

Abstract
We recently reported on a new H/D exchange- and matrix-assisted laser desorption/ionization (MALDI) mass spectrometry-based technique, termed SUPREX, that removes several important limitations associated with measuring the thermodynamic stability of proteins. In contrast to conventional spectroscopy-based techniques for characterizing the equilibrium unfolding behavior of proteins, SUPREX is amenable to the thermodynamic analysis of both purified and unpurified proteins using mg to ng quantities of material. Here we report on the application of SUPREX to the analysis of multimeric protein systems. Included in this work are the SUPREX results we obtained in studies on six model multimeric proteins including the GCN4p1 dimer, the coil-VaLd trimer, the 4-oxalocrotonate tautomerase (4-OT) hexamer, the Trp repressor (TrpR) dimer, the Arc repressor (ArcR) dimer, and an ArcR mutant (the (DOA20)ArcR) dimer which contained two destabilizing mutations including an Asp to Ala mutation at position 20 and an amide to ester bond mutation between amino acid (aa) residues 19 and 20. As part of the work described here, we present a new method for the analysis of SUPREX data that is generally applicable to both monomeric and multimeric protein systems. Our results on the model proteins in this study indicate that this new method can be used to determine folding free energies for proteins with the accuracy and precision of conventional spectroscopy-based methods.
Keywords: H/D exchange, MALDI, thermodynamic stability, protein folding
Conventional, spectroscopy-based methods for measuring the thermodynamic stability of proteins have the disadvantage that they require relatively large amounts of pure protein. This limits the thermodynamic analysis of proteins to those that can be purified in large quantities. We recently reported a new H/D exchange- and matrix-assisted laser desorption/ionization (MALDI) mass spectrometry-based technique, termed SUPREX, that can be used to quantitate the thermodynamic stability of proteins (Ghaemmaghami et al. 2000). In contrast to conventional methods, the SUPREX technique can be used to quantitate the stability of mg to ng quantities of both purified and unpurified proteins (Powell and Fitzgerald 2001). In SUPREX, protein samples are subjected to H/D exchange by dilution into a series of deuterated exchange buffers containing different concentrations of a chemical denaturant such as guanidinium chloride (GdmCl). After a specified exchange time, the deuterium content of each protein sample is determined using MALDI mass spectrometry. Ultimately, the change in mass relative to the fully protonated sample is plotted as a function of [GdmCl] to generate a SUPREX curve.
SUPREX curves can be used to extract accurate thermodynamic parameters for a protein's folding reaction provided that the protein's equilibrium unfolding behavior is well modeled by a two-state process (i.e., partially folded intermediate state(s) are not significantly populated under equilibrium conditions) and that the protein under study is under EX2 H/D exchange conditions (i.e., the refolding rate of the protein is faster than the average H/D exchange rate for an unprotected amide proton; see Hvidt and Nielsen 1966). Initial reports on SUPREX have included experiments with ribonuclease A, maltose binding protein, and eight variants of a monomeric λ repressor construct (Ghaemmaghami et al. 2000; Ghaemmaghami and Oas 2001; Powell and Fitzgerald 2001). The data obtained in these previous reports indicate that the SUPREX technique can be used to determine folding free energies of monomeric proteins with the precision of conventional techniques (typically < 5%). Moreover, the folding free energies determined by SUPREX are generally consistent (i.e., within 15%) with those obtained by conventional GdmCl-induced equilibrium unfolding techniques.
Here we report on the extension of SUPREX to the thermodynamic analysis of multimeric protein systems. We investigated the SUPREX behavior of six model multimeric protein systems: GCN4p1, coil-VaLd, 4-oxalocrotonate tautomerase (4-OT), Trp repressor (TrpR), Arc repressor (ArcR), and an ArcR mutant ((DOA20)ArcR) which contained two destabilizing mutations including an Asp to Ala mutation at position 20 and an amide to ester bond mutation between aa residues 19 and 20. The GCN4p1, TrpR, and ArcR proteins are homodimeric proteins that are comprised of 33-, 107-, and 53-aa polypeptide chains, respectively. The coil-VaLd construct is a homotrimer of three identical 29 aa-containing polypeptide chains, and 4-OT is a homohexamer of six identical 62 aa-containing polypeptide chains. The chemical denaturant-induced equilibrium unfolding properties of the model, multimeric protein systems above have all been studied using conventional spectroscopy-based approaches (Bowie and Sauer 1989; Gittleman and Matthews 1990; Zitzewitz et al. 1995; Boice et al. 1996; Gloss and Matthews 1997; Silinski et al. 2001; Wales and Fitzgerald 2001). These studies showed that the chemical denaturant-induced equilibrium unfolding properties of these proteins are all well described by two-state (i.e., folded oligomers and unfolded monomers) models, a prerequisite for quantitative SUPREX analyses.
Results
SUPREX analysis of model multimeric proteins
Shown in Figure 1 ▶ are representative data (ΔMass versus [GdmCl] plots) obtained in SUPREX experiments on five of the model proteins examined. The lines in Figure 1 ▶ represent the best fit of each data set to the conventional SUPREX equation [equation (7) in Materials and Methods]. Summarized in Table 1 are the thermodynamic parameters that we extracted from the SUPREX data in Figure 1 ▶ using equation (7). Our results with GCN4p1, ArcR, and coil-VaLd indicate that the SUPREX transition midpoints (C1/2SUPREX values) and the folding free energies (ΔGf values) determined for these multimeric proteins increased with increasing protein concentration. This was true for each protein with the exception of the (DOA20)ArcR construct, which was difficult to analyze using equation (7) as it was not possible to record a complete SUPREX curve (i.e., the pretransition region was absent) for this construct under the conditions of our experiment.
Fig. 1.

Typical SUPREX data obtained on the five model multimeric proteins studied in this work. The proteins included: (A) the GCN4p1 dimer, (B) the ArcR dimer, (C) the (DOA20)ArcR dimer, (D) the coil-VaLd trimer, and (E) the 4-OT hexamer. The protein concentrations [i.e., (n-mer)] in these experiments were 10 μM (closed circles) and 56 μM (open circles) for GCN4p1, 5 μM (closed circles) and 18 μM (open circles) for ArcR, 190 μM for (DOA20)ArcR, 12 μM (closed circles) and 28 μM (open circles) for coil-VaLd, and 1.7 μM for 4-OT. In each case the line represents the best fit of the data to equation (7).
Table 1.
Thermodynamic parameters for model multimeric proteins as determined by conventional SUPREX analysis (Method 1)
| Protein (oligomeric state) | Protein concentration (μM)a | C1/2SUPREX ([GdmCl] M) | mapp (kcal mol−1 M−1)b | ΔGf (kcal mol−1)b |
| GCN4pl (Dimer) | 10 | 0.75 | 1.0 ± 0.2c | −2.3 ± 0.2 |
| 56 | 1.14 | 1.0 ± 0.2c | −2.9 ± 0.2 | |
| ArcR (Dimer) | 5 | 0.35 | 1.9d | −3.7 ± 0.1 |
| 12 | 0.41 | 1.9d | −3.8 ± 0.2 | |
| 18 | 0.67 | 1.9 ± 0.2c | −4.4 ± 0.2 | |
| 25 | 0.64 | 1.9d | −4.4 ± 0.1 | |
| 50 | 0.76 | 1.9d | −4.6 ± 0.1 | |
| (DOA20)ArcR (Dimer) | 72 | 0.13 | 1.0d | −3.4 ± 0.1 |
| 72 | 0.03 | 1.0d | −3.3 ± 0.2 | |
| 72 | 0.26 | 1.0d | −3.5 ± 0.1 | |
| 190 | −0.08 | 1.0 ± 0.2c | −3.2 ± 0.1 | |
| Coil-VaLd (Trimer) | 12 | 1.02 | 0.8 ± 0.2c | −3.9 ± 0.3 |
| 28 | 1.52 | 0.9 ± 0.2c | −4.6 ± 0.3 | |
| 4-OT (Hexamer) | 1.7 | 2.16 | 1.8 ± 0.3c | −11.8 ± 0.6 |
a Protein concentrations are expressed as [n-mer].
b Errors are the standard errors of curve fitting generated by SigmaPlot.
c Value was allowed to vary in non-linear least squares fit of SUPREX data to equation 7.
d Value was fixed in non-linear least squares fit of SUPREX data to equation 7.
The protein concentration-dependent results we obtained in our SUPREX analyses of GCN4p1, ArcR, and coil-VaLd are consistent with the general behavior of multimeric proteins in conventional GdmCl-induced equilibrium unfolding studies. Multimeric proteins are more stable to GdmCl denaturation at higher protein concentrations due to the law of mass action; therefore, conventional circular dichroism (CD) and fluorescence unfolding transitions are shifted to higher [GdmCl] with increasing protein concentration. Thus, it is not surprising that the SUPREX transitions recorded for the model multimeric proteins in this work were also shifted to higher [GdmCl] with increasing protein concentration. In fact, the SUPREX transitions that we obtained for the model multimeric proteins in this study were essentially identical to CD transitions recorded under similar solution conditions (i.e., buffer pH, and protein concentration) with the exception that the SUPREX transition midpoints were shifted to lower [GdmCl] (see Fig. 2 ▶). As we noted in our earlier studies on monomeric proteins, such shifts are a function of the exchange time and solution pH; in addition, they can be predicted by equation (5) below (Ghaemmaghami et al. 2000).
Fig. 2.

A comparison of the SUPREX (ΔM versus [GdmCl]) and CD (Fapp versus [GdmCl]) unfolding transitions obtained for ArcR and 4-OT. (A) The SUPREX and CD unfolding transitions (closed and open circles, respectively) for ArcR obtained under similar buffer conditions and protein concentrations (5 and 18 μM, respectively). (B) The SUPREX and CD unfolding transitions (closed and open circles, respectively) for 4-OT obtained under similar buffer conditions and enzyme concentrations (2 μM).
Significantly, the mapp values we determined for the GCN4p1 and coil-VaLd constructs using equation (7) did not vary with protein concentration. This observation is similar to what is observed in conventional GdmCl-induced equilibrium unfolding experiments. However, it is noteworthy that the mapp values we determined for the five model proteins in our study using equation (7) were all substantially smaller than m values previously reported for these proteins in conventional GdmCl-induced equilibrium unfolding experiments. These discrepancies are due to the fact that true m values in conventional GdmCl-induced equilibrium unfolding experiments are extracted from fraction folded (Fapp) versus [GdmCl] plots using data analysis procedures which take into account the total protein concentration and the reaction order of the folding reaction. Such factors are not accounted for in equation (7), and they clearly must be taken into consideration to calculate meaningful m and ΔGf values for multimeric proteins. It is interesting to note that when the mapp values we determined in Table 1 using equation (7) are multiplied by a factor corresponding to the oligomeric state of each folded protein, then the values are comparable to m values previously reported for these multimeric proteins (see Table 2 and references therein). Based on the data presented in Table 2, this appears to be a useful way in which to empirically derive true m values directly from SUPREX data on multimeric proteins.
Table 2.
Thermodynamic parameters for model multimeric proteins as determined by transition midpoint analysis of SUPREX curves (Method 2)
| Protein (oligomeric state) | Protein concentration (μM)a | m1b (kcal mol−1 M−1) | m2c (kcal mol−1 M−1) | Literature m (kcal mol−1 M−1) | ΔGf1°d (kcal mol−1) | ΔGf2°e (kcal mol−1) | Literature ΔGf° (kcal mol−1) | ||
| GCN4p1 (Dimer) | 10 | 2.0 ± 0.4 | 1.7 | 1.9f | −9.4 ± 0.6 | −9.2 ± 0.5 | −10.5f | ||
| 56 | −9.2 ± 0.7 | −8.8 ± 0.5 | |||||||
| ArcR (Dimer) | 5 | 3.8 ± 0.4 | 2.8 | 3.6g | −11.4 ± 0.3 | −11.0 ± 0.2 | −10.6g | ||
| 12 | −11.1 ± 0.5 | −10.6 ± 0.3 | |||||||
| 18 | −11.8 ± 0.6 | −11.1 ± 0.4 | |||||||
| 25 | −11.5 ± 0.4 | −10.9 ± 0.3 | |||||||
| 50 | −11.6 ± 0.5 | −10.8 ± 0.3 | |||||||
|
|
||||||||
| (DOA20)ArcR (Dimer) | 72 | 2.0 ± 0.4 | 2.8 | 3.0i | −8.7 ± 0.2 | −8.8 ± 0.3 | −8.6i | ||
| 72 | −8.6 ± 0.4 | −8.6 ± 0.6 | |||||||
| 72 | −9.0 ± 0.2 | −9.2 ± 0.3 | |||||||
| 190 | −7.7 ± 0.2 | −7.7 ± 0.3 | |||||||
|
|
||||||||
| Coil-VaLd (Trimer) | 12 | 2.6 ± 0.6 | 2.3 | 2.74j | −18.2 ± 1.3 | −17.9 ± 1.0 | −18.4j | ||
| 28 | −18.5 ± 1.5 | −18.0 ± 1.1 | |||||||
| 4-OT (Hexamer) | 1.7 | 11.1 ± 1.8 | 9.7 | 11.7k | −66.8 ± 6.6 | −63.7 ± 5.1 | −61.7k |
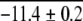
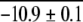
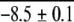
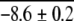
a Protein concentrations are expressed as [n-mer]. bm1 = mapp (from Table 1) × # subunits; reported with standard error. cm2 = # residues in n-mer × 0.026 kcal mol−1 M−1 residue−1 (Myers et al. 1995). dCalculated using m1 with equation 8; reported with standard error. eCalculated using m2 with equation 8; reported with standard error assuming a 10% error in m2. fZitzewitz et al. 1995. gWales and Fitzgerald 2001. hWeighted mean and standard error. iWales and Fitzgerald, unpublished results. jBoice et al. 1996. kSilinski et al. 2001
The SUPREX data shown in Figure 1 ▶ were also analyzed by a second method to determine folding free energies for each model protein. In this method, the transition midpoints of each SUPREX curve, that is, the C1/2SUPREX values given in Table 1, were used in equation (8) (see Materials and Methods) to generate a folding free energy under standard-state conditions (1 M n-mer). The standard-state folding free energies, ΔGf° values, that we determined for each protein using equation (8) are summarized in Table 2. Two sets of ΔGf° values, ΔGf1° and ΔGf2°, are presented in Table 2. These two ΔGf° values were calculated using two slightly different m values in equation (8): ΔGf2° values were calculated using an m value estimated from the number of amino acids in each protein complex, and ΔGf1° values were calculated using an empirically derived m value from our SUPREX data. It is important to note that there is little difference between the ΔGf1° values and the ΔGf2° values in Table 2. The ΔGf1° and ΔGf2° values we calculated for the model proteins in Table 2 were all within ∼15% of ΔGf° values previously reported for these proteins. We should also note that the small uncertainties associated with our m value assignments did not affect the precision of our measurements.
SUPREX analysis of a binary mixture of ArcR analogs
We utilized SUPREX to measure the relative stabilities of two closely related ArcR constructs, wild-type ArcR and (DOA20)ArcR, in a binary mixture of the two proteins. Using conventional CD denaturation techniques, we determined that (DOA20)ArcR is destabilized by 2.0 kcal mol−1 relative to wild-type ArcR (T. Wales and M. Fitzgerald, unpubl.). The molecular weights of ArcR and (DOA20)ArcR, 6227.3 and 6184.2 respectively, are readily resolved by MALDI; therefore, it was possible to simultaneously record SUPREX curves for the ArcR and (DOA20)ArcR proteins directly from a binary mixture of the two constructs. Shown in Figure 3 ▶ are the SUPREX data we obtained on a binary mixture of ArcR and (DOA20)ArcR. The mixture we analyzed contained equimolar concentrations (24 μM each) of the 53-aa polypeptide chains of ArcR and (DOA20)ArcR. The MALDI mass spectrum in Figure 3A ▶ is typical in quality (i.e., signal intensity and resolution) to those that we used to generate the SUPREX curves shown in Figure 3B ▶.
Fig. 3.

Results from the SUPREX analysis of a binary mixture of ArcR and (DOA20)ArcR. (A) Representative MALDI mass spectrum acquired in the SUPREX analysis of the binary mixture. (B) Typical SUPREX data obtained for ArcR (open circles) and (DOA20)ArcR (closed circles) when a binary mixture of the two proteins was analyzed. The concentration of each protein was 24 μM. The lines represent the best fit of the data to equation (7).
The lines in Figure 3B ▶ represent the best fit of each data set to equation (7) using a nonlinear least squares analysis. The ΔGf values that we obtained from our analyses of the SUPREX data shown in Figure 3B ▶ and the data obtained in two additional SUPREX experiments on similar binary mixtures of ArcR and (DOA20)ArcR were −4.7, −4.4, and −4.4 kcal mol−1 for ArcR and −3.3, −3.3, and −3.0 kcal mol−1 for (DOA20)ArcR. These ΔGf values calculated using equation (7) were found to be in reasonably good agreement with the ΔGf values determined above for ArcR and (DOA20)ArcR when they were each subjected to similar SUPREX analyses in separate experiments (see Table 1).
The ArcR and (DOA20)ArcR SUPREX curves in Figure 3B ▶ were also analyzed using equation (8) to calculate ΔGf° values. The weighted average ΔGf° values we obtained from three replicate measurements on similar binary mixtures of ArcR and (DOA20)ArcR are summarized in Table 3. Two sets of ΔGf° values are presented in Table 3. The ΔGf1° values were generated using m values of 3.8 and 2.0 kcal mol−1 M−1 for ArcR and (DOA20)ArcR, respectively. These m values were empirically derived from the mapp values reported for ArcR and (DOA20)Arc in Table 1. The ΔGf2° values were generated using an estimated m value of 2.8 kcal mol−1 M−1 for both ArcR and (DOA20)ArcR. This m value was estimated from the size of these dimeric protein complexes according to the data in Myers et al. (1995). The average ΔGf1° and ΔGf2° values we calculated are similar in magnitude to each other; significantly, they are comparable to average values we calculated for these same constructs when they were analyzed individually (see Table 2).
Table 3.
SUPREX results from the analysis of binary mixtures of ArcR and (DOA20)ArcR
| Protein | ΔGf1°a,c (kcal mol−1) | ΔGf2°b,c (kcal mol−1) |
| ArcR | −11.8 ± 0.2 | −11.1 ± 0.2 |
| (DOA20)ArcR | −9.1 ± 0.1 | −9.1 ± 0.2 |
a Calculated using m1 (Table 2) and equation 8.
b Calculated using m2 (Table 2) and equation 8.
c The weighted mean from three replicate determinations is reported with the standard error.
SUPREX analysis of Trp repressor
A partially purified sample of TrpR was subjected to SUPREX analysis in the absence and in the presence of its cognate ligand, L-tryptophan. The TrpR sample used in these experiments was isolated from a crude cell lysate of E. coli cells using SEC chromatography. An RP-HPLC chromatogram and a MALDI mass spectrum of the "semipure" TrpR sample that we isolated are shown in Figure 4A and B ▶, respectively. The results of our RP-HPLC and MALDI analyses of the TrpR sample both indicate that other proteins are present in our TrpR sample; however, TrpR does appear to be the major protein component in the sample. The SUPREX curves recorded for the TrpR sample in the absence and in the presence of L-tryptophan (230 μM in the deuterated exchange buffers) are shown in Figure 4C ▶.
Fig. 4.

Results from the SUPREX analysis of TrpR. (A) RP-HPLC chromatogram of "semipure" TrpR sample using an acetonitrile gradient (20–80% buffer B over 30 min). (B) Representative MALDI mass spectrum acquired in the SUPREX analysis of the "semipure" TrpR sample in the absence of L-tryptophan. The peaks labeled with an * are attributed to protein impurities in the sample. The peaks labeled with a # are attributed to SA matrix adducts. (C) SUPREX data obtained on TrpR in the absence (closed circles) and presence (open circles) of L-tryptophan. The lines represent the best fit of the data to equation (7).
The lines in Figure 4C ▶ represent the best fit of each data set to equation (7) using a nonlinear least squares analysis. In our nonlinear least squares analysis of each TrpR data set in Figure 4C ▶, the values we used for t and <kint> were defined by the parameters of the experiment; the mapp value was defined as 2.8 kcal mol−1 M−1, and values for ΔM0, ΔM∞, and ΔGf were allowed to vary. The steep slope of the transitions in the TrpR SUPREX curves shown in Figure 4C ▶ made it difficult to acquire data points in these regions. This ultimately made it necessary to define an mapp value in our analysis of the SUPREX curves in Figure 4C ▶. Myers et al. showed that m values can be estimated based on a protein's size. Using an estimate of 0.026 kcal mol−1 M−1 per residue, an approximate m value of 5.6 kcal mol−1 M−1 can be calculated for the 214-aa TrpR homodimer. Our SUPREX results with GCN4p1, ArcR, and (DOA20)ArcR indicated that the mapp values for these homodimers were approximately half the value of those expected from the literature (see Tables 1 and 2). Therefore, the mapp value we employed in our analysis of the SUPREX curves in Figure 4C ▶ was 2.8 kcal mol−1 M−1. Ultimately, our analysis of the data in Figure 4C ▶ using equation (7) yielded ΔGf values of 14.0 and 15.6 kcal mol−1 for TrpR in the absence and in the presence of L-tryptophan ligand, respectively. This corresponds to a ΔGBinding value of −1.6 kcal mol−1. Using equation (9), we calculated a Kd of 82 μM for L-tryptophan binding to TrpR. This Kd value is somewhat larger than previously reported Kd values for L-tryptophan binding to TrpR, 15–42 μM (Joachimiak et al. 1983; Arvidson et al. 1986; Marmorstein et al. 1987; He and Matthews 1990; Jin et al. 1993).
The SUPREX curves in Figure 4C ▶ were also analyzed using equation (8). It was not possible to determine the exact concentration of TrpR in the "semipure" samples of TrpR that were subjected to SUPREX analyses in our experiment. Therefore it was not possible to obtain a specific ΔGf° value for the "semipure" TrpR samples in the presence and absence of L-tryptophan ligand using equation (8), as the equation includes a protein concentration term. However, it is important to note that the TrpR concentration was constant in our SUPREX analyses of TrpR with and without ligand. This made it possible to extract a meaningful free energy value for the binding reaction (i.e., a ΔΔGf° or ΔGBinding value), as the protein concentration term in equation (8) is eliminated in calculations of ΔΔGf° values if the protein concentration is constant. Ultimately, our analysis of the data in Figure 4C ▶ using equation (8) yielded a ΔGBinding value of −3.2 kcal mol−1. Using this ΔGBinding value, we calculated a Kd of 17 μM for L-tryptophan binding to TrpR using equation (9). This Kd value is comparable to those previously reported for TrpR and L-tryptophan binding (15–42 μM).
Discussion
SUPREX analysis of multimeric proteins
The ΔMass versus [GdmCl] plots in Figure 1 ▶ show that it is possible to record SUPREX curves for multimeric proteins. However, the data in Table 1 reveal that the conventional SUPREX equation [equation (7)] cannot be directly used to extract meaningful m and ΔGf values from SUPREX data obtained on multimeric proteins. The inability of equation (7) to adequately describe the SUPREX behavior of multimeric proteins is due to the lack of appropriate terms that account for the protein concentration and the reaction order in the folding reactions of multimeric proteins.
In theory, a protein concentration term could be incorporated into the conventional SUPREX equation, and the reaction order of the folding reaction could be accounted for in SUPREX analyses. As described by Ghaemmaghami et al. (2000) the conventional SUPREX equation was derived from the following first-order rate equation:
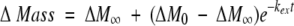 |
(1) |
where ΔMass is the increase in mass of the protein due to H/D exchange, ΔM∞ is the ΔMass measured after complete exchange, ΔM0 is ΔMass measured before global exchange, kex is the observed exchange rate for each amide hydrogen, and t is the time of H/D exchange. Under EX2 conditions, the following expression can be obtained for kex:
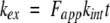 |
(2) |
where Fapp is the apparent fraction of unfolded protein, and kint is the intrinsic exchange rate of an unprotected amide proton. For a two-state unfolding reaction, the following relationship can be derived for Fapp and Kopen:
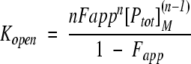 |
(3) |
where [Ptot]M is the total protein concentration in monomer equivalents and n is the number of subunits. In the case of a monomeric protein, n = 1 and equation (3) reduces to:
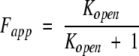 |
(4) |
Substitution of equations (2) and (4) into equation (1) ultimately yields the conventional SUPREX equation if Kopen is defined as 1/Kfold [see equation (7) in Materials and Methods]. In cases where n = 2, a unique expression for Fapp in terms of [Ptot]M and Kopen can also be derived from equation (3); in addition, this expression can in turn be used to generate a modified SUPREX equation. Unfortunately, it is difficult to derive such an expression for Fapp in terms of [Ptot]M and Kopen when n > 2, due to the high order of equation (3) in these cases. This ultimately complicates the direct application of the conventional SUPREX equation to higher order, multimeric proteins.
Here we describe a simplified approach for analyzing SUPREX data on multimeric proteins. The approach, which is based on a transition midpoint analysis, employs equation (8) for the quantitative analysis of SUPREX curves. The approach is analogous to the transition midpoint analysis technique that has been described in the literature for the analysis of conventional equilibrium unfolding data on multimeric proteins (Backmann et al. 1998). Equation (8) is a combination of equations (5) and (6), below.
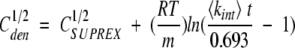 |
(5) |
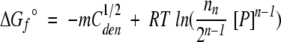 |
(6) |
In equations (5) and (6), C1/2den and C1/2SUPREX are the [GdmCl] values at the transition midpoints of a conventional denaturation curve and a SUPREX curve, respectively; m is defined as δΔGf°/δ[GdmCl], R is the gas constant, T is the temperature in Kelvin, <kint> is the average intrinsic exchange rate of an amide proton, t is the H/D exchange time, ΔGf° is the standard-state folding free energy in the absence of chemical denaturant, n is the number of subunits in the protein, and [P] is the protein concentration expressed in n-mer equivalents. Equation (5) was derived by Ghaemmaghami et al. (2000) for monomeric proteins to describe the relationship between the denaturation midpoint of a SUPREX denaturation curve and a traditional denaturation curve. Equation (6), which is taken from Backmann et al. (1998) relates the midpoint of a conventional denaturation curve to ΔGf°. Therefore, equation (8) effectively converts a SUPREX denaturation midpoint to a conventional denaturation midpoint and then converts that conventional denaturation midpoint into the free energy of unfolding in the absence of denaturant.
Our results indicate that equation (8) accurately predicts the protein concentration-dependent, transition midpoint shifts we observed in our SUPREX analysis of the model multimeric proteins in this study. This is evidenced by the protein concentration-independent ΔGf° values that we obtained for each model protein using this method of analysis. Our results also indicate that the ΔGf° values we calculated by SUPREX for all five of the model proteins in this study using equation (8) are all within ∼15% of ΔGf° values previously reported for these proteins in conventional studies (Table 2). Moreover, we should add that equation (8) is also applicable to the analysis of monomeric proteins. When the SUPREX data on the monomeric proteins reported on by Ghaemmaghami et al. (2000) were reanalyzed using equation (8), the ΔGf values obtained were identical to those determined in that work using the conventional SUPREX equation (data not shown).
The quantitative analysis of SUPREX data using equation (8) requires that the protein of interest is under EX2 exchange conditions. The tendency of some proteins to enter the EX1 exchange regime at high denaturant concentrations and/or pH can preclude the accurate determination of ΔGf° values from SUPREX data using equation (8). Under EX1 conditions, the observed H/D exchange rate becomes a function of the unfolding rate rather than the folding equilibrium constant (Hvidt and Nielson 1966; Clarke and Fersht 1996; Loh et al. 1996; Yi et al. 1997). Therefore, it is important that the experimental conditions for SUPREX experiments on multimeric proteins (i.e., buffer pH, protein concentration, and exchange time) be chosen such that the SUPREX transition be at a sufficiently low [GdmCl] that EX2 conditions prevail. We have found that the EX1 exchange behavior of proteins in SUPREX buffers is easily detected by the presence of two distinct ion signals in the MALDI analysis step of SUPREX. This behavior is diagnostic of the EX1 exchange regime in H/D exchange experiments using a mass spectrometric readout (Miranker et al. 1993; Smith et al. 1997; Arrington et al. 1999). We note that such EX1 exchange behavior was detected for the coil-VaLd construct in SUPREX buffers containing as low as 1.5 M GdmCl at pD 6.0. Therefore, as noted in the Materials and Methods section, it was necessary to lower the buffer pD and increase the exchange time so that the SUPREX transition was shifted to a [GdmCl] < 1.5 M. Under these conditions, only one protein ion signal was detected for the coil-VaLd construct at each [GdmCl] in the SUPREX curve, suggesting EX2 exchange conditions.
Mixture analysis with SUPREX
Thermodynamic stability measurements on individual species in a multicomponent mixture are difficult to acquire using traditional spectroscopy-based equilibrium unfolding techniques. This is because optical spectroscopies, such as those involving CD and fluorescence measurements, only record the bulk properties of a sample. The SUPREX technique is based on a mass spectrometric readout; therefore, provided the molecular masses of different components in a mixture are resolvable by MALDI mass spectrometry, one can utilize SUPREX to directly investigate the thermodynamic properties of individual components in a mixture (Ghaemmaghami et al. 2000; Ghaemmaghami and Oas 2001).
Our results with the "semipure" TrpR samples and the binary mixtures of ArcR constructs in this work indicate that multimeric proteins are amenable to analysis in multicomponent mixtures. The accuracy of our thermodynamic stability measurements on the TrpR system was not compromised by the low purity of the sample. However, we should note that the accurate calculation of ΔGf° values from SUPREX data on proteins in complex mixtures requires that there is no appreciable interaction between the protein of interest and other mixture components. Our results with the binary mixtures of ArcR and (DOA20)Arc also indicate that SUPREX can be used to accurately rank the stability of two closely related multimeric protein analogs in a multicomponent mixture. The weighted average ΔGf2° values we determined for ArcR and (DOA20)Arc in the binary mixture (−11.1 ± 0.2 and −9.1 ± 0.2 kcal mol−1, respectively) were essentially identical to those we determined by SUPREX for each protein individually, −10.9 ± 0.1 and −8.6 ± 0.2 kcal mol−1 (respectively).
SUPREX analysis of protein–ligand binding interactions in multimeric protein systems
Trp repressor (TrpR) from E. coli is a homodimeric DNA-binding protein involved in the L-tryptophan biosynthetic pathway. For high-affinity binding to its operator DNA, TrpR must bind two molecules of L-tryptophan (He and Matthews 1990). As part of the work described here, we found that the Kd value for L-tryptophan binding to TrpR can be accurately determined from SUPREX measurements on TrpR in the absence and in the presence of L-tryptophan. Moreover, our results indicate that pure samples of TrpR are not required for such an analysis.
One of the major advantages of the SUPREX technique over conventional spectroscopy-based methods is that unpurified protein solutions can be analyzed. In the SUPREX analysis of multimeric proteins, the use of impure samples precludes an accurate determination of ΔGf° because such a calculation requires that the protein concentration be accurately known. Typically, it is difficult to determine the exact concentration of a specific protein in a multicomponent protein mixture. However, our results with the TrpR system indicate that accurate ΔΔGf° values for unpurified multimeric protein samples can be obtained by SUPREX as long as the protein concentration is held constant.
Materials and methods
Reagents
Deuterium oxide (99.9 atom %D), sodium deuteroxide, deuterium chloride, L-tryptophan, and α-cyano-4-hydroxy cinnamic acid (4HCCA) were purchased from Aldrich (Milwaukee, WI). Deuterated phosphoric acid was obtained from Cambridge Isotope Laboratories (Andover, MA), and GdmCl (OmniPur) was from EM Science (Gibbstown, NJ). Sinapinic acid (SA) was either from Acros Organics (Pittsburgh, PA) or from Aldrich. Trifluoroacetic acid (TFA) was obtained from Halocarbon (River Edge, NJ), and acetonitrile (MeCN) was from Fisher (Pittsburgh, PA). The protein mass standards (bovine insulin, bovine ubiquitin, and hen egg lysozyme), the isopropyl β-D-thiogalactopyranoside (IPTG), and the phenylmethylsulfonyl fluoride (PMSF) were purchased from Sigma (St. Louis, MO).
General methods and instrumentation
MALDI mass spectra were acquired on a Voyager DE Biospectrometry Workstation (PerSeptive Biosystems, Framingham, MA) in the linear mode using a nitrogen laser (337 nm). SUPREX samples were prepared for MALDI analysis as described below. In our experiments with coil-VaLd we used 4HCCA as the MALDI matrix; all other experiments were performed using SA as the matrix. MALDI mass spectra for coil-VaLd experiments were collected in the negative ion mode, while spectra for all other proteins were collected in the positive ion mode. All experiments utilized an acceleration voltage of 25 kV, a grid voltage between 23 and 24 kV, a guide wire voltage of 75 V, and a delay time of 225 ns. Each spectrum obtained was the sum of between 16 and 32 laser shots. The raw intensity versus time data in each mass spectrum was smoothed using a Savitsky-Golay smoothing routine prior to mass calibration using internal standards.
UV/Vis absorbance measurements for protein concentration determinations were recorded on a Hewlett Packard 8452A Diode Array UV/Vis Spectrophotometer. All GdmCl concentrations were determined using a Bausch & Lomb (Rochester, NY) refractometer by the method of Nozaki (1972). All pH measurements were recorded using a Jenco 6072 pH meter (San Diego, CA) equipped with a Futura calomel pH electrode from Beckman Instruments (Fullerton, CA). Deuterated exchange buffer pD values were determined from pH measurements by adding 0.4 to the measured pH (Glasoe and Long 1960).
The CD denaturation curve for 4-OT was recorded by monitoring the far UV-CD signal of the enzyme in 50 mM phosphate buffers (pH 7.4) containing different amounts of GdmCl using a Jasco J-710 Spectropolarimeter as described previously (Silinski et al. 2001). The concentration of enzyme in this experiment was 1.7 μM (based on hexamer equivalents). The CD denaturation curve for ArcR was recorded in buffer containing 50 mM Tris-HCl (pH 7.5), 100 mM KCl, 0.2 mM EDTA, and varying amounts of GdmCl using an Aviv Circular Dichroism Spectrometer (Model 202) as described (Wales and Fitzgerald 2001). The concentration of ArcR was 5 μM (based on dimer equivalents).
Our RP-HPLC system included two Rainin Dynamax SD-200 pumps and a Rainin Dynamix UV-C absorbance detector (Woburn, MA). Analytical RP-HPLC was performed using either a Vydac (Hysperia, CA) C18 column (5 μm particle size, 4.6 × 150 mm) or a Vydac C4 column (5 μm particle size, 4.6 × 150 mm) at a flow rate of 1 mL min−1. Semipreparative RP-HPLC was performed using an Alltech (Deerfield, IL) C18 column (10 μm particle size, 10 × 250 mm) at a flow rate of 3 mL min−1. Analytical and semipreparative RP-HPLC separations were performed using linear gradients of buffer B in A (buffer A = 0.1% TFA in water, buffer B = 90% acetonitrile in water containing 0.09% TFA). Detection was at 214 nm for analytical separations and at 230 nm for semipreparative separations. The size exclusion chromatography (SEC) system consisted of a Rainin Dynamax SD-200 pump, a Superdex 75 HR 10/30 size exclusion column from Pharmacia Biotech (Piscataway, NJ), and a Rainin Dynamix UV/Vis absorbance detector. Electrospray ionization (ESI) mass spectra were acquired on a PE Sciex API 150EX (Foster City, CA).
Protein samples
GCN4p1 was obtained by total chemical synthesis using standard solid-phase peptide synthesis (SPSS) protocols for Boc chemistry. The sequence of our GCN4p1 construct (Ac-RMKQLEDKVEE LLSKNYHLENEVARLKKLVGER-CONH2) was identical to that reported by Zitzewitz et al. (1995). The crude synthetic product we obtained after 33 cycles of SPPS was purified by RP-HPLC under the following conditions: 3 mL/min−1 flow rate, and 30–50% linear gradient of B in A over 30 min. Pure fractions (as judged by ESI mass spectrometry) were pooled, frozen, and lyophilized to a dry white solid. The pure, lyophilized protein product was folded by dissolution of the sample (∼5 mg/mL) in a folding buffer that contained 20 mM sodium phosphate (pH 6.0) and 200 mM KCl. The concentration of GCN4p1 peptide was ultimately determined spectrophotometrically in a 6 M GdmCl solution using its molar extinction coefficient of 1500 M−1 cm−1 at 280 nm (Edelhoch 1967).
Purified coil-VaLd was a gift from Dr. William F. DeGrado's laboratory at the University of Pennsylvania (Boice et al. 1996). The lyophilized peptide was refolded in water. Coil-VaLd protein concentrations were ultimately determined by the Waddell method (Waddell 1956; Wolf 1983).
A pET24a plasmid containing the wild-type 4-OT gene within the SalI and NdeI restriction sites was kindly provided by Professor Christian P. Whitman (University of Texas–Austin). The protein was overexpressed and purified as described elsewhere (Silinski et al. 2001).
ArcR was obtained by total chemical synthesis using highly optimized SPPS protocols for Boc chemistry as described elsewhere (Wales and Fitzgerald 2001). The (DOA20)ArcR analog was also prepared by SPPS protocols for Boc chemistry. The protocols employed for the SPPS preparation of the (DOA20)ArcR analog which contains an Asp to Ala and amide to ester bond mutation at position 20 in Arc repressor's 53-aa polypeptide chain were similar to those employed for wild-type ArcR except that L-lactic acid was coupled in place of Asp-20. The procedure used to couple the L-lactic acid to the growing peptide chain was similar to that described by Lu et al. (1997). Subsequent coupling of Leu-19 was also performed according to the procedure described by Lu et al. The products from our ArcR and (DOA20)ArcR syntheses were purified by RP-HPLC, as described (Wales and Fitzgerald 2001). Pure RP-HPLC fractions, as judged by ESI-MS, were pooled, frozen, and lyophilized. This pure, lyophilized protein product was reconstituted in a folding buffer containing 50 mM Tris, 100 mM KCl, 0.2 mM EDTA (pH 7.5) prior to SUPREX analyses. The binary mixture of ArcR and (DOA20)ArcR was prepared by first equilibrating an equimolar solution of the two proteins in ice-cold folding buffer for 30 min, then lyophilizing the sample to a dry white solid, and ultimately reconstituting the proteins in water. ArcR and (DOA20)ArcR concentrations were determined spectrophotometrically using a molar extinction coefficient of 6756 M−1 cm−1 at 280 nm (Brown et al. 1990).
TrpR was obtained by recombinant DNA methods. A plasmid containing the Trp repressor gene was kindly provided by Dr. C. Robert Matthews (University of Massachusetts, Worcester). The plasmid was transformed into Epicurian Coli® BL21-Gold(DE3)pLysS cells (Stratagene) following the manufacturer's instructions, and colonies were grown on LB/Amp plates (ampicillin = 50 μg mL−1 in this and all further procedures) at 37°C. A single colony was used to inoculate 5 mL of LB/Amp broth which was that shaken at 37°C overnight. The overnight growth was used to inoculate 1 L of LB/Amp broth in a 2 L Erlenmeyer flask which was that shaken at 37°C. When the A600 reached about 0.6, IPTG was added to a final concentration of 0.5 mM, and protein expression was allowed to continue for 4 h. At that time, the cells were centrifuged for 10 min at about 4700 g at 4°C. The cell pellets were resuspended in a 100 mM Tris, 20 mM MgCl2 (pH 7.6) buffer and lysed by sonication. The lysed cells were centrifuged at 10,000 g for 10 min to remove cellular debris, and PMSF was added to the supernatant to a final concentration of 2 mM. The cell lysate was fractionated by SEC using 20 mM sodium phosphate buffer (pH 7.4). TrpR eluted in a broad peak with many other cellular components (data not shown). Semipure SEC fractions containing TrpR (as detected by MALDI) were pooled. The TrpR protein in this sample pool was concentrated using Microcon YM-3 Centrifugal Filter Devices (Millipore, Bedford, MA), and analyzed by SUPREX without any further purification.
SUPREX data collection
The protocols used to generate the SUPREX curves in this work were similar to those described (Ghaemmaghami et al. 2000). Briefly, H/D exchange reactions were initiated by combining 1-μL aliquots of a protein stock solution (fully protonated) with 9-μL volumes of several deuterated exchange buffers containing concentrations of GdmCl that varied from 0 to 6 M. The exchange buffer constituents (i.e., salts and pD) differed slightly for different proteins, and are discussed in detail below. After a specified exchange time (see below for exact exchange times), a 1-μL aliquot of each exchange reaction was combined with at least 9 μL of a MALDI matrix solution. The MALDI matrix solution consisted of an ice-cold, saturated, aqueous solution of either SA or 4HCCA containing 45% MeCN and 0.1% TFA (pH 3.0). The MALDI matrix solution effectively quenched the H/D exchange reaction and prepared the sample for MALDI analysis. The matrix solution also included proteins of known mass that were used as internal mass standards. Ultimately, 1 μL of the quenched exchange reaction was spotted on a stainless steel MALDI sample stage at room temperature. The solvent was evaporated (typically in less than 1 min) using a gentle flow of air.
Five to ten replicate MALDI mass spectra were collected and analyzed to determine an average change in mass relative to the fully protonated sample (ΔMass) at each [GdmCl]. These spectra were generated by sampling different regions of a single MALDI sample preparation. The mass of the deuterated protein of interest was determined with a two-point calibration utilizing proteins of known mass as internal standards. It should be noted that no correction for back exchange in the matrix solution or for back exchange in the matrix crystals was necessary, because the samples were prepared and analyzed such that the elapsed time for each stage of the experiment was exactly the same. Furthermore, typically only about 5 min elapsed between spotting the sample on the MALDI plate and the completion of data acquisition for each data point. The number of deuterons that back exchanged during this short time was minimal.
All of the deuterated exchange buffers contained 20 mM sodium phosphate; however, the exact composition of the deuterated exchange buffers varied slightly for the different proteins we analyzed. The phosphate exchange buffers used in the GCN4p1 experiments were adjusted to pD 6.0 and contained 200 mM KCl. The phosphate exchange buffers used in the coil-VaLd experiments were adjusted to pD 4.8. At higher pDs, our MALDI analyses of coil-VaLd indicated that this protein exhibited EX1 exchange behavior (i.e., the refolding rate was slower than the intrinsic H/D exchange rate of the amide protons in the protein's polypeptide backbone) (Miranker et al. 1993; Smith et al. 1997; Arrington et al. 1999). As we previously noted, EX1 exchange conditions complicate the quantitative analysis of SUPREX data (Ghaemmaghami et al. 2000). Under such conditions, the measured H/D exchange rate becomes a function of the unfolding rate rather than the folding equilibrium constant (Loh et al. 1996). At pH 4.8, where the rate of H/D exchange is relatively slow, our MALDI analyses of coil-VaLd were consistent with the protein being under EX2 conditions. The phosphate exchange buffers used in the 4-OT and TrpR experiments were adjusted to pD 7.4. ArcR and (DOA20)ArcR were subjected to SUPREX analyses using phosphate exchange buffers at pD 6.0 with either 200 mM or 1 M KCl. Our MALDI analyses of 4-OT, TrpR, and ArcR indicated that EX2 conditions prevailed for these proteins under the above conditions.
The exchange times used in our SUPREX experiments on GCN4p1, coil-VaLd, 4-OT, both ArcR constructs, and TrpR were 5, 150, 1000, 5, and 45 min, respectively. Proteins can enter the EX1 exchange regime at high concentrations of denaturant. As noted above, this can complicate the quantitative analysis of SUPREX data. Therefore, the exchange times employed in the experiments described here generally corresponded to those at which C1/2SUPREX values were at relatively low [GdmCl] (typically about 1 M GdmCl). In our SUPREX analyses of the 4-OT enzyme and the ArcR constructs in this study, our choices of exchange times (1000 and 5 min, respectively) were especially critical. When long exchange times (> 5 min) were used for the SUPREX analysis of the ArcR constructs, it was not possible to record SUPREX curves, as even at 0 M GdmCl, H/D exchange was complete (i.e., ΔMass was equivalent to ΔM∞). In the case of 4-OT, we found that the exchange time had to be relatively long compared to the 60 to 120 min it took the enzyme to equilibrate in the SUPREX buffers.
SUPREX data analysis
Method 1
A nonlinear least squares analysis routine in Sigma Plot was used to fit the data in our SUPREX curves to equation (7), below:
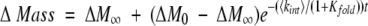 |
(7) |
where Kfold = e−(ΔGf+mapp[GdmCl])/RT, ΔM0 is ΔMass measured before global exchange, ΔM∞ is ΔMass measured after complete exchange, t is the time of exchange, <kint> is the average intrinsic H/D exchange rate for an amide proton determined using the program SPHERE (Zhang 1995; Bai et al. 1993), ΔGf is the folding free energy in the absence of GdmCl, [GdmCl] is the GdmCl concentration, mapp is the apparent δΔGf/δ[GdmCl], R is the gas constant, and T is the temperature in Kelvin.
In our analyses of the SUPREX data sets in this work using equation (7) values for <kint>, t, and T were fixed according to the specific parameters of each SUPREX experiment; and values for mapp, ΔM0, ΔM∞, and ΔGf were typically allowed to vary. However, in the case of the (DOA20)ArcR curve (Fig. 1E ▶), ΔM0 was also fixed, as the beginning baseline region of the SUPREX curve generated for this analog was not defined under the conditions of our experiment (i.e., protein concentration, exchange time, and buffer pD). However, we reasoned that the beginning baseline regions of the ArcR and (DOA20)ArcR SUPREX curves should be the same for these closely related analogs. Therefore, the ΔM0 value that we determined for ArcR, 20.6 Da, was used in our analysis of the (DOA20)ArcR data in Figure 1E ▶.
Method 2
Equation (8) was used to determine folding free energy values under standard-state conditions (1 M n-mer) from our SUPREX data.
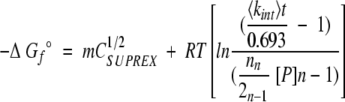 |
(8) |
In Equation (8), m is defined as δΔGf°/δ[GdmCl], C1/2SUPREX is the [GdmCl] at the SUPREX transition midpoint, R is the gas constant, T is the temperature in Kelvin, <kint> is the average intrinsic exchange rate of an amide proton, t is the H/D exchange time, n is the number of subunits in the protein, and [P] is the protein concentration expressed in n-mer equivalents. Equation (8) is valid for finite values of t such that C1/2SUPREX values are greater than or equal to 0 M GdmCl. In our application of equation (8) to the SUPREX data in this work, the values we used for <kint> and t were defined by the parameters of the experiment, n was defined by the multimeric state of the protein under study, and T was 298 K (283 K for GCN4p1). Typically, C1/2SUPREX values were extracted from our ΔMass versus [GdmCl] plots by fitting the data to a sigmoidal function such as equation (7). The m values used in our calculation of ΔGf° values using equation (8) were estimated either by using a value of 0.026 kcal mol−1 M−1 per amino acid residue as the data in Myers et al. (1995) suggests, or by using an empirically derived value from our SUPREX data. Empirically derived values from our SUPREX data were determined by multiplying mapp values by a factor corresponding to the multimeric state of the protein (i.e., 2 for dimeric proteins, 3 for trimeric proteins, etc.). The mapp values were derived from SUPREX data using equation (7) as described above.
Kd determinations
Dissociation constants, that is, the Kd values, were calculated from ΔΔGf values (i.e., ΔGBinding values) using equation (9) (Schellman 1975):
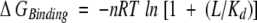 |
(9) |
In Equation (9), n is the number of independent binding sites, L is the molar concentration of ligand, R is the gas constant, and T is the temperature in Kelvin. Equation (9) is valid when excess ligand is used in the binding reaction. This was the case in all of the protein–ligand binding studies in this work.
Acknowledgments
Financial support was provided by an NSF grant CHE0094224 to M.C.F. K.D.P. was supported in part by an NIH Biological Chemistry Training Grant to Duke University. We thank Profs. C.P. Whitman and C.R. Matthews for making recombinant DNA clones of 4-OT and TrpR (respectively) readily available to us. A purified synthetic coil-VaLd construct was kindly provided by Prof. W. DeGrado. We thank Prof. T.G. Oas and Peter Silinski for helpful discussions throughout the course of this work. Peter Silinski also collected the CD denaturation data for 4-OT.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.3820102.
References
- Arrington, C.B., Teesch, L.M., and Robertson, A.D. 1999. Defining protein ensembles with native-state NH exchange: Kinetics of interconversion and cooperative units from combined NMR and MS analysis. J. Mol. Biol. 285 1265–1275. [DOI] [PubMed] [Google Scholar]
- Arvidson, D.N., Bruce, C., and Gunsalus, R.P. 1986. Interaction of the Escherichia coli trp aporepressor with its ligand, L-tryptophan. J. Biol. Chem. 261 238–243. [PubMed] [Google Scholar]
- Backmann, J., Schäfer, G., Wyns, L., and Bönisch, H. 1998. Thermodynamics and kinetics of unfolding of the thermostable trimeric adenylate kinase from the Archaeon Sulfolobus acidocaldarius. J. Mol. Biol. 284 817–833. [DOI] [PubMed] [Google Scholar]
- Bai, Y., Milne, J.S., Mayne, L., and Englander, S.W. 1993. Primary structure effects on peptide group hydrogen exchange. Proteins 17 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boice, J.A., Dieckmann, G.R., DeGrado, W.F., and Fairman, R. 1996. Thermodynamic analysis of a designed three-stranded coiled coil. Biochemistry 35 14480–14485. [DOI] [PubMed] [Google Scholar]
- Bowie, J.U. and Sauer, R.T. 1989. Equilibrium dissociation and unfolding of the arc repressor dimer. Biochemistry 28 7139–7143. [DOI] [PubMed] [Google Scholar]
- Brown, B.M., Bowie, J.U., and Sauer, R.T. 1990. Arc repressor is tetrameric when bound to operator DNA. Biochemistry 29 11189–11195. [DOI] [PubMed] [Google Scholar]
- Clarke, J. and Fersht, A.R. 1996. An evaluation of the use of hydrogen exchange at equilibrium to probe intermediates on the protein folding pathway. Fold. Des. 1 243–254. [DOI] [PubMed] [Google Scholar]
- Edelhoch, H. 1967. Spectroscopic determination of tryptophan and tyrosine in proteins. Biochemistry 6 1948–1954. [DOI] [PubMed] [Google Scholar]
- Ghaemmaghami, S., Fitzgerald, M.C., and Oas, T.G. 2000. A quantitative, high-throughput screen for protein stability. Proc. Natl. Acad. Sci. 97 8296–8301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaemmaghami, S. and Oas, T.G. 2001. Quantitative protein stability measurement in vivo. Nat. Struct. Biol. 8 879–882. [DOI] [PubMed] [Google Scholar]
- Gittelman, M.S. and Matthews, C.R. 1990. Folding and stability of trp aporepressor from Escherichia coli. Biochemistry 29 7011–7020. [DOI] [PubMed] [Google Scholar]
- Glasoe, P.K. and Long, F.A. 1960. Use of glass electrodes to measure acidities in deuterium oxide. J. Phys. Chem. 64 188–190. [Google Scholar]
- Gloss, L.M. and Matthews, C.R. 1997. Urea and thermal equilibrium denaturation studies on the dimerization domain of Escherichia coli trp repressor. Biochemistry 36 5612–5623. [DOI] [PubMed] [Google Scholar]
- He, J.-j. and Matthews, K.S. 1990. Effect of amino acid alterations in the tryptophan-binding site of the trp repressor. J. Biol. Chem. 265 731–737. [PubMed] [Google Scholar]
- Hvidt, A. and Nielsen, S.O. 1966. Hydrogen exchange in proteins. Adv. Prot. Chem. 21 287–386. [DOI] [PubMed] [Google Scholar]
- Jin, L., Yang, J., and Carey, J. 1993. Thermodynamics of ligand binding to trp repressor. Biochemistry 32 7302–7309. [DOI] [PubMed] [Google Scholar]
- Joachimiak, A., Kelley, R.L., Gunsalus, R.P., Yanofsky, C., and Sigler, P.B. 1983. Purification and characterization of trp aporepressor. Proc. Natl. Acad. Sci. 80 668–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh, S.N., Rohl, C.A., Kiefhaber, T., and Baldwin, R.L. 1996. A general two-process model describes the hydrogen exchange behavior of RNase A in unfolding conditions. Proc. Natl. Acad. Sci. 93 1982–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, W., Qasim, M.A., Laskowski, M., and Kent, S.B.H. 1997. Probing intermolecular main chain hydrogen bonding in serine proteinase-protein inhibitor complexes: Chemical synthesis of backbone-engineered turkey ovomucoid third domain. Biochemistry 36 673–679. [DOI] [PubMed] [Google Scholar]
- Marmorstein, R.Q., Joachimiak, A., Sprinzl, M., and Sigler, P.B. 1987. The structural basis for the interaction between L-tryptophan and the Escherichia coli trp aporepressor. J. Biol. Chem. 262 4922–4927. [PubMed] [Google Scholar]
- Miranker, A., Robinson, C.V., Radford, S.E., Aplin, R.T., and Dobson, C.M. 1993. Detection of transient protein folding populations by mass spectrometry. Science 262 896–900. [DOI] [PubMed] [Google Scholar]
- Myers, J.K., Pace, C.N., and Scholtz, J.M. 1995. Denaturant m values and heat capacity changes: Relation to changes in accessible surface areas of protein unfolding. Protein Sci. 4 2138–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozaki, Y. 1972. The preparation of guanidine hydrochloride. Methods Enzymol. 26 43–50. [DOI] [PubMed] [Google Scholar]
- Powell, K.D. and Fitzgerald, M.C. 2001. Measurements of protein stability by H/D exchange and matrix-assisted laser desorption/ionization mass spectrometry using picomoles of material. Anal. Chem. 73 3300–3304. [DOI] [PubMed] [Google Scholar]
- Schellman, J.A. 1975. Macromolecular binding. Biopolymers 14 999–1018. [Google Scholar]
- Silinski, P., Allingham, M.J., and Fitzgerald, M.C. 2001. Guanidine-induced equilibrium unfolding of a homo-hexameric enzyme 4-oxalocrotonate tautomerase (4-OT). Biochemistry 40 4493–4502. [DOI] [PubMed] [Google Scholar]
- Smith, D.L., Deng, Y., and Zhang, Z. 1997. Probing the non-covalent structure of proteins by amide hydrogen exchange and mass spectrometry. J. Mass Spectrom. 32 135–146. [DOI] [PubMed] [Google Scholar]
- Waddell, W.J. 1956. A simple ultraviolet spectrophotometric method for the determination of protein. J. Lab. Clin. Med. 48 311–314. [PubMed] [Google Scholar]
- Wales, T.E. and Fitzgerald, M.C. 2001. The energetic contribution of backbone-backbone hydrogen bonds to the thermodynamic stability of a hyperstable P22 arc repressor mutant. J. Am. Chem. Soc. 123 7709–7710. [DOI] [PubMed] [Google Scholar]
- Wolf, P. 1983. A critical reappraisal of Waddell's technique for ultraviolet spectrophotometric protein estimation. Anal. Biochem. 129 145–155. [DOI] [PubMed] [Google Scholar]
- Yi, Q., Scalley, M.L., Simons, K.T., Gladwin, S.T., and Baker D. 1997. Characterization of the free energy spectrum of peptostreptococcal protein L. Fold. Des. 2 271–280. [DOI] [PubMed] [Google Scholar]
- Zhang, Y.-Z. 1995. "Protein and peptide structure and interactions studied by hydrogen exchange and NMR." Ph.D. thesis, Structural Biology and Molecular Biophysics, University of Pennsylvania, PA.
- Zitzewitz, J.A., Bilsel, O., Luo, J., Jones, B.E., and Matthews, C.R. 1995. Probing the folding mechanism of a leucine zipper peptide by stopped-flow circular dichroism spectroscopy. Biochemistry 34 12812–12819. [DOI] [PubMed] [Google Scholar]