very high affinity (Ka>108 M−1) (Izadi et al. 1997). The crystal structure of the holo-HasASM contains an original α/β fold and an unusual pattern of b heme ligation (Arnoux et al. 1999). Heme, which is highly exposed to solvents, is held by two loops and coordinated to iron by two residues, His 32 and Tyr 75. This histidine/tyrosine ligand pair of b heme iron has only been observed in alkaline ferric Chlamydomonas chloroplast hemoglobin (Das et al. 1999) and in a few mutated proteins (Nagai et al. 1989; Maurus et al. 1994). A third residue, His 83, which forms a hydrogen bond with Tyr 75, was shown recently to play a crucial role in heme ligation in an extensive study of HasASM mutant proteins (Létoffé et al. 2001).
Histidine residues often serve critical functional roles, acting either as nucleophiles or as electrophiles. The pKa of free imidazole rings is close to the physiological pH, allowing the ionization state of this side chain to be modulated readily by its environment within the protein (Nozaki and Tanford 1967; Tanokura 1983). The protonation state and hydrogen bonding of the His 32 and His 83 heme pocket histidines residues in HasASM might be essential for heme capture and release processes. Therefore, the 15N and 1H histidine imidazole signals in both apo- and holo-HasASM were followed with 15N heteronuclear correlation NMR spectroscopy as a function of pH to determine their pKa values and their protonation state.
Heme iron within the heme-HasASM complex is in a low spin ferric state and is, therefore, paramagnetic (Izadi et al. 1997). Moreover, its very low redox potential value (−550 mV vs. standard hydrogen electrode) precludes the reduction of the holo-protein in aerobic conditions. We thus chose a diamagnetic non-iron metalloporphyrin to form the holo-protein, the gallium-protoporphyrin IX (GaPPIX). This prevented any resonance shifts and relaxation effects induced by paramagnetism in the holo-protein and allowed us to observe and to compare the heme binding pocket histidines in apo- and holo-HasASM directly.
In this study, we measured the affinity constant of HasASM for GaPPIX. We assigned the imidazole side-chain 15N and 1H resonances, and determined the pKa values and the predominant tautomeric state according to the pH of all six histidines of HasASM in both its apo- and holo-forms. Protonation features of the histidines are discussed. Further insights into the structural factors of the heme pocket in both the apo- and holo-HasASM proteins are given; these allow us to address the mechanistic issues of the heme binding and release processes.
Results and Discussion
General features
Most of the protons attached directly to imidazole ring nitrogen atoms could not be observed by NMR because they exchange with water too quickly. In contrast, the nonexchangeable Hδ2 and Hɛ1 protons, which are linked to the nitrogens via weak two- and three-bond couplings, could be detected in 1H{15N}SBC and 1H{15N}MBC spectra. The cross-peak pattern enables the unambiguous assignment of the Nδ1 and Nɛ2 nitrogen as well as of the Hδ1 and Hɛ2 proton frequencies. Cross-peak intensities reflect the magnitude of the coupling constants between correlated 1H and 15N atoms. The magnitude of the three-bond 3JNδ1-Hδ2 coupling constant is lower than that of the 2JNH coupling constants in both the protonated and the neutral histidine ring. Thus, the 3JNδ1-Hδ2 coupling produces only weak or even unobservable cross-peak (Blomberg et al. 1977).
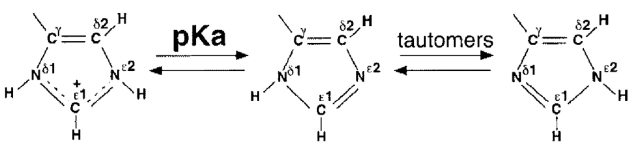 |
Specific assignment of the 15N and 1H resonances of the six histidine rings of HasASM were determined from 1H{15N}SBC and 1H{15N}MBC cross peak patterns (Table 1). Spin systems were assigned to His 32 and His 83 in apo-HasASM and in GaPPIX-HasASM by comparing the spectra recorded for the wild-type protein and for the His32Ala and His83Ala mutant proteins. The remaining histidines (His 17, His 128, His 133, and His 179) in apo-HasASM were identified from the correlations observed in 13C NOESY-HSQC spectra between histidines δ and β protons. These four histidines in GaPPIX-HasASM were assigned from the similarities between the apo-HasASM and the complexed HasA spectra. The protonation states of individual nitrogens were then inferred from 15N chemical shifts; in a positively charged ring, both protonated and partially charged nitrogens resonate at 176.5 ppm (type-α+) and in a neutral ring, an unprotonated nitrogen typically resonates at 249.5 ppm (type-β), whereas a protonated nitrogen resonates at 167.5 ppm (type-α), that is at 82 ppm upfield. Intermediate values are indicative of a fast exchange between the two tautomers and the α-type or β-type character of a nitrogen can be estimated from the differences between the observed and the theoretical chemical shift values (Van Dijk et al 1992; Pelton et al. 1993; Bhattacharya et al. 1997). An imidazole ring with an equal population of 15Nɛ and 15Nδ-protonated neutral tautomers would yield an average chemical shift of 210 ppm. However, the local environment and the hydrogen bonding of the histidine may induce compensatory shifts. Thus, a type-β nitrogen moves upfield (in the direction of the protonation) when behaving as an acceptor in a hydrogen bond, whereas a type α or α+ nitrogen moves downfield (in the direction of deprotonation) when behaving as a donor. The magnitude of these shifts can reach 10 ppm. Hence, information on the tautomeric equilibrium and the hydrogen bond status from the nitrogen chemical shifts must be collected carefully. Note that free histidine forms a mixture with appoximately a 4:1 preference for Nɛ2H protonation and displays an ∼50 ppm difference between its two imidazole nitrogen chemical shifts. Nɛ2 and Nδ1 atoms have distinct microscopic pKa; the Nɛ2 pKa being slightly more basic than the Nδ1 pKa (Tanokura 1983).
