

Abstract
The peripheral subunit-binding domain (PSBD) of the dihydrolipoyl acetyltransferase (E2, EC 2.3.1.12) binds tightly but mutually exclusively to dihydrolipoyl dehydrogenase (E3, EC 1.8.1.4) and pyruvate decarboxylase (E1, EC 1.2.4.1) in the pyruvate dehydrogenase multienzyme complex of Bacillus stearothermophilus. Isothermal titration calorimetry (ITC) experiments demonstrated that the enthalpies of binding (ΔH°) of both E3 and E1 with the PSBD varied with salt concentration, temperature, pH, and buffer composition. There is little significant difference in the free energies of binding (ΔG° = −12.6 kcal/mol for E3 and = −12.9 kcal/mol for E1 at pH 7.4 and 25°C). However, the association with E3 was characterized by a small, unfavorable enthalpy change (ΔH° = +2.2 kcal/mol) and a large, positive entropy change (TΔS° = +14.8 kcal/mol), whereas that with E1 was accompanied by a favorable enthalpy change (ΔH° = −8.4 kcal/mol) and a less positive entropy change (TΔS° = +4.5 kcal/mol). Values of ΔCp of −316 cal/molK and −470 cal/molK were obtained for the binding of E3 and E1, respectively. The value for E3 was not compatible with the ΔCp calculated from the nonpolar surface area buried in the crystal structure of the E3-PSBD complex. In this instance, a large negative ΔCp is not indicative of a classical hydrophobic interaction. In differential scanning calorimetry experiments, the midpoint melting temperature (Tm) of E3 increased from 91°C to 97.1°C when it was bound to PSBD, and that of E1 increased from 65.2°C to 70.0°C. These high Tm values eliminate unfolding as a major source of the anomalous ΔCp effects at the temperatures (10–37°C) used for the ITC experiments.
Keywords: Pyruvate dehydrogenase;, microcalorimetry;, protein, protein interaction;, thermodynamics;, multienzyme complex
The pyruvate dehydrogenase (PDH) complex is a member of a widespread family of multienzyme complexes responsible for the oxidative decarboxylation of 2-oxo acids, which have structural and mechanistic features in common (for reviews, see de Kok et al. 1998, Perham 2000, and references therein). The PDH complex (Mr = 5–10 × 106) is made up of multiple copies of three different enzymes, one of which, dihydrolipoyl acetyltransferase (E2, EC 2.3.1.12), self-assembles with either octagonal (24-mer) or icosahedral (60-mer) symmetry depending on the source, to form the structural core of the complex. Multiple copies of the other two enzymes, pyruvate decarboxylase (E1, EC 1.2.4.1) and dihydrolipoyl dehydrogenase (E3, EC 1.8.1.4), bind tightly but independently to this E2 core.
The E2 polypeptide chain consists of several separately folded domains joined by extended, flexible linker regions. At the N-terminus are one or more lipoyl domains (up to three, again depending on the source of the E2 chain), each of which contains approximately 80 amino acid residues folded as an eight-stranded β-barrel. In each lipoyl domain is a lipoyl-lysine residue prominently displayed in an exposed β-turn, which acts as a `swinging arm' in the swinging domain that serves to transfer the substrate between the three different active sites (Reed and Hackert 1990; de Kok et al. 1998; Jones et al. 2000; Perham 2000). Following the lipoyl domain(s) and linker regions is a much smaller domain (∼35 amino acid residues) involved in binding the peripheral enzymes (E1 and E3), the structure of which is dominated by two parallel α-helices connected by a loop region and a helical turn (Robien et al. 1992; Kalia et al. 1993). This peripheral subunit-binding domain (PSBD) in turn is separated by another linker region from the large (28kD) C-terminal domain that contains the acetyltransferase active site. It is this acetyltransferase domain that aggregates to form the inner structural core (octahedral or icosahedral) of the PDH complex (Reed and Hackert 1990; Perham 2000). The structures of the octahedral inner core of the Azotobacter vinelandii PDH complex (Mattevi et al. 1993) and the icosahedral inner core of the Bacillus stearothermophilus and Enterococcus faecalis PDH complexes (Izard et al. 1999) have been solved by means of X-ray crystallography.
In octahedral PDH complexes, the PSBD provides the binding site for E3, whereas E1 is thought to bind principally to the C-terminal acyltransferase domain (de Kok et al. 1998; Perham 2000). In contrast, the PSBD of the icosahedral PDH complex of B. stearothermophilus is responsible for binding both E1 and E3. The situation in the icosahedral PDH complexes of yeast and mammals is different again, owing to the presence of 6–12 copies of an additional type of subunit, protein X, in the E2 core (Sanderson et al. 1996). Protein X resembles the N-terminal half of the E2 chain, with a lipoyl domain capable of participating in PDH complex activity and a PSBD-like domain responsible for binding E3 (Patel and Roche 1990; Lawson et al. 1991). In such complexes it appears that the PSBD in the E2 chain binds only E1.
A recombinant di-domain (DD) representing the lipoyl domain and PSBD of the E2 chain of the B. stearothermophilus PDH complex is capable of binding to the dimeric E3 (Hipps et al. 1994) and tetrameric E1 (α2β2) (Lessard and Perham 1995) components from the same complex. In both instances, the stoichiometry of the interaction was surprisingly found to be 1:1, suggesting that the binding sites for the PSBD on E1 and E3 must lie on, or close to, the two-fold axis of symmetry of the E3 dimer or E1 tetramer (Lessard and Perham 1995). The binding of B. stearothermophilus E3 and E1 to the PSBD is also mutually exclusive, but exhibits similar free energies (ΔG°bind) of −12.6 and −12.9 kcal/mol, respectively (Lessard et al. 1996) (Fig. 1A ▶).
Fig. 1.

(A) MolScript (Kraulis 1991) representation of the competitive interaction between E3 (green, monomer A; blue, monomer B; orange and ball-and-stick model, cofactor FAD) and E1 (green and blue, α subunits; yellow and purple, β subunits; gray sphere, magnesium ion; red ball-and-stick model, thiamin diphosphate) and the PSBD (red) of E2 in the B. stearothermophilus PDH complex. The crystal structure of the E3-PSBD at 2.6Å resolution is that of Mande et al. (1996). The structure of E1 from the B. stearothermophilus PDH complex is unknown, but crystal structures of the E1 components of P. putida and human branched chain 2-oxo acid dehydrogenase complexes have been reported (Ævarsson et al. 1999a,1999b). A three-dimensional model (H.I. Jung, H. Chauhan, M. Fries, and R.N. Perham, unpubl.) of the B. stearothermophilus E1 derived from homology modeling (Guex and Peitsch 1997) is displayed here; the orientation of the PSBD with respect to the E1β subunits is arbitrary. (B) The binding interface of the E3-PSBD complex (Mande et al. 1996) is shown in detail. The electrostatic `zipper' is formed between acidic residues (AspB344 and GluB431) of E3 and basic residues (Arg135 and Arg139) of the PSBD.
A crystal structure of the B. stearothermophilus E3-PSBD complex has revealed that the binding is dominated by electrostatic interactions between the negatively charged side chains of Asp and Glu residues derived from one E3 subunit, and positively charged side chains of Arg residues contributed by the PSBD (Mande et al. 1996). The binding site on E3 is indeed so close to the two-fold axis that the binding of one PSBD precludes the binding of a second (Fig. 1B ▶). There is no crystal structure of B. stearothermophilus E1 or of the E1-PSBD complex. However, crystal structures of the homologous E1 (α2β2) components of Pseudomonas putida (Ævarsson et al. 1999a) and human (Ævarsson et al. 1999b) branched chain 2-oxo acid dehydrogenase complexes are available. Details of the binding site for PSBD on E1 are not clear, but biochemical studies indicate that the E1β subunit is chiefly responsible (Stepp and Reed 1985; Lessard and Perham 1995).
Recent advances in biological microcalorimetry have made it possible to explore directly, and in some detail, the thermodynamics of protein–protein interactions. In the present study, two calorimetric methods, isothermal titration calorimetry (ITC) and differential scanning calorimetry (DSC), were applied to determine the thermodynamic properties of the competitive interaction of E3 and E1 with the PSBD of B. stearothermophilus E2. The results have important general implications for protein–protein interaction when interpreted in the light of the E3-PSBD structure (Mande et al. 1996) and offer a deeper understanding of the assembly of this vast multifunctional enzyme complex.
Results
Binding enthalpies of E3 and E1 with PSBD
The heats of interaction of PSBD with E3 and E1 were determined using ITC. Figure 2 ▶ shows typical titrations for the interaction of DD with E3 and E1 in HBS buffer (10 mM Hepes, 150 mM NaCl, 3.4 mM EDTA) at pH 7.4 and 25°C, conditions identical to those used for surface plasmon resonance (SPR) measurements of the binding constant (Ka) (Lessard et al. 1996). Under these conditions, complexation of DD with E3 (Fig. 2A ▶) was endothermic (positive peaks in the ITC output), saturating at a stoichiometry of roughly 1:1. In contrast, under the same conditions, binding of DD to E1 was exothermic, but exhibited the same stoichiometry.
Fig. 2.

Isothermal titration calorimetric analysis of the interaction of the PSBD (180–230μM) with E3 (7.2μM) and E1 (13.5μM) components in HBS buffer (10 mM Hepes, 150 mM NaCl, and 3.4 mM EDTA, pH 7.4), at 25°C. (A) Raw data obtained over a series of injections of PSBD and plotted as heat (μcal/sec) versus time (min). Dashed line, binding of E3 showing the trace of positive peaks (endothermic); solid line, binding of E1 showing the trace of much larger negative peaks (exothermic). (B) Binding isotherms created by plotting the areas under the peaks in panel A against the molar ratio of the PSBD injected. The lines through the best-fit values obtained by least-squares regression using a one-site model give the stoichiometry (n) and enthalpy (ΔH). For E3 (open circles and dashed line), n = 1.12, ΔH = +2.2 kcal/mol; for E1 (closed rectangles and solid line), n = 0.98 and ΔH = −8.6 kcal/mol.
Integrated heat data (Fig. 2B ▶) yield differential thermal binding curves which may be analyzed by standard nonlinear regression methods to give estimates of the binding stoichiometry (n), equilibrium constant for association (Ka), and enthalpy of binding (ΔH), assuming a single stoichiometric binding equilibrium. However, as shown in a previous study using SPR detection (Lessard et al. 1996), the binding of DD to E3 and E1 is very tight (Ka = 1.7 × 109 M−1 for E3 and Ka = 3.1 × 109 M−1 for E1). For such tight binding (Ka > 108 M−1), only the enthalpy change upon binding (ΔH) and the stoichiometry of the association (n) can be precisely determined by isothermal titration microcalorimetry (Wiseman et al. 1989). As shown in Fig. 2B ▶, the binding stoichiometry calculated for each interaction was close to unity, which is in a good agreement with previous studies (Hipps et al. 1994; Lessard et al. 1996; Mande et al. 1996). The binding enthalpies at 25°C were +2.2 and −8.4 kcal/mol for E3 and E1, respectively.
The effect of salt on the binding enthalpy
Previous kinetic measurements of the interaction of E1 and E3 with DD were performed in the presence of 150 mM NaCl to minimize any nonspecific binding during SPR detection (Lessard et al. 1996). To evaluate the effect of salt on the binding enthalpies, we titrated E1 and E3 with DD in the presence of four different concentrations of NaCl (up to 250 mM) at 30°C (Table 1). As the salt concentration was increased, the enthalpy of binding became less favorable, suggesting that charged groups play an important part in the interactions. In the case of E3, no heat of binding was observed at higher salt concentrations (≥150mM) although the formation of an E3-DD complex under the same conditions was confirmed (data not shown) by means of nondenaturing PAGE (Lessard and Perham 1995). For experimental consistency and to allow comparison with other measurements, the concentration of the salt was fixed at 150 mM for further calorimetric measurements, unless stated otherwise.
Table 1.
The enthalpy changes of the interaction of E3 and E1 with the PSBD at different temperatures and salt concentrations
| E3 | E1 | ||||
| pH | Temp. (°C) | Buffer | NaCl (mM) | ΔH° (kcal/mol) | ΔH° (kcal/mol) |
| 7.4 | 30 | Hepes | 0 | −7.7 (±0.8) | −15.6 (±1.2) |
| 7.4 | 30 | Hepes | 50 | −3.8 (±0.4) | −13.3 (±0.5) |
| 7.4 | 30 | Hepes | 150 | 0* | −11.3 (±0.0) |
| 7.4 | 30 | Hepes | 250 | 0* | −10.2 (±0.1) |
| 7.4 | 10 | HBS | 150 | +6.6 (±0.6) | −1.7 (±0.0) |
| 7.4 | 25 | HBS | 150 | +2.2 (±0.1) | −8.4 (±0.1) |
| 7.4 | 30 | HBS | 150 | 0* | −11.3 (±0.0) |
| 7.4 | 37 | HBS | 150 | −1.8 (±0.3) | −14.3 (±0.4) |
All the values are the average of three or more repeated measurements, from which the standard error was calculated.
* No heat change detected.
E3 and E1 bind competitively to PSBD with different thermodynamic parameters
The standard free energy of binding (ΔG°) was calculated from the equilibrium constant of the association (Ka), determined previously by means of SPR analysis at 25 °C (Lessard et al. 1996), using the following expression:
 |
(1) |
The change of entropy on binding (TΔS°) was subsequently calculated from the relationship:
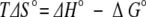 |
(2) |
where ΔH° is the enthalpy change of binding.
As shown in Table 2, the interaction of E3 with PSBD was found to be characterized by an enthalpically unfavorable (ΔH° = +2.2 kcal/mol) but entropically favorable (TΔS° = +14.8 kcal/mol) process. In contrast, the enthalpy effect for E1 had the opposite sign to that for E3 under the same conditions, despite the small difference in the free energies of binding (ΔG° = −12.6 kcal/mol for E3 and ΔG° = −12.9 kcal/mol for E1). The association of E1 with PSBD was exothermic (ΔH° = −8.4 kcal/mol), this strongly favorable enthalpy change being associated with an entropy change (TΔS°) of only +4.5 kcal/mol. These contrasting values exemplify, for E1 and E3, enthalpy–entropy compensation phenomena (Table 2) that now appear characteristic of many macromolecular interactions (Cooper et al. 2001).
Table 2.
Comparison of the thermodynamic parameters of the interaction of E3 and E1 with the PSBD of E2
| Enzyme | Kaa (M−1) | ΔG°b (kcal/mol) | ΔH°c (kcal/mol) | TΔS°d (kcal/mol) | ΔCpc (kcal/molK) | Protons linkedc (n) |
| E3 | 1.7 × 109 | −12.6 | +2.2 | +14.8 | −316 | −0.02 |
| E1 | 3.1 × 109 | −12.9 | −8.4 | +4.5 | −470 | +0.11 |
All the kinetic and thermodynamic parameters are taken from experiments in HBS buffer, pH 7.4 at 25°C.
a Obtained from SPR analysis.
b Calculated from ΔG° = −RT lnKa.
c Calculated from ITC measurments.
d Calculated from TΔS° = ΔH° − ΔG°.
Heat capacity changes
This apparent difference in thermodynamic signature (exothermic versus endothermic) between binding at 25°C becomes less clear cut when we consider the temperature dependence. Isothermal microcalorimetric titrations were carried out over a range of temperatures (10–37°C) in HBS buffer, pH 7.4, to obtain an estimate of heat capacity change, ΔCp, upon formation of the E3-PSBD and E1-PSBD complexes. This was derived from the slope of ΔH° versus T plots, assumed linear (Fig. 3 ▶), using the standard thermodynamic relationship:
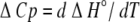 |
(3) |
Fig. 3.

Temperature dependence of enthalpy changes. The heat capacity (ΔCp) values were determined from the slopes of the fitted lines. For E3 (closed circles and solid line), ΔCp = −316 (±16) cal/molK; for E1 (closed rectangles and solid line), ΔCp = −470 (±14) cal/molK in HBS buffer (10 mM Hepes, 150 mM NaCl, and 3.4 mM EDTA, pH 7.4). The dashed line (closed triangles) is for E3 in phosphate buffer (50 mM sodium phosphate, pH 7.0) without salt, giving ΔCp = −475 (±2) cal/molK.
Both complexes showed a significant temperature dependence of ΔH° (Fig. 3 ▶), complex formation becoming more exothermic as the temperature rose. The data fit well to a linear function, whose slope gives the heat capacity change for binding of E1 or E3 to the PSBD. Values of ΔCp of −316 (±16) cal/molK for E3 and −470 (±14) cal/molK for E1 were obtained, which are around the average value (−333 cal/molK) of ΔCp for protein–protein interactions (Stites 1997).
A large negative ΔCp has been thought to be characteristic of the classical hydrophobic interaction between nonpolar groups in aqueous systems (Kauzmann 1959), and empirical correlations between ΔCp and changes in exposed surface area have been proposed (Spolar et al. 1992). From studies of model compounds (Murphy and Gill 1991), the change in heat capacity in a protein folding process was found to depend on buried polar and nonpolar surface area, according to the following empirical relationship (Murphy et al. 1993):
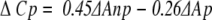 |
(4) |
where ΔAnp and ΔAp are the changes in the nonpolar and polar surface area buried upon folding, respectively. This relationship has been proposed to apply also to protein–protein interactions, as comparable with a folding process (Gómez and Freire 1995). Since a buried surface area has a negative sign in equation (4), a negative ΔCp is derived from the nonpolar buried surface area. Thus, a large negative ΔCp has been used as an indication of hydrophobic interaction in complex formation.
From the crystal structure of the E3-PSBD complex (Mande et al. 1996), the nonpolar and polar surface areas buried in the interface are 674.9 Å2 and 449.2 Å2, respectively, calculated using the algorithm of Lee and Richards (1971) in the program NACCESS (Hubbard and Thornton 1993). Using equation (4), the calculated ΔCp is −187 cal/molK, significantly different from the value of −316 cal/molK observed in the ITC measurements (see above). At lower ionic strength (50 mM sodium phosphate buffer, pH 7.0), an even greater temperature dependence of the heat of binding and, consequently, an even larger ΔCp (−475±2 cal/molK), was observed (Fig. 3 ▶). Unless the separate E3 and PSBD and/or the E3-PSBD complex have markedly different conformations at different ionic strengths (for which there is no evidence), it is difficult to reconcile such ΔCp differences with changes in accessible surface area alone. The dependence of the absolute ΔH values on ionic strength (Table 1, Fig. 3 ▶) indicate that electrostatic effects must be playing a significant role here. It is now becoming clear that large ΔCp effects, together with associated entropy–enthalpy compensations, are a much more general phenomenon to be anticipated in any system comprising a multiplicity of weak interactions (Dunitz 1995), and this has been quantitatively demonstrated in some systems (Cooper et al. 2001).
Protonation changes during complex formation
One possible source of anomalous enthalpy or heat capacity effects is uptake or release of protons during complexation (Stites 1997), and this can be examined by ITC experiments in different buffer systems (Bradshaw and Waksman 1998). For any process that embodies uptake or release of protons, the experimentally observed enthalpy change (ΔHobs) will include contributions from the heat of protonation/deprotonation of the buffer employed. Thus it is essential that effects of buffer ionization (ΔHion) should be allowed for to obtain the true enthalpy change of binding (ΔHbind). The relationship between these enthalpies is expressed by the following equation:
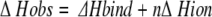 |
(5) |
where n is the number of protons taken up (positive sign) or released (negative sign) by the buffer in the formation of the complex.
A further set of ITC experiments was therefore performed in several different buffers with different heats of ionization (ΔHion) and at four different pHs (pH 5.5, 6.5, 7.4, and 8.5). The enthalpies of ionization of the buffers used in this experiment were as follows: cacodylate, −0.56 kcal/mol; MES, +3.73 kcal/mol; Bistris, +6.75 kcal/mol; Aces, +7.47 kcal/mol; Hepes, +5.00 kcal/mol; Tricine, +7.76 kcal/mol; Tris-HCl, +11.51 kcal/mol (Bradshaw and Waksman 1998). Experiments below pH 5.5 were not attempted, owing to the aggregation of both E3 and E1 in sodium acetate buffer at pH 4.5. The plots of ΔHobs versus ΔHion for the formation of the E3-PSBD and E1-PSBD complexes are presented in Figure 4 ▶.
Fig. 4.

Variation of the binding enthalpies (ΔH°) for the interaction of E3 (A) and E1 (B) with PSBD as a function of the ionization enthalpy (ΔH°ion) of the buffer. All the experiments were at 25°C: pH 5.5 (▪), pH 6.5 (▴), pH 7.4 (•) and pH 8.5 (♦). The average value of at least two estimates is depicted. Arrows indicate the ΔH°ion values for the buffers employed. The number of protons linked to the complex formation is obtained from the slope of the best-fit line at a given pH.
Neither E3 nor E1 binding to PSBD showed significant protonation effects at pHs of 7.4 and 8.5, but there were clear buffer-dependent effects for the E3-PSBD complex at pH 6.5 and for both complexes at pH 5.5. The number of protons linked to the association event was obtained from the slope of the best-fit line at a specific pH. It appeared that there was release of up to one proton during binding to PSBD at acidic pH (Table 3). This suggests that one (or more) protein group(s) with acidic pKa is (are) required in an unprotonated state for effective complex formation. Given the structural information about the E3-PSBD complex (Mande et al. 1996), the protonation observed in the E3-PSBD interaction may be due to two ionizable groups, Asp344 and Glu431, at the binding site on E3 (Fig. 1B ▶). In the absence of a structure for E1-PSBD, it is impossible to speculate on the origins of any protonation effect there.
Table 3.
The enthalpy changes of the interaction of E3 and E1 with PSBD in different buffers at different pH values
| E3 | E1 | ||||
| pH | Buffer | ΔH°obs (kcal/mol) | Protons linkeda (n) | ΔH°obs (kcal/mol) | Protons linkeda (n) |
| 5.5 | cacodylate | 3.9 | −1.14 ± 0.14 | −7.5 | −0.64 ± 0.21 |
| MES | −1.8 | −11.5 | |||
| Bistris | −4.3 | −12.0 | |||
| 6.5 | cacodylate | 2.9 | −0.36 ± 0.04 | −9.2 | 0.1 ± 0.01 |
| MES | 1.6 | −8.8 | |||
| Aces | 0 | −8.4 | |||
| 7.4 | cacodylate | 2.0 | −0.02 ± 0.05 | −9.4 | 0.11 ± 0.08 |
| MES | 1.6 | −8.2 | |||
| Hepes | 2.1 | −8.5 | |||
| Aces | 1.8 | −8.6 | |||
| 8.5 | Hepes | 1.9 | 0.08 ± 0.07 | −10.8 | −0.02 ± 0.18 |
| Tricine | 1.7 | −9.8 | |||
| Tris-HCl | 2.4 | −10.8 | |||
All experiments were repeated twice and the average values are listed. The buffer conditions were 20 mM buffer, 150 mM NaCl and 3.4 mM EDTA at 25°C.
a The number of protons exchanged upon binding was obtained from the slopes of plots of ΔHobs versus ΔHion in Figure 4 ▶.
Thermal stability of the complexes
Anomalous ΔCp effects could also arise from perturbation of the protein structure, especially at temperatures close to the natural unfolding transition temperature, Tm. This possibility was tested by DSC experiments on the isolated E1 and E3 and their complexes with DD. Typical DSC curves are shown in Figure 5 ▶. Two distinct transitions were observed in a DSC experiment with DD alone, implying that the lipoyl domain and PSBD are unfolding independently, as expected for two domains linked by a long flexible segment of polypeptide chain. E1 and E3 were found to unfold in single cooperative transitions, with a Tm of 91.0°C and 65.2°C for E3 (3.6 μM) and E1 (4.6 μM), respectively. Both Tm values were shifted higher (to 97.1°C for E3 and 70.0°C for E1) in the presence of DD at a saturating concentration of 22.3 μM, representing the enhanced thermal stability that accompanies tight complex formation in both instances. It is clear that no significant unfolding of E1, E3, or DD occurs at the much lower temperatures used for the ITC measurements, so perturbation of the protein structures is extremely unlikely as a major source of the anomalous ΔCp effects described above.
Fig. 5.

DSC trace of the thermal denaturation of E3 (Tm = 91°C) and E1 (Tm = 65.2°C) in the absence (dotted line) and presence (solid line) of PSBD. Inset, DSC traces of the PSBD (Tm = 67.8°C), LD (Tm = 88.7°C), and DD.
Discussion
In general, an unfavorable enthalpy change and large positive entropy change associated with biomolecular complex formation might be characteristic of electrostatic (Ortiz-Salmerón et al. 1998) and/or hydrophobic interactions (Velazquez-Campoy et al. 2000). In the case of polar interactions such as hydrogen bonding, salt bridge formation, and other electrostatic effects, the unfavorable enthalpy change results from a net loss of such bonds, in forming the complex itself, or between ordered solvent molecules on the biomolecular surface. An unfavorable enthalpy change in hydrophobic interactions is due to the burial of hydrophobic groups, which also requires disruption of an ordered solvation network. In most hydrophobic interactions, the net enthalpy change is low and the entropy change is high. However, in both polar and hydrophobic interactions, most of the entropy gain is related to the liberation of water molecules from a binding interface in the course of complex formation, outweighing the loss of entropy associated with the formation of the complex itself. Consequently it is very difficult to disentangle the various components of molecular interactions from thermodynamic data alone (Cooper 1999; Cooper et al. 2001).
According to the 2.6Å crystal structure (Mande et al. 1996), the E3-PSBD complex is stabilized chiefly by electrostatic interactions between positively charged residues from the binding domain and negatively charged residues from the interface domains of both E3 monomers. Specifically, residues Arg135 and Arg139 of PSBD interact with Asp344 and Glu431 of E3 (monomer B), and no water molecule is visible in the binding interface (Fig. 1B ▶). These charged sidechains of E3 and PSBD are likely to form hydration shells with water molecules when they are not in the complex. Thus, it appears that the thermodynamic parameters observed in the formation of the E3-PSBD complex reflect the presence of water molecules bound to the future binding site in the two proteins when they are uncomplexed in solution and the release of such water molecules from the newly formed interface as the binding proceeds.
Other examples of water exclusion from interfaces caused by salt bridge formation can be found in biomolecular interactions such as protease–inhibitor, antigen–antibody and protein–DNA interactions (Janin 1999 and references therein). Many principles of molecular interