

Abstract
The hyperthermophilic bacterium Thermotoga maritima encodes a gene sharing sequence similarities with several known genes for alkaline phosphatase (AP). The putative gene was isolated and the corresponding protein expressed in Escherichia coli, with and without a predicted signal sequence. The recombinant protein showed phosphatase activity toward the substrate p-nitrophenyl-phosphate with a kcat of 16 s−1 and a Km of 175 μM at a pH optimum of 8.0 when assayed at 25°C. T. maritima phosphatase activity increased at high temperatures, reaching a maximum kcat of 100 s−1, with a Km of 93 μM at 65°C. Activity was stable at 65°C for >24 h and at 90°C for 5 h. Phosphatase activity was dependent on divalent metal ions, specifically Co(II) and Mg(II). Circular dichroism spectra showed that the enzyme gains secondary structure on addition of these metals. Zinc, the most common divalent metal ion required for activity in known APs, was shown to inhibit the T. maritima phosphatase enzyme at concentrations above 0.3 moles Zn: 1 mole monomer. All activity was abolished in the presence of 0.1 mM EDTA. The T. maritima AP primary sequence is 28% identical when compared with E. coli AP. Based on a structural model, the active sites are superimposable except for two residues near the E. coli AP Mg binding site, D153 and K328 (E. coli numbering) corresponding to histidine and tryptophan in T. maritima AP, respectively. Sucrose-density gradient sedimentation experiments showed that the protein exists in several quaternary forms predominated by an octamer.
Keywords: Alkaline phosphatase, Thermotoga maritima, metalloenzymes, heat stable proteins
Alkaline phosphatase (AP) (EC 3.1.3.1) catalyzes the nonspecific hydrolysis of phosphomonoesters. This ubiquitous enzyme is found in organisms from all three biological kingdoms, although most of the current data on AP has been determined with enzymes isolated from prokaryotic organisms such as Bacillus subtilis and Escherichia coli. These APs have a high degree of primary sequence homology, especially at the active site, and require divalent metal ions for catalysis and active site structure (Holtz and Kantrowitz 1999; Hulett et al. 1991). Because E. coli AP is a well-characterized homodimeric metalloenzyme, it can serve as a basis for comparison to enzymes with similar homology.
Proteins from extremophilic organisms are of interest for their unique characteristics and stability. Thermotoga maritima, isolated from geothermal heated marine sediment, represents the first hyperthermophile found from bacterial, not archaeal, origins (Huber et al. 1986). Its genome is the result of lateral gene transfer because 52% of predicted coding sequences are most similar to proteins in bacterial species, namely, B. subtilis, and 24% of predicted coding sequences are most similar to proteins in archaeal species (Nelson et al. 1999). T. maritima grows optimally at a temperature of 80°C and a salt concentration of 2.5 M. It is gram-negative and has a unique outer sheath structure (Huber et al. 1986).
From sequence alignment, the T. maritima AP gene is closely related to the B. subtilis phoAIII gene, which produces a homodimeric AP requiring Co(II) for activity. T. maritima AP also shows strong homology with the B. subtilis phoAIV gene product, the monomeric Co(II)-requiring AP, and the E. coli enzyme. We have determined the activity, quaternary structure, thermostability, and metal preference of T. maritima AP. The preference for Co(II) in T. maritima AP is analogous to the metal dependence found in the B. subtilis enzymes. On comparison with the Zn(II)-requiring E. coli AP, it can be shown that this change in metal preference may be attributed to specific amino acid substitutions near the metal-binding positions in the active site.
Results and Discussion
Alignment, sequence comparison, and modeling
A protein BLAST search was performed to find sequences that most closely matched the T. maritima AP sequence (Table 1). The gene products from B. subtilis phoAIII and phoAIV genes were among the most homologous proteins. These were chosen for further study because the signal sequence cleavage point and metal requirement for these enzymes had already been characterized (Hulett et al. 1990; Hulett et al. 1991). Although the E. coli AP was not among the most similar, it is the only AP from a bacterial source whose structure has been solved (Kim and Wyckoff 1991); therefore, it was also used for comparisons to T. maritima AP.
Table 1.
Closest matches to the T. maritima AP sequence from the BLAST databasea
| E. coli | |||||
| Organism of AP origin | Identity to T. maritima AP | Metals | 153 | 328 | Reference |
| T. maritima | 100% | Co(II) Mg(II) | H | W | Nelson et al. 1999 |
| P. abyssi | 44% | Unknown | H | H | Zappa et al. 2001 |
| B. halodurans | 42% | Unknown | H | W | Takami et al. 2000 |
| Lactobacillus | 40% | Unknown | H | W | AAG33874b |
| E. faecalis | 39% | Unknown | H | W | Lee et al. 1999 |
| TAB5 | 38% | Mg(II) | H | W | Rina et al. 2000 |
| B. subtilis phoAIII | 37% | Co(II) | H | W | Hulett et al. 1991 |
| S. aureus | 35% | Unknown | D | K | Kuroda et al. 2001 |
| B. subtilis phoAIV | 35% | Co(II) | H | W | Hulett et al. 1991 |
| B. lichenformis | 33% | Unknown | H | W | Kim et al. 1998a |
| Vibrio sp. | 32% | Unknown | H | W | Asgeirsson and Andresson 2001 |
| E. coli | 28% | Zn(II) Mg(II) | D | K | Kim and Wyckoff 1991 |
a Only residues that represent major changes from the E. coli AP metal-binding and active-site residues are indicated.
b NCBI database accession number.
A primary sequence alignment was performed using the T. maritima AP sequence and three homologous AP enzymes (Fig. 1 ▶). As seen from the alignment, T. maritima AP is very similar to both the B. subtilis AP proteins and the well-characterized E. coli AP, especially in the active site and metal-binding regions. This sequence alignment was also used to generate a structural model of the monomeric T. maritima AP using the structure 1ED9 (Stec et al. 2000) with the program SWISS-MODEL (Peitsch 1995; Guex et al. 1999; Guex and Peitsch 1997). The sequence alignment and structural model were used to compare the active sites of T. maritima and E. coli AP (Fig. 2A ▶), as well as the overall predicted structure of the monomer (Fig. 2B ▶).
Fig. 1.
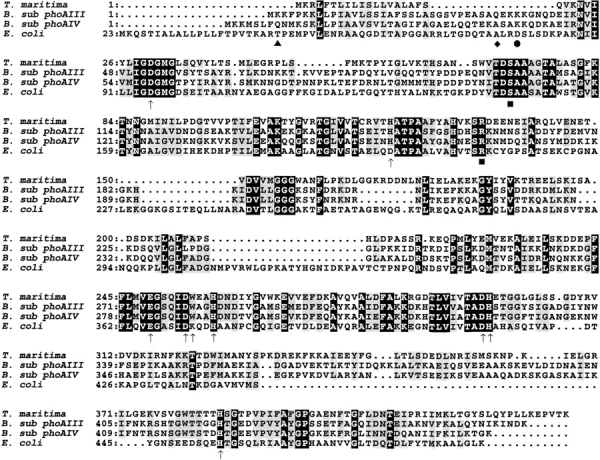
Sequence alignment of Bacillus subtilis phoAIII, phoAIV gene products, Thermotoga maritima, and Escherichia coli alkaline phosphatases (APs). Residues associated with metal binding in the E. coli AP active sites are labeled with (↑). These residues are completely conserved in all four of these examples of AP, with the exception of D153H and K328W (mature E. coli AP numbering). The N-terminus of the B. subtilis phoAIII AP mature protein is indicated with (♦), the N-terminus of the B. subtilis phoAIV AP mature protein is indicated with (•), the N-terminus of the E. coli mature AP is labeled with (▴), and the serine nucleophile and conserved arginine are labeled with (▪).
Fig. 2.

SWISS-MODEL of T. maritima (purple) on E. coli (elementally colored [top] or blue [bottom]) produced using the sequence alignment generated for Fig. 1 ▶ and the ED9A E. coli AP coordinates (Stec et al. 2000). (Top) T. maritima and E. coli AP active site overlay showing metal-binding residues highlighted in Fig. 1 ▶. The two zinc ions (blue) and magnesium (cyan) are from the E. coli AP coordinates (Stec et al. 2000). Green dotted lines indicate the salt bridge between E. coli AP residues D153 and K328 and the residue-metal interactions; water-ligand interactions to the magnesium have been eliminated for clarity. (Bottom) Monomeric model of T. maritima AP on E. coli AP. The putative signal sequence has been eliminated from the T. maritima protein as predicted by SignalP and the sequence alignment as stated in the text. The termini and differing regions are highlighted with E. coli (EC) or T. maritima (TM) numbering. T. maritima AP is numbered from the first amino acid including the putative signal sequence.
Residues implicated in the reaction mechanism, S1021 and R166, are completely conserved in all proteins for which sequence data are available. Most residues involved in binding the metal ions, directly or indirectly, are conserved. In E. coli AP, Zn1 is bound by the imidazole nitrogens of H412 and H331 and the carboxyl of D327 (Kim and Wyckoff 1991). All these positions are conserved in T. maritima AP, as well as in all other sequences shown in these alignments. E. coli AP residues D369 and H370 along with one carboxyl oxygen from D51 are involved in binding Zn2 (Kim and Wyckoff 1991). Again, these residues are conserved in the sequence alignment with other AP enzymes. The Mg ion is bound by the E. coli AP residue D51, one carboxyl oxygen of E322, and the hydroxyl of T155 (Kim and Wyckoff 1991). These three residues are also conserved in the AP enzymes compared here.
Sequence comparisons yielded some notable differences. E. coli AP has an aspartate at position 153 that binds the Mg through a water molecule. This residue is a histidine in T. maritima AP, as well as in the B. subtilis AP sequences and in some forms of the mammalian enzyme (Murphy et al. 1993). E. coli AP has a lysine at position 328 that forms a salt bridge to the 153 aspartate. The analogous position in T. maritima AP is a tryptophan, and this difference from E. coli recurs in the other three sequences. A different substitution in this position is a histidine, which occurs in Pyrococcus abyssi AP (Zappa et al. 2001), as well as in most mammalian forms of AP (Murphy et al. 1993).
As a consequence of the high homology in the primary sequence alignment, there are corresponding similarities in the derived structural model, with most of the differences occurring in the loop regions. One striking difference is the addition of an extra loop near the location of the E. coli AP dimer interface. This extra loop has no counterpart in E coli AP to use as a basis for structural modeling, so the exact position and interactions cannot be determined. There are also differences observed in the sequence that lies closer to the center of the E. coli dimer interface; these interface substitutions could have implications for the quaternary structure of the T. maritima AP.
Signal sequence
The SignalP program designated for protein sequences from gram-negative bacteria (Nielsen et al. 1997a; Nielsen et al. 1997b; Nielsen et al. 1999) located an N-terminal signal sequence consisting of 19 amino acids, which would be cleaved between a serine and glutamine. The sequence alignment to the most similar enzymes showed a portion of the N-terminus of the T. maritima enzyme in alignment with the signal sequences of the two B. subtilis AP enzymes. In the T. maritima AP sequence there is a gap following the sequence mentioned above, and then the alignment continues at glutamine 20 in the region of the mature B. subtilis and E. coli AP proteins (Fig. 1 ▶). This observation indicates that the small N-terminal region in alignment with the signal sequences of the E. coli and B. subtilis AP enzymes may represent a signal sequence in T. maritima AP. Because the cleavage point from the SignalP prediction and the gap in the alignment occur at the same residue, the first 19 amino acids were considered a putative signal sequence.
AP from the gram-positive bacteria B. subtilis has a signal sequence to transport the protein out of the cell. The signal sequence in the gram-negative E. coli targets the AP to the periplasmic space. Because of the unusual nature of the outer sheath structure of the T. maritima cells, the AP signal could be unique, not matching known patterns for periplasmic space or extracellular targeting sequences. Given this uncertainty about the actual function of the N-terminal region, T. maritima AP was produced with and without the putative N-terminal signal sequence so that the two forms could be compared.
T. maritima alkaline phosphatase production
Because the T. maritima genes contain archaea-like codons, the plasmid pSJS1240 (Kim et al. 1998b) was included to enhance protein expression. This plasmid encodes genes conferring spectinomycin resistance, as well as tRNA genes for the arginine codons AGA and AGG and isoleucine codon AUA, which are not expressed at high levels in E. coli (Kim et al. 1998b). Expression plasmids and pSJS1240 were cotransformed into the appropriate E. coli strain for protein production.
Two expression systems were used for the production of T. maritima AP. The protein with the putative signal sequence was produced using the T7 RNA polymerase system (Studier et al. 1990), whereas the enzyme without the putative signal sequence was produced using the IMPACT-CN system from New England Biolabs. This last method uses a self-cleaving intein fusion protein, consisting of a chitin-binding domain fused to the T. maritima AP.
To express T. maritima AP with the putative signal sequence, the strain/plasmid combination pEK453/EK1597 was used. Detectable but relatively small amounts of the T. maritima AP protein were produced but not transported to the periplasmic space efficiently. The majority of the activity appeared in the whole cell extracts, rather than the periplasmic space where it should be found if the signal had been correctly recognized and used by E. coli cellular machinery. The quantity of protein isolated was not enough to allow detection of a possible cleavage of the signal sequence. In contrast with protein expression from the T7 promoter, significantly more AP was produced from the IMPACT-CN system as outlined below.
For the T. maritima AP without the putative signal sequence, the strain/plasmid combination ER2566/pEK491 was used. A solution of cell-free extract was applied to the chitin column, and following washing, T. maritima AP was released from the fusion protein by treatment with DTT. As seen in Figure 3 ▶, a single band at a molecular mass of 46 kD was observed. Typically, 2–5 mg of pure protein was produced per liter culture. The no-signal IMPACT-CN construct was used to produce the T. maritima AP used in these studies, unless otherwise indicated.
Fig. 3.

SDS-gel electrophoresis of the purification of T. maritima AP. Lanes correspond to (1) standards, (2) cell lysis, (3) column load, (4) cleavage product, and (5) SDS wash.
Protein structure
Immediately after isolation from the chitin column, the molecular mass of the T. maritima AP was determined by size-exclusion chromatography and by sucrose-density gradient sedimentation. Based on calibration curves, the majority of the T. maritima AP had an approximate molecular mass of 45,000 Da. The peak at the monomeric mass was not clearly separated but rather contained a tail region of higher molecular masses. Most of the enzymatic activity was associated with larger molecular masses, including dimeric and higher. After the protein was incubated at 90°C with the optimum complement of metals, the majority of the protein was assembled into the high molecular mass species. To confirm the quaternary structure after heat and metal treatment, the protein was subjected to sucrose-density gradient sedimentation. Analysis of the gradients showed several peaks of activity, including species corresponding to monomeric, dimeric, tetrameric, and octameric forms. The majority of the protein was found as the octameric quaternary structure (Fig. 4 ▶).
Fig. 4.

Analysis of sucrose-density gradient trace of active T. maritima AP (4), represented by a thick line. The standards carbonic anhydrase (1), E. coli AP (2) and aspartate transcarbamoylase (3) are shown in thin lines. Sucrose-density gradient sedimentation experiments were performed according to the protocol in Materials and Methods. Absorbance measurements were taken continuously as the protein-containing solution was pumped through a UV detector.
Circular dichroism spectra were taken before and after the heat and metal treatment to examine the secondary structure (Fig. 5 ▶). The spectrum of the enzyme taken after activation showed an increase in organized secondary structure compared with the spectrum taken before heat and metal treatment. Spectra of the T. maritima enzyme following heat and metal treatment were almost superimposable with the spectrum of wild-type E. coli AP. From these results, it is evident that T. maritima AP has more random coil character before the metals bind. Metal binding, facilitated by heating the enzyme, allows full secondary structure to form, organizing the active site and possibly loops that interact with other subunits. The reorganization of the secondary structure on the metal binding also induces the change in the quaternary structure.
Fig. 5.

Circular dichroism spectra of T. maritima inactive AP (1), E. coli AP (2), and T. maritima active AP (3). These spectra were determined as described in Materials and Methods. For clarity, the inactive T. maritima AP spectrum (1) was shifted up from its original position overlaying the other two spectra.
Activity of the T. maritima alkaline phosphatase
A monomer of T. maritima AP was encoded in the IMPACT-CN T. maritima AP fusion protein, thus the monomeric form was released immediately after cleavage. This was confirmed by the molecular mass measurements reported in the previous section. This form of T. maritima AP had a specific activity of ∼2 U/mg. Activity was increased on addition of Co(II) and Mg(II) and exposure to heat to a maximum value of 88 U/mg at 25° C. Once active, the protein was stable for several weeks if stored at either 4°C or 25°C. Metal incubation at room temperature also activated the enzyme, but the activation was much slower (on the order of days). The gain in activity coincided with the assembly to the quaternary structure as discussed above. Dialysis to a metal-free buffer did not alter the activity or affect the secondary or quaternary structure. However, incubation with 1 mM EDTA at 25°C diminished the activity by more than 90%, and all activity was abolished on exposure to EDTA at 90°C. This result further showed the metal dependence of the enzyme and the heat enhancement of metal binding and loss.
The optimum temperature for activity with metals added was 65°C (kcat; 100 s−1, Km; 93 μM) and the enzyme was more active at 75°C (kcat; 58 s−1, Km; 90 μM) than at 25°C (kcat; 16 s−1, Km; 175 μM). T. maritima AP maintained maximal activity for >18 h when incubated at 65°C and remained active when incubated at 90°C for 5 h. The temperature optimum was the same for the enzyme with the putative signal sequence. If metals were not added to the enzyme solution before the activity measurements, the specific activity remained low but continued to increase with increasing temperature. In this case, the maximal activity obtained was 9.0 U/mg at 75°C. This phenomenon might be attributable to a residual amount of metal ions in the enzyme preparation.
The optimal pH of the T. maritima AP was determined at 25°C and 65°C for both the enzymes, with and without the putative signal sequence. Before the activity measurements, the protein was activated with heat and the optimal metal complement. In all cases, the pH optimum of the enzyme was 8.0, although activity did not drop off significantly between pH 8.0 and pH 9.5.
Metal dependence
The protein attained a maximal activity of 289 U/mg with the optimum complement of Co(II) and Mg(II) when assayed at 65°C. Metals tested were Co(II), Fe(II), Mn(II), Mg(II), Zn(II), Ca(II), Na, and K (Fig. 6 ▶). Activity was enhanced over the activity of the apoenzyme when the protein was incubated with each of these metals. The activity was enhanced further when the incubation took place at 90°C as opposed to 37°C or 25°C. Various combinations of metals were investigated and the optimum complement was Co(II) and Mg(II). As shown in Figure 6 ▶, Co(II) and Mg(II) used in combination enhanced activity only slightly more than Co(II) addition alone. The activity attained with Mn(II) did not differ much from that attained with Mg(II). Different concentrations of Co(II) and Mg(II) were tested to determine the optimum ratio of metal to monomeric protein. Initially, the Mg(II) concentration was held constant, whereas the Co(II) was varied to find the metal to monomeric protein ratio that yielded the highest specific activity. Next, the Co(II) concentration was held constant at the optimum value, whereas the Mg(II) concentration was varied. Optimal activity for T. maritima AP was achieved with a molar ratio of ∼35 moles Co(II) : 1 mole monomer. The enzyme was optimally active as long as the Mg(II) concentration was at least 7 moles Mg(II) : 1 mole monomer. Assayed at 25°C, this metal combination yielded a specific activity of 88 U/mg. The experiment was repeated for Mn(II) concentration dependence. For Mn(II), activity was concentration dependent and peaked at a ratio of 10 : 1, Mn(II) : monomeric unit. Because zinc is a common metal found in APs, and the Co(II) and Mn(II) activation of the T. maritima AP were concentration dependent, zinc was also tested at varying concentrations. Initially, addition of Zn(II) at very low concentrations enhanced activity slightly more than the apoenzyme levels. At low concentrations, the metal may occupy one binding site and somewhat stabilize the active site. However, at concentrations above 0.3 moles Zn(II) : 1 mole monomeric unit, Zn(II) had an inhibitory effect. Zn(II) completely inhibited the enzyme at the concentrations that Co(II) and Mg(II) yielded the maximal T. maritima AP activity. This result could be attributable to the Zn(II) occupying all metal-binding positions and hindering catalysis. If Zn(II) does not naturally fit these sites, then forcing the metal into the active site would cause a distortion.
Fig. 6.

Metal preference of T. maritima AP. Experiments were performed at 25°C at 6 mM PNPP for 5 min. Metals were added to the enzyme before heating to 90°C, then the solution was slowly cooled. Twenty-five μL of this preparation containing 0.041 μg of enzyme was added to the PNPP assay solution to start the reaction.
Although the metal-binding sites are well conserved between T. maritima and E. coli AP, T. maritima AP activity is dependent on cobalt, not zinc, as in E. coli AP. Notably, both the B. subtilis AP enzymes require cobalt for activity (Hulett et al. 1991). A comparison of the active sites of the four enzymes shows that the only substitutions are D153H and K328W. These two amino acid differences are close to the Mg binding site in E. coli. However, they may alter the active site cavity by either slightly changing the electrostatic potential or the exact conformation in such a way that the E. coli zinc metal-binding sites are optimized for the binding of cobalt. Both Zn(II) and Co(II) can adopt a tetrahedral coordination site, but Co(II) is smaller and may fit into a more confined space. Could these two differences from the E. coli sequence be solely responsible for the change in metal dependence? Previous work has already shown that a single amino acid substitution is sufficient to alter the specificity of a metal-binding site. In E. coli AP, residue D153 was mutated to a histidine (Murphy et al. 1993). This change resulted in the conversion of the M3 site from an Mg to a Zn, enhanced catalytic activity, and a shift in the optimal pH for activity. This result shows that a single amino acid substitution is sufficient to alter metal specificity at one site. The changes D153H and K328W and a known cobalt metal dependency of the B. subtilis enzymes lend support to the theory that the substitutions at D153H and K328W may be responsible for the cobalt dependence of the T. maritima AP.
Conclusions
T. maritima AP is the first AP isolated from a hyperthermophilic bacterium. T. maritima AP is a thermostable metal-dependent enzyme requiring Co(II) rather than Zn(II) for activity. A Co(II) preference over Zn(II) in the AP from this organism, and possibly B. subtilis, may be explained by the key substitutions at the active site, D153H and K328W. On metal activation with Co(II) and Mg(II), T. maritima AP gains secondary structure and assembles into oligomeric form.
Materials and methods
Materials
Agar, dibasic sodium phosphate, potassium chloride, sodium chloride, sucrose, ampicillin, spectinomycin, Triton-X 100, metal salts, carbonic anhydrase, bromophenol blue, and PNPP were purchased from Sigma Chemical Company. Tris was purchased from ICN Biomedicals. Bacto tryptone and yeast extract were obtained from Difco Laboratories. 5-bromo-4-chloro-3-indolyl-β-D-galactoside (X-gal) and isopropyl-β-D-thiogalactoside (IPTG) were purchased from USBiological. PCR was performed with the GeneAmp PCR kit from Applied Biosystems. The T-tail vector pGEM-T used in cloning PCR fragments was purchased from Promega. Kits for isolation and purification of DNA fragments from agarose gels and for plasmid purification were from Qiagen. The IMPACT-CN system, as well as restriction endonucleases, was from New England Biolabs. Electrophoresis-grade agarose, Source 15Q strong anion exchange resin, the SE-100 molecular weight column, and protein standards for calibration of the molecular weight column were purchased from Bio-Rad Laboratories. E. coli AP and aspartate transcarbamoylase were prepared in this laboratory (Macol et al. 2001). Amicon YM-10 centricon protein concentrators were from Millipore.
Strains and plasmids
T. maritima genomic DNA was kindly provided by M. Adams, University of Georgia. The E. coli strain EK1597, used for expression of the T. maritima AP, is the λDE3 lysogenized version of E. coli strain SM547 from J. Beckwith (Δ[phoA-proC], phoR, tsx::Tn5, Δlac, galK, galU, leu, kanr, strr). The E. coli strain ER2566, from New England Biolabs, was used for expression with plasmids based on the IMPACT-CN system. Plasmid pSJS1240 was provided by S. Sandler. λDE3 and the pET23a expression vector were from Novagen.
Construction of the expression systems
For expression in the IMPACT-CN system, plasmid pEK491 was created harboring the T. maritima AP gene without the putative signal sequence. The primers used were 5`-GGG GGG TGC TCT TCC AAC CAG GTG AAG AAC GTT ATC TAC-3` and 5`-CCC CCC GCG GCC GCT CAT TTC GTT ACG GGT TCT TTC-3`, which introduced restriction sites for Sap I and Not I. The PCR product obtained was ligated to pGEM-T, removed with Sap I and Not I, and ligated to the IMPACT-CN expression vector pTBY11 previously cut with the same enzymes. The T. maritima AP gene was sequenced by submission to the Molecular Medicine Unit, DNA Sequencing Facility at Beth Israel Deaconess Medical Center using prescribed amounts and volumes.
For expression in the T7 RNA polymerase system, the T. maritima AP gene was amplified by PCR with the primers 5`-GGG GGG GGG CAT ATG AAA AGG CTT TTT ACA-3` and 5`-GGG GGG GAG CTC TCA TTT CGT TAC GGG TTC-3`, which also introduced restriction sites for Nde I and Sac I. The PCR fragment was ligated to the pGEM-T vector. Correct clones were identified by blue/white screen and then confirmed by restriction digestion. The AP gene was removed from the intermediate construct with Nde I and Sac I and ligated to pET23a previously cut with the same enzymes. The resulting plasmid, pEK453, was verified by restriction digestion.
PCR
Genomic PCR was performed with the GeneAmp kit. Four μL of 5 μM of each primer and 0.5 μL of ∼1 μg/mL genomic DNA were used in a final reaction volume of 50 μL. Twenty-five cycles of PCR were used; each cycle involved denaturation at 94°C for 1 min, annealing at 55°C for 1 min, and elongation at 72°C for 2 min.
Expression
Once the correct constructs were identified by restriction digestion, the plasmids were cotransformed with pSJS1240 to either EK1597 or ER2655 for T7 or IMPACT-CN expression, respectively. The plasmid pSJS1240 confers spectinomycin resistance and encodes the arginine and isoleucine tRNA genes that are not well expressed in E. coli (Kim et al. 1998b). To start the growth, 5 mL overnight cultures were prepared from these transformations in LB with 150 μg/mL ampicillin and 50 μg/mL spectinomycin. The overnight culture was used to inoculate 1 L LB media with the same antibiotics. This culture was grown to an A600 between 0.7 and 1.0 and then induced with 0.1 mM IPTG. The ER2655/pEK491 cells were transferred to 15°C after induction. For both expressions, induction continued for 12–16 h before harvesting.
Purification
ER2566/pEK491 cultures were centrifuged and resuspended in 20 mM Tris, 500 mM NaCl pH 8.5 (buffer A), and lysed by sonication. After centrifugation, the supernatant was saved and the pellet resuspended in buffer A with 0.1% Triton-X 100. The second suspension was centrifuged, and the supernatant was combined with the first. The mixture was dialyzed against buffer A and then applied to a chitin column (2 cm diameter × 3 cm length). The column was washed with 200 mL of buffer A at a flow rate of 3 mL/min. DTT was introduced to induce intein cleavage by passing 60 mL of buffer A with 50 mM DTT through the column before incubation at room temperature for 48 h. Fresh DTT was critical for high yields of protein. Cleaved protein was then eluted with 40 mL of the above buffer. The protein was dialyzed against buffer A extensively to remove the DTT. The purity was checked by SDS polyacrylamide gel electrophoresis.
Activity assay
A spectrophotometric assay was used to determine the activity of the AP (Garen and Levinthal 1960). Standard assays were performed at saturating PNPP (6 mM) in 1 M Tris, pH 8.0. The solution was equilibrated and the assay performed at the temperatures indicated. The reaction was started on addition of 10–25 μL of enzyme solution and monitored at 410 nm for at least 5 min. A unit is defined as 1 μmole PNPP hydrolyzed per minute.
Metal removal and activation
Enzyme preparations (<1.5 mg/ml) were dialyzed twice against 20 mM Tris, 50 mM NaCl, 1 mM EDTA pH 8.0 for 12 h, then twice to 20 mM Tris, 50 mM NaCl pH 8.0 for 12 h to remove any remaining EDTA. CoCl2 was then added to the enzyme solution in a 35 : 1 ion : monomer ratio and MgCl2 in a 10 : 1 ion : monomer ratio. The mix was heated to 90°C, then cooled slowly for an hour or longer to room temperature.
Secondary and quaternary structure determination
Circular dichroism spectra were recorded on an Aviv Circular Dichroism spectrometer Model 202. Spectra were collected from 190 nm to 300 nm on 2 mL samples of 0.05 mg/mL protein in 10 mM K2HPO4 pH 8.0 at 25°C.
Initial determination of molecular mass was performed by gel filtration using a SE-100 column. After active protein was obtained, molecular mass measurements were performed by sucrose-density gradient sedimentation using a Beckman L70 ultracentrifuge and SW55Ti rotor. Two-hundred μL of an ∼5 mg/mL protein solution was layered on top of a 4.6 mL 6%–25% sucrose gradient in buffer A. The standards carbonic anhydrase (29 kD), E. coli AP (94 kD), and E. coli aspartate transcarbamoylase (310 kD) were used for calibration. The gradients were centrifuged at 167,000 × g for 20 h at 10°C and decelerated with no brake. On completion of the run, each sucrose gradient was fractionated. A 40% sucrose 1% bromophenol blue solution was drawn at 1 mL/min into a Brandel BR-9620 fractionator by pump. The liquid was pumped from the fractionator to a UV detector (Gilson Model 112) to monitor changes in absorbance. The increase in absorbance, caused by the presence of bromophenol blue, was used to determine the end of gradient collection.
Acknowledgments
We are grateful to Dr. M. Adams at the University of Georgia for the generous gift of T. maritima genomic DNA. We thank Dr. C. Hoffman, Dr. M. Roberts, Dr. M.M. Teeter, and Dr. M. Clarke for helpful discussions, and Dr. E. Pastra-Landis for critical reading of the manuscript. This work was supported by the National Institutes of Health (Grant GM42833).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
AP, alkaline phosphatase
PNPP, p-nitrophenylphosphate
DTT, dithiothreitol
BSA, bovine serum albumin
T. maritima, Thermotoga maritima
B. subtilis, Bacillus subtilis
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.4260102.
References
- Asgeirsson, B. and Andresson, O.S. 2001. Primary structure of cold-adapted alkaline phosphatase from a Vibrio sp., as deduced from the nucleotide sequence. Biochim. Biophys. Acta. 1549 99–111. [DOI] [PubMed] [Google Scholar]
- Garen, A. and Levinthal, C. 1960. A fine-structure genetic and chemical study of the enzyme alkaline phosphatase of E. coli. Biochim. Biophys. Acta. 38 470–483. [DOI] [PubMed] [Google Scholar]
- Guex, N., Diemand, A., and Peitsch, M.C. 1999. Protein modeling for all. Trends Biochem. Sci. 24 364–367. [DOI] [PubMed] [Google Scholar]
- Guex, N. and Peitsch, M.C. 1997. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 18 2714–2723. [DOI] [PubMed] [Google Scholar]
- Holtz, K.M. and Kantrowitz, E.R. 1999. The mechanism of the alkaline phosphatase reaction: Insights from NMR, crystallography and site-specific mutagenesis. FEBS Lett 462 7–11. [DOI] [PubMed] [Google Scholar]
- Huber, R., Langworthy, T.A., Konig, H., Thomm, M., Woese, C.R., Sleytr, U.B., and Stetter, K.O. 1986. Thermotoga maritima sp. nov. represents a new genus of unique extremely thermophilic eubacteria growing up to 90°C. Arch. Microbiol. 144 324–333. [Google Scholar]
- Hulett, F.M., Bookstein, C., and Jensen, K. 1990. Evidence for two structural genes for alkaline phosphatase in Bacillus subtilis. J. Bacteriol. 172 735–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulett, M.F., Kim, E.E., Bookstein, C., Kapp, N.V., Edwards, C.W., and Wyckoff, H.W. 1991. Bacillus subtilis alkaline phosphatases III and IV. J. Biol. Chem. 266 1077–1084. [PubMed] [Google Scholar]
- Kim, E.E. and Wyckoff, H.W. 1991. Reaction mechanism of alkaline phosphatase based on crystal structures. J. Mol. Biol. 218 449–464. [DOI] [PubMed] [Google Scholar]
- Kim, J.W., Peterson, T., Bee, G., and Hulett, F.M. 1998a. Bacillus licheniformis MC14 alkaline phosphatase I gene with an extended COOH-terminus. FEMS Microbiol. Lett. 159 47–58. [DOI] [PubMed] [Google Scholar]
- Kim, R., Sandler, S.J., Goldman, S., Yokota, H., Clark, A.J., and Kim, S. 1998b. Overexpression of archaeal proteins in Escherichia coli. Biotech. Lett. 20 207–201. [Google Scholar]
- Kuroda, M., Ohta, T., Uchiyama, I., Baba, T., Yuzawa, H., Kobayashi, I., Cui, L., Oguchi, A., Aoki, K., and Nagai, Y., et al. 2001. Whole genome sequencing of meticillin-resistant Staphylococcus aureus. Lancet 357 1225–1240. [DOI] [PubMed] [Google Scholar]
- Lee, M.H., Nittayajarn, A., Ross, R.P., Rothschild, C.B., Parsonage, D., Claiborne, A., and Rubens, C.E. 1999. Characterization of Enterococcus faecalis alkaline phosphatase and use in identifying Streptococcus agalactiae secreted proteins. J. Bacteriol. 181 5790–5795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macol, C., Tsuruta, H., Stec, B., and Kantrowitz, E. 2001. Direct structural evidence for a concerted allosteric transition in Escherichia coli aspartate transcarbamoylase. Nat. Struct. Biol. 5 423–426. [DOI] [PubMed] [Google Scholar]
- Murphy, J.E., Xu, X., and Kantrowitz, E.R. 1993. Conversion of a magnesium binding site into a zinc binding site by a single amino acid substitution in Escherichia coli alkaline phosphatase. J. Biol. Chem. 268:29, 21497–21500. [DOI] [PubMed] [Google Scholar]
- Nelson, K.E., Clayton, R.A., Gill, S.R., Gwinn, M.L., Dodson, R.J., Haft, D.H., Hickey, E.K., Peterson, J.D., Nelson, W.C., and Ketchum, K.A., et al. 1999. Evidence for lateral gene transfer between Archaea and Bacteria from genome sequence of Thermotoga maritima. Nature 399 323–329. [DOI] [PubMed] [Google Scholar]
- Nielsen, H., Brunak, S., and von Heijne, G. 1999. Machine learning approaches to the prediction of signal peptides and other protein sorting signals. Protein Engineering 12 3–9. [DOI] [PubMed] [Google Scholar]
- Nielsen, H., Engelbrecht, J., Brunak, S., and von Heijne, G. 1997a. Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng. 10 1–6. [DOI] [PubMed] [Google Scholar]
- ———. 1997b. A neural network method for identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Int. J. Neural Syst. 8 581–599. [DOI] [PubMed] [Google Scholar]
- Peitsch, M.C. 1995. Protein modeling by E-mail. Bio/Technology 13 658–660. [Google Scholar]
- Rina, M., Pozidis, C., Mavromatis, K., Tzanodaskalaki, M., Kokkinidis, M., and Bouriotis, V. 2000. Alkaline phosphatase from the Antarctic strain TAB5. Eur. J. Biochem. 267 1230–1238. [DOI] [PubMed] [Google Scholar]
- Stec, B., Holtz, K.M., and Kantrowitz, E.R. 2000. A revised mechanism for the alkaline phosphatase reaction involving three metal ions. J. Mol. Biol. 299 1303–1311. [DOI] [PubMed] [Google Scholar]
- Studier, F.W., Rosenberg, A.H., Dunn, J.J., and Duberdorff, J.W. 1990. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 185 60–69. [DOI] [PubMed] [Google Scholar]
- Takami, H., Nakasone, K., Takaki, Y., Maeno, G., Sasaki, R., Masui, N., Fuji, F., Hirama, C., Nakamura, Y., and Ogasawara, N., et al. 2000. Complete genome sequence of the alkaliphilic bacterium Bacillus halodurans and genomic sequence comparison with Bacillus subtilis. Nucleic Acids Res. 28 4317–4331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zappa, S., Rolland, J.-L., Flament, D., Gueguen, Y., Boudrant, J., and Dietrich, J. 2001. Characterization of a highly thermostable alkaline phosphatase from the euryarchaeon Pyrococcus abyssi. Appl. Environ. Microbiol. 67 4504–4511. [DOI] [PMC free article] [PubMed] [Google Scholar]