Table 1.
Observed 15N (left) and 1H (right) chemical shifts (ppm) of HasASM in apo- and holo-forms
| apo-HasASM | holo-HasASM | apo-HasASM | holo-HasASM | |||||||||
| Residue | Atom | δNAH+ | δAN | |δAN| | δNAH+ | δAN | |δAN| | Atom | δHAH+ | δAH | δHAH+ | δAH |
| His 17 | Nδ1 | 183.4 | nd | — | 184.1 | 172.2 | 71 | Hδ2 | 7.32 | nd | 7.34 | 6.85 |
| Nɛ2 | 171.0 | nd | 170.3 | 243.2 | Hɛ1 | 8.51 | nd | 8.52 | 7.63 | |||
| His 32 | Nδ1 | 173.6 | 254.4 | 78.3 | 164.6 | 163.4 | 70.3 | Hδ2 | 6.61 | 6.43 | 0.40 | 0.40 |
| Nɛ2 | 180.7 | 167.1 | 229.0 | 233.7 | Hɛ1 | 8.79 | 7.76 | 1.31 | 1.21 | |||
| His 83 | Nδ1 | nd | 227.3 | 59.4 | 185.2 | 165.4 | 76.9 | Hδ2 | 7.36 | 7.05 | 6.89 | 6.15 |
| Nɛ2 | 172.2 | 167.9 | 168.3 | 242.3 | Hɛ1 | 8.54 | 7.59 | 4.71 | 4.93 | |||
| His 128 | Nδ1 | 175.1 | 231.5 | 67.1 | 175.1 | 244.1 | 79.3 | Hδ2 | 6.63 | 6.35 | 6.60/6.47 | 6.33/6.2 |
| Nɛ2 | 169.4 | 164.4 | 170.1 | 164.8 | Hɛ1 | 8.66 | 7.62 | 9.04 | 7.95 | |||
| His 133 | Nδ1 | 253.5 | 253.5 | 92 | 257.2 | 256.5 | 95.5 | Hδ2 | 6.81 | 6.74 | 7.20 | 7.09 |
| Nɛ2 | 162.3 | 161.5 | 162.6 | 161 | Hɛ1 | 7.40 | 7.46 | 8.83 | 8.95 | |||
| His 179 | Nδ1 | 176.6 | 245.1 | 63.8 | 175.2 | 220.6 | 31.7 | Hδ2 | 7.33 | 6.97 | 7.28 | 6.92 |
| Nɛ2 | 172.6 | 181.3 | 172.1 | 188.9 | Hɛ1 | 8.64 | 7.64 | 8.61 | 7.67 | |||
The 15N and 1H chemical shifts of the protonated and deprotonated states were obtained from the experimental data by a least-square fitting procedure as described in the text. As only partial curve titration could be obtained for His 83 nitrogens, their chemical shifts are reported for the lowest and highest pH at which peaks were observed in the heteronuclear correlation NMR spectra. The same procedure was applied for His 133 in both HasASM apo- and holofoms and for His 32 in the HasASM holo-form, as their nitrogens and protons did not titrate within the pH range studied.
For titration with protoporphyrin, gallium seemed to be an apropriate substitute for ferric iron because Ga3+ and Fe3+ have a similar charge, atomic radius (0.62 Å vs. 0.65 Å) and coordination preferences (Martin 1988). Ga3+ is the only biochemically accessible redox state. Also, it has already been shown that enzymes or proteins that use heme as a cofactor can incorporate GaPPIX into their catalytic centres (Stojilkovic et al. 1999). Absorption spectoscopy showed that wild-type HasA binds GaPPIX with a stoichiometry of one and with a Ka value of 5.3(± 1.3).107 M−1. Moreover, when HasASM is complexed with gallium, it is able to interact with its receptor, HasR, and can deliver GaPPIX to the cell (S. Létoffé and C. Wandersman, unpubl.). Lastly, we recently determined the backbone assignment of GaPPIX-HasAsm (Deniau et al. 2001) and showed that the secondary structures and the overall fold of the protein are similar with those of heme-HasAsm.
The NMR spectra of GaPPIX-HasASM revealed a splitting of the heme pocket histidine peaks, with largest differences in chemical shift of 0.15 ppm in proton and 1 ppm in nitrogen. Two protein forms, in which the orientation of the heme differs by a 180° rotation around the αγ meso axis, have been found in most myoglobins, hemoglobins, and other b-type hemoproteins (Yamamoto et al. 1998). The functional consequence of the two forms has not yet been established. The X-ray crystal structure of HasASM at a resolution of 1.77 Å also showed that heme could take these two orientations in the crystal, with a ratio of about 1:1 (Arnoux et al. 2000). These data and the fact that the relative intensities of each couple of peaks were similar (∼40:60) for the heme pocket histidines, suggest that GaPPIX can also take two orientations in slow exchange on the NMR chemical shift time scale. The chemical environment of the protein atoms in the binding site (electronic structure of the pyrroles, metal-centered dipolar magnetic field, propionate side chains conformation) would only be influenced slightly by the heme rotation, explaining the low differences in chemical shifts observed between the two sets of peaks.
His 32
According to the X-ray structure, His 32 was almost completely buried in the holo-form of the protein. Its Nɛ2 was coordinated to the heme iron and its Nδ1 formed a tight hydrogen bond with the backbone carbonyl group of Asn 41.
The pKa of His 32 in apo-HasASM was neutral, slightly higher than the value of ∼6.8 found for a histidine residue in an unstructured peptide and consistent with a rather large accessibility to solvents (Table 2). The cross peak pattern observed in the 1H-15N NMR spectra at basic pH showed three connectivities, indicating the formation of the Nɛ2H tautomer. The missing Nδ1-Hδ2 cross peak appears in the protonated form of the residue, when the pH is lowered. The chemical shift values of type α and type β nitrogens were similar to the values for pure α and β types, with a difference of 78 ppm. These data show that the Nɛ2H tautomeric form is highly predominant and that it precludes in apo-HasASM the Asn 41 CO–His 32 Nδ1 interaction observed in the holo-HasASM structure.
Table 2.
Histidine pKa values in wild-type apo- and holo-HasASM and in Tyr75Ala mutant apo-HasASM
| pKa | ||||
| Residue | apo-HasA | GaPPIX-HasA | apo-Tyr75Ala | Accessible surface (Å2) |
| His 17 | >8.1 | 8.7 | >8.5 | 84/84 |
| His 32 | 7.3 | <4.8 | 7.5 | 7/43 |
| His 83 | 5.6 | 9.7 | 7.0 | 25/81 |
| His 128 | 7.1 | 7.4 | 7.2 | 92/106 |
| His 133 | <4.6 | <4.8 | <5.7 | 12/27 |
| His 179 | 7.1 | 7.4 | 7.2 | nd |
The pKa values were determined from fitting the pH titration curves of 15N and the 1H chemical shifts to a modified Henderson-Hasselbalch equation by nonlinear least-squares analysis.
The accessible surface (Å2) were calculated with and without (./.) b heme in the HasASM X-ray structure.
Spectra of GaPPIX-HasASM presented connectivities that are consistent with the formation of the His 32 Nδ1H tautomer. Thus, coordination to gallium occurs through the Nɛ atom as does coordination to iron. His 32 did not titrate over the pH range studied. Its pKa value, below 4.8, indicates that protonation of Nɛ and disruption of the ligation to gallium requires a very low pH. A slight chemical shift deviation was observed for Hɛ1 and Nɛ2 when the pH was above 9. As seen below, His 83 has a basic pKa of 9.7 in GaPPIX-HasASM, and this deviation probably results in a secondary effect due to His 83 titration. His 32 Hɛ1 and Hδ2 protons display highly upfield shifted chemical shifts, at 1.21 and 0.40 ppm, respectively (Fig. 1 ▶). Similar shifts caused by the protoporphyrin ring current have also been observed for the imidazole protons of iron coordinated histidines in other diamagnetic hemoproteins as follows: reduced cytochrome b5, hemoglobin-CO, cytochrome cd1 nitrite reductase (Guiles et al. 1990; Alam et al. 1998; Steensma et al. 2001). Although nitrogen chemical shifts are perturbed to a lesser extent than proton chemical shifts, they may also be affected by the protoporphyrin ring current. Thus, this would preclude the identification of hydrogen bonds from their sole chemical shifts values; an upfield shift of 4.5 ppm caused by heme ring current was thus observed in ferrocytochrome c2 of Rodospirillum rubrum for the Nδ1 of the coordinated histidine (Yu and Smith 1990). Therefore, the His 32 Nδ1 chemical shift value did not allow us to determine unambiguously the hydrogen bonding of His 32 to Asn 41, although the bond between His 32 Hδ1 and the backbone carbonyl of Asn 41 is likely to exist in GaPPIX-HasASM as in the heme-HasASM complex. In other hemoproteins with an iron coordinated histidine, a hydrogen bond was also observed between the axial histidine Hδ1 and a carbonyl group of the backbone or an amino acid side chain of the protein (Rousseau and Rousseau 1992). This bond makes the histidine more anionic and therefore reinforces the strength of the coordination.
Fig. 1.

Portions of 1H{15N}MBC spectra of uniformly 15N labeled HasA in the apo-form (left) and in the holo-form (right) in 20 mM sodium phosphate buffer (pH 5.6) and 30°C. Only His 17, His 32, His 83, His 128, and His 133 cross peak patterns are indicated.
His 83
The structure of holo-HasASM showed that His 83 is located at the edge of the heme pocket, exposed to solvent, and that it forms a hydrogen bond via its Nδ1 with the hydroxyl group of Tyr 75. In most apo-HasASM spectra, the His 83 resonances are weak or undetectable and only partial 15N and 1H titration curves could be obtained. As the JNH coupling constant values only varied slightly with the protonation state of the imidazole ring, the cross-peaks for His 83 probably disappear due to line broadening of the 1H and/or 15N signals as a result of intermediate exchange of tautomerization (Blomberg et al. 1977). The magnitude of the rate constant in the tautomeric equilibrium is critical for the spectral appearance of the resonance. The His 83 cross-peak pattern indicated the formation of a predominant Nɛ2H tautomeric form. The nitrogen chemical shift separation, 59 ppm, was low compared with that of pure α and β types. However, as nitrogen chemical shifts could not be obtained over the entire pH range (Fig. 2 ▶), it is therefore difficult to comment on Nδ1H–Nɛ2H tautomerization. The low value obtained for Nδ1 (227.3 ppm) could be partially conferred to the hydrogen bonding between His 83, the hydrogen acceptor via its Nδ1, and Tyr 75 acting as a hydrogen donor through its hydroxyl proton Hη. This hydrogen bond that stabilizes the neutral form of His 83 may account for the low pKa of this residue (5.6).
Fig. 2.

Heme pocket histidine 15N chemical shifts of HasASM for the apo-form (left) and for the holo-form (right) as a function of pH. The solid lines represent the least-squares best fit to the data. The titration curve for the His 83 1H resonance in the apo-HasASM is shown in the bottom right corner because only partial 15N titration curves didn't allow to determine the pKa value by fitting the data for this residue.
To investigate His 83–Tyr 75 hydrogen bonding, we followed the titration of the histidines as a function of pH in the Tyr75Ala mutant apo-protein. The only histidine that was significantly affected by the loss of the tyrosine was His 83. Its pKa increased by 1.4 unit (7.0), which is close to the value for a free histidine. This strongly suggests the direct influence of Tyr 75 on its protonation state (Table 2). The Nɛ2H tautomer was predominant but, as in the wild-type protein, the Nδ1 chemical shift limits could not be reached. The highest value obtained, 236 ppm, did not allow to state that H83 is involved in a hydrogen bond.
GaPPIX binding highly affected the cross peak pattern and the pKa value of His 83. Connectivities and nitrogen resonance separation in GaPPIX-HasASM spectra indicate the formation of the Nδ1H tautomer instead of Nɛ2H in the apo-form of the protein. Hɛ1 and Hδ2 protons are upfield shifted (4.93 and 6.15 ppm, respectively), reflecting the protoporphyrin ring current effect, although this was less pronounced than for His 32. The pKa became extremely basic (9.7), thus, His 83 remained in a protonated state over a large pH range. The proximity of the protoporphyrin prevented us from determining the hydrogen bonding of the imidazole from its Nδ1 and Nɛ2 chemical shifts. However, the very high and unusual pKa of His 83 is probably due to a strong interaction between the proton Nδ1H and the deprotonated phenolic oxygen, Oη. The low redox potential of holo-HasASM (−540 mV) and the ferric state of the iron (Izadi et al. 1997) were consistent with localized Oη negative charge favoring the binding to the metallic ion.
We observed two sets of peaks for His 83 in the GaPPIX-Tyr75Ala mutant protein. The imidazole proton chemical shifts in both sets were shifted compared with those of the apo-protein. This is in agreement with the proximity of the protoporphyrin. At neutral pH, one set was consistent with a neutral imidazole ring, and the other with a protonated one. All other histidines behaved in the same manner in the GaPPIX-Tyr75Ala protein and in the GaPPIX-wild-type protein. A recent study on HasASM mutant proteins (Létoffé et al. 2001) found that His 83 appears to be an alternative iron coordination residue. The neutral form of His 83 observed at neutral pH might correspond to an axial ligand of the metal via its Nδ1 in absence of the natural ligand, Tyr 75. However, coordination of histidines to b heme iron in hemoproteins were thought to occur through their Nɛ2. Therefore, we have to confirm this type of Nδ1 ligation. The protonated form may correspond to HasASM molecules with just one remaining axial ligand (His 32).
His 128 and His 133
As shown by the crystal structure of holo-HasASM, His 128 and His 133 are located at the bottom of the heme pocket. His 128 is very accessible to solvent and its Nɛ2 forms a hydrogen bond with the backbone carbonyl group of Ala 82. His 133 is completely buried and its Nɛ2 forms a hydrogen bond with the backbone carbonyl group of His 83. These two imidazole rings are parallel to each other and separated by ∼4 Å.
His 128 has a neutral pKa in apo- and GaPPIX-HasASM (7.1 and 7.4, respectively). It predominantly adopts the Nɛ2H tautomeric form in both apo-HasASM and GaPPIX-HasASM. These results are consistent with the interaction between His 128 Nɛ2 and Ala 82 CO observed in the crystal structure, but the nitrogen chemical shift difference did not allow us to conclude about the formation of a hydrogen bond.
His 133 does not titrate over the pH range studied in apo-HasASM (pH 4 to pH 10) or in GaPPIX-HasASM (pH 4.8 to pH 11). The cross peak pattern and nitrogen chemical shifts clearly indicate that the residue is in the neutral state and adopts the Nɛ2H tautomeric form. These data suggest that the side chain of His 133 forms a strong hydrogen bond and/or this residue is located in an environment that is particularly resistant to the protonation. It is likely that in both apo-HasASM and GaPPIX-HasASM, His 133 is less accessible to solvent and is hydrogen bonded through its Nɛ2H to the carbonyl group of His 83.
GaPPIX binding induces moderate (<1.5 ppm) downfield shifts of the proton chemical shifts of both His 128 and His 133 rings. These shifts reflect a weak protoporphyrin ring current effect, which is consistent with distances of over 7 Å between histidine ring and the metallic ion.
His 17 and His 179
According to the X-ray structure of holo-HasASM, His 17 belongs to helix α2. It is located at the interface between helix α2 and helix α3 and is highly exposed to solvents. Its Nδ1 is hydrogen bonded to the side chain carbonyl of Glu 148. His 179 is within the C-terminal region, which is shown to be highly flexible in a previous NMR study on apo-HasASM and belongs to the C-terminal 14 residues not observed in the X-ray structure (Izadi-Pruneyre et al. 1999b). As predicted by the distance of His 17 and His 179 from the binding site, the cross peak patterns and the pKa values of His 17 and His 179 are fairly unaffected by the GaPPIX binding. The spectra indicate the formation of predominant Nδ1H and Nɛ2H tautomers for His 17 and His 179, respectively. The predominance of the His 17 Nδ1H tautomer supports the existence of the hydrogen bond observed in the crystal structure between its Hδ1 proton and Glu 148 side-chain carbonyl group, regardless of pH. The basic pKa of imidazole is probably due to the stabilization of the protonated ring through this interaction. The Nɛ2H tautomeric state of His 179 is more populated in the apo-form of HasASM and less populated in the GaPPIX complex than in free histidine, although no obvious hydrogen bonding account for these data.
Conclusion
The NMR data for the GaPPIX-HasASM complex are consistent with the topology of the heme-binding pocket that was determined by X-ray diffraction (Fig. 3 ▶) and with our study on the heme binding of HasASM and its mutants. As shown by the cross-peak patterns and by the titration curves, GaPPIX binding has a major effect on the protonation state and the ring atom chemical shifts of the two histidines that are directly implicated in the heme-binding process: His 32 and His 83. Their tautomeric states change when GaPPIX binds and they show the largest differences in the pKa values between apo- and GaPPIX-HasA forms, resulting in distinct hydrogen bond networks. GaPPIX binding only has a minor effect on the four other histidines.
Fig. 3.

(A) Ribbon representation of the overall HasA X-ray crystal structure with the location of b heme, histidines, and Tyr 75 residues (PDB entry 1B2V; Arnoux et al. 1999). (B) Detailed view of the b heme structural environment. Nitrogens are shown in green, oxygen in light blue, hydrogens in black, carbons in red, and backbone in yellow.
His 32 must be in a neutral state for heme or GaPPIX binding, because coordination occurs through its unprotonated nitrogen. According to its pKa, His 32 is deprotonated in apo-HasASM for pH values higher than 7.3 and predominantly adopts a Nɛ2H tautomeric form. At these pH, His 83 is also deprotonated (pKa = 5.6) and can form a hydrogen bond via its Nδ1 with the phenolic proton Hη of Tyr 75. As HasASM binds heme, the tautomeric equilibrium of His 32 is displaced toward the formation of the Nδ1 tautomer, Nɛ2 coordinating to the metallic ion. His 83 is protonated (pKa = 9.7) and can act as a hydrogen donor to form a hydrogen bond with the Oη of Tyr 75 via its Nδ1H, thus enhancing the nucleophilic character of the phenolate and increasing the strength of the coordination to the metallic ion (Fig. 4 ▶). This result is consistent with our previous data on HasASM mutant proteins showing that Tyr 75 is a stronger ligand than His 32 and, therefore, is in an unprotonated state (Létoffé et al. 2001). The study on the Tyr75Ala mutant apo-protein confirmed the direct influence of Tyr 75 on the His 83 protonation state. Our results strongly support that His 83 may control the protonation state of the Tyr 75 side chain. It is noteworthy that, unlike His 32, the couple Tyr 75/His 83 is conserved in the three other known hemophores: Pseudomonas fluorescens (HasAPF), Pseudomonas aeruginosa (HasAPA), and Yersinia pestis (HasAYP). A hydrogen bond between Tyr 75 and His 83 was observed in the crystal structure of heme-HasAsm at pH 4.6 without presuming of the hydrogen donor. We have shown here that His 83 is protonated up to pH 4.8 and thus can act as the hydrogen donor. Tyr 75 is therefore deprotonated up to pH 4.8. The strong interaction between the negative tyrosinate ion and the positive imidazole could account for the very low pKa value of Tyr 75. Unusual acid stability of tyrosinate ion was already observed in other proteins. In ferric Chlamydomonas hemoglobin, it was proposed that the low pKa of the tyrosine ligand, about four units lower than that of free tyrosine, resulted from an interaction of the tyrosinate with a neighboring lysine (Das et al. 1999). Similarly, in UDP-galactose 4-Epimerase, a positive electrostatic field was proposed to strongly stabilize the phenolate form of a tyrosine in the active site (Liu et al. 1997).
Fig. 4.

Schematic representation of the Tyr 75–His 83 interaction in apo-HasSM (left) and in GAPPIX-HasSM (right) at neutral pH. The dotted lines represent the hydrogen bond between the two residues.
Disruption of the metal coordination to His 32 by protonation of the imidazole needs an acidic pH, because His32Nɛ2 still is deprotonated at pH 4.8. The break of the metal coordination to Tyr 75 may occur via the protonation of the phenolate, which is controlled by His 83 at very basic pH values, His 83 remaining protonated up to 9.7. However, the release of heme involves a protein–protein interaction between HasASM and its receptor HasRSM, which probably induces conformational and dynamics changes, and variations in the heme environment. Analysis of the HasASM–HasRSM complex, which is in progress in our laboratories, will allow us to complete the description of the heme release mechanism.
Materials and methods
Expression and purification of the HasASM proteins
Wild-type HasASM was expressed in Escherichia coli strain POP3 transformed with plasmid pSYC34 (pAM238) (Létoffé et al. 1994b). Mutant hasA genes were constructed by in vitro site-directed mutagenesis (Létoffé et al. 2001). They were cloned in pAM238 and introduced into strain POP3 (pSYC15O) (Létoffé et al. 1994a). Uniformly labeled samples were produced in M9 minimal medium containing 15NH4Cl as the sole nitrogen source, as reported previously (Izadi-Pruneyre et al. 1999a). Wild-type and mutant apo-HasASM proteins (His32Ala, Tyr75Ala, His83Ala, His32Ala–Tyr75Ala) were purified as described previously (Izadi et al. 1997). Their heme content, determined from the absorbance of the Soret band wavelength, was always <.0.5 %.
Absorption spectrometry and Ka determination
Heme-binding experiments were carried out in 50 mM sodium phosphate buffer (pH 7.3) at room temperature by measuring the absorbance in the Soret region. Aliquots of GaPPIX solution (Adler et al. 1970) were successively added to the cell containing the apo-protein. Fresh GaPPIX solution was prepared immediately prior to the titration as follows: GaPPIX was dissolved in a minimal volume of 0.1 N NaOH and diluted with phosphate buffer to the desired concentration using the ɛ413, DMSO value of 249000 M−1.cm−1 (V. Kumar and I. Stojiljkovic, pers. comm.). Additional experimental details are given in reference Létoffé et al. (2001). The Soret band absorbance, ASoret, was measured with a base line drawn between 300 and 450 nm and reported as a function of the GaPPIX concentration in the cell. The curve was fitted using the kaleidaGraph 3.5 software and the Ka determined.
NMR sample preparation
When necessary, saturating amounts of gallium-protoporphyrin IX (GaPPIX) solution were added to the apo-HasASM wild-type and mutant proteins by following the absorbance in the Soret region. Samples were dialyzed against water, freeze dried, and dissolved in 20 mM phosphate buffer, 99,97% 2H2O (pH 5.6). The concentrations of the wild-type and mutant proteins in both apo- and GaPPIX forms were ∼1 and 0.3 mM, respectively. pH were adjusted to the desired values with 0.1 M 2HCl or NaO2H. pH were measured at room temperature before and after NMR data collection. The values were not corrected for the deuterium isotope effect.
NMR spectroscopy
NMR spectra were recorded at 30°C on a Varian Inova spectrometer operating at 500 MHz for protons and equipped with a triple resonance z-gradient probe. Data processing was performed with standard Varian NMR software. 1H{15N}SBC (Bodenhausen and Ruben 1980; Sklenar et al. 1994) and 1H{15N}MBC (Bax and Summer 1986) the 2D spectra were recorded for the wild-type and mutant proteins at each pH value. The delay for observation of the long-range proton-nitrogen correlation of histidine was 30 ms (JN-H.∼15 Hz), which gave the best signal to noise ratio. Experimental parameters were as follows: 1H and 15N sweep widths of 10000 Hz and 8500 Hz, respectively, 64 and 512 complex t1 and t2 points, respectively, and 128 or 256 scans. Prior to Fourier transformation, each data set was zero filled to 512 and 2048 points in F1 and F2 dimensions, respectively, and the data were multiplied in both dimensions by a phase-shifted, skewed, sinebell window function. FIDs were Fourier transformed without baseline correction. Chemical shifts were measured relative to the external DSS for 1H and calculated assuming γN/γH = 0.101329118.
pH titration and calculation of pKa
NMR spectra were recorded at 20 pH values between pH 4.0 and pH 10.0 for the wild-type apo-HasASM and between pH 4.8 and pH 11.0 for the GaPPIX-HasASM. Experiments were performed at 10 pH values between pH 4.9 and pH 9.0 for the apo-Tyr75Ala mutant. Proteins precipitated below pH 4.5.
The pKa values of the six HasASM histidines were determined from the pH dependencies of their 15N and 1H chemical shifts. The pH titration curves were fitted to a modified Henderson-Hasselbalch equation by nonlinear least-squares analysis:
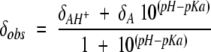 |
in which δobs is the chemical shift observed at each pH value and δAH+ and δA are the chemical shifts for the protonated and deprotonated histidines, respectively. Curve fits were performed using the KaleidaGraph3.5 Software (Synergy Software).
Structure analysis
The X-ray holo-HasA structure (PDB filecode 1B2V, Arnoux et al. 1999) was analyzed. This allowed us to calculate the solvent accessibility of histidines and to check the hydrogen-bonding pattern involving these residues by use of Insight II Biosym/MSI.
Acknowledgments
We thank Drs Philippe Delepelaire and Laurent Debarbieux for fruitful discussions and for critical reading.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
HasA, Haem acquisition system A
GaPPIX, gallium-protoporphyrin IX
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.3630102.
References
- Adler, A.D., Longo, F.R., Kampas, F., and Kim, J. 1970. Preparation of metalloporphyrins. J. Inorg. Nucl. Chem. 32 2443. [Google Scholar]
- Alam, S.L., Volkman, B.F., Markley, J.L., and Satterlee, J.D. 1998. Detailed NMR analysis of the heme-protein interactions in component IV Glycera dibranchiata monomeric hemoglobin-CO. J. Biomol. NMR 11 119–133. [DOI] [PubMed] [Google Scholar]
- Arnoux, P., Haser, R., Izadi, N., Lecroisey, A., Delepierre, M., Wandersman, C., and Czjzek, M. 1999. The crystal structure of HasA, a hemophore secreted by Serratia marcescens. Nat. Struct. Biol. 6 516–520. [DOI] [PubMed] [Google Scholar]
- Arnoux, P., Haser , R., Izadi-Pruneyre, N., Lecroisey, A., and Czjzek, M. 2000. Functional aspects of the heme bound hemophore HasA by structural analysis of various crystal forms. Proteins 41 202–210. [DOI] [PubMed] [Google Scholar]
- Bax, A. and Summer, M. 1986. H-1 and C-13 assignments from sensitivity-enhanced detection of heteronuclear multiple-bond connectivity by 2D multiple quantum NMR. J. Am. Chem. Soc. 108 2093–2094. [Google Scholar]
- Bhattacharya, S., Sukits, S., MacLaughlin, K., and Lecomte, J. 1997. The tautomeric state of histidine in myoglobin. Biophys. J. 73 3230–3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomberg, F., Maurer, W., and Rüterjans, H. 1977. Nuclear magnetic resonance investigation of 15N-labeled histidine in aqueous solution. J. Am. Chem. Soc. 99 8149–8159. [DOI] [PubMed] [Google Scholar]
- Bodenhausen, L. and Ruben, D.J. 1980. Natural abundance N-15 NMR by enhanced heteronuclear spectroscopy. Chem. Phys. Lett. 69 185–189. [Google Scholar]
- Braun, V. and Killmann, H. 1999. Bacterial solutions to the iron-supply problem. Trends Biochem Sci 24 104–109. [DOI] [PubMed] [Google Scholar]
- Das, T.K., Couture, M., Lee, H.C., Peisach, J., Rousseau, D.L., Wittenberg, B.A., Wittenberg, J.B., and Guertin, M. 1999. Identification of the ligands to the ferric heme of Chlamydomonas chloroplast hemoglobin: Evidence for ligation of tyrosine-63 (B10) to the heme. Biochemistry 38 15360–15368. [DOI] [PubMed] [Google Scholar]
- Deniau, C., Couprie, J., Simenel, C., Kumar, V., Stojiljkovic, I., Wandersman, C., Delepierre, M., and Lecroisey A. 2001. J. Biomol. NMR 21 189–190. [DOI] [PubMed] [Google Scholar]
- Ghigo, J. M., Létoffé, S., and Wandersman, C. 1997. A new type of hemophore-dependent heme acquisition system of Serratia marcescens reconstituted in Escherichia coli. J. Bacteriol. 179 3572–3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guiles, R.D., Altman, J., Kuntz, I.D., and Waskell, L. 1990. Structural studies of cytochrome b5: Complete sequence-specific resonance assignments for the trypsin-solubilized microsomal ferrocytochrome b5 obtained from pig and calf. Biochemistry 29 1276–1289. [DOI] [PubMed] [Google Scholar]
- Izadi, N., Henry, Y., Haladjian, J., Goldberg, M. E., Wandersman, C., Delepierre, M., and Lecroisey, A. 1997. Purification and characterization of an extracellular heme-binding protein, HasA, involved in heme iron acquisition. Biochemistry 36 7050–7057. [DOI] [PubMed] [Google Scholar]
- Izadi-Pruneyre, N., Wolff, N., Castagne, C., Czisch, M., Wandersman, C., Delepierre, M., and Lecroisey, A. 1999a. Backbone NMR assignment and secondary structure of the 19 kDa hemophore HasA. J. Biomol. NMR 14 193–194. [DOI] [PubMed] [Google Scholar]
- Izadi-Pruneyre, N., Wolff, N., Redeker, V., Wandersman, C., Delepierre, M., and Lecroisey, A. 1999b. NMR studies of the C-terminal secretion signal of the haem-binding protein, HasA. Eur. J. Biochem. 261 562–568. [DOI] [PubMed] [Google Scholar]
- Létoffé, S., Ghigo, J.M., and Wandersman, C. 1994a. Secretion of the Serratia marcescens HasA protein by an ABC transporter. J. Bacteriol. 176 5372–5377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Létoffé, S., Ghigo, J.M., and Wandersman, C. 1994b. Iron acquisition from heme and hemoglobin by a Serratia marcescens extracellular protein. Proc. Natl. Acad. Sci. 91 9876–9880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Létoffé, S., Deniau, C., Wolff, N., Dassa, E., Delepelaire, P., Lecroisey, A., and Wandersman, C. 2001. Haemophore-mediated bacterial haem transport: Evidence for a common or overlapping site for haem-free and haem-loaded haemophore on its specific outer membrane receptor. Mol. Microbiol. 41 439–450. [DOI] [PubMed] [Google Scholar]
- Liu Y., Thoden, J.B., Berger, E., Gulick, A.M., Ruzicka, F.J., Holden, H.M.., and Frey, P.A. 1997. Mechanistic roles of tyrosine 149 and serine 124 in UDP-galctose 4-epimerase from Escherichia coli. Biochemistry 36 10675–10684. [DOI] [PubMed] [Google Scholar]
- Martin, R.B. 1988. Bioinorganic chemistry of aluminium. Met. Ions Biol. Syst. 24 1–57. [Google Scholar]
- Maurus, R., Bogumil, R., Luo, Y., Tang, H.L., Smith, M., Mauk, A.G., and Brayer, G.D. 1994. Structural characterization of heme ligation in the His64→Tyr variant of myoglobin. J. Biol. Chem. 269 12606–12610. [DOI] [PubMed] [Google Scholar]
- Nagai, M., Yoneyama, Y., and Kitagawa, T. 1989. Characteristics in tyrosine coordinations of four hemoglobins M probed by resonance Raman spectroscopy. Biochemistry 28 2418–2422. [DOI] [PubMed] [Google Scholar]
- Nozaki, Y. and Tanford, C. 1967. Examination of titration behavior. Methods Enzymol. 11 715–734. [Google Scholar]
- Pelton, J.G., Torchia, D A., Meadow, N.D., and Roseman, S. 1993. Tautomeric states of the active-site histidines of phosphorylated and unphosphorylated IIIGlc, a signal-transducing protein from Escherichia coli, using two-dimensional heteronuclear NMR techniques. Protein Sci. 2 543–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau, D.G. and Rousseau, D.L. 1992. Hydrogen bonding of iron-coordinated histidine in heme proteins. J. Struct. Biol. 109 13–17. [DOI] [PubMed] [Google Scholar]
- Sklenar, V., Peterson, R.D., Rejante, M.R., and Feigon, J. 1994. Correlation of nucleotide base and sugar protons in a 15N-labeled HIV-1 RNA oligonucleotide by 1H-15N HSQC experiments. J. Biomol. NMR 4 117–122. [DOI] [PubMed] [Google Scholar]
- Steensma, E., Gordon, E., Öster, L., Ferguson, S.J., and Hajdu, J. 2001. Heme ligation and conformational plasticity in the isolated c domain of cytochrome cd1 nitrite reductase. J. Biol. Chem. 276 5846–5855. [DOI] [PubMed] [Google Scholar]
- Stojiljkovic, I., Kumar, V., and Srinivasan, N. 1999. Non-iron metalloporphyrins: Potent antibacterial compounds that exploit haem/Hb uptake systems of pathogenic bacteria. Mol. Microbiol. 31 429–442. [DOI] [PubMed] [Google Scholar]
- Tanokura, M. 1983. 1H-NMR study on the tautomerism of the imidazole ring of histidine residues. I. Microscopic pK values and molar ratios of tautomers in histidine-containing peptides. Biochim. Biophys. Acta 742 576–585. [DOI] [PubMed] [Google Scholar]
- Van Dijk, A.A., Scheek, R.M., Dijkstra, K., Wolters, G.K., and Robillard, G.T. 1992. Characterization of the protonation and hydrogen bonding state of the histidine residues in IIAmtl, a domain of the phosphoenolpyruvate-dependent mannitol-specific transport protein. Biochemistry 31 9063–9072. [DOI] [PubMed] [Google Scholar]
- Yamamoto, Y., Nakashima, T., Kawano, E., and Chûjô, R. 1998. 1H-NMR investigation of the influence of the heme orientation on functional properties of myoglobin. Biochim. Biophys. Acta 1388 349–362. [DOI] [PubMed] [Google Scholar]
- Yu, L.P. and Smith, G.M. 1990. Characterization of pH-dependent conformational heterogeneity in Rhodospirillum rubrum cytochrome c2 using 15N and 1H NMR. Biochemistry 29 2920–2925. [DOI] [PubMed] [Google Scholar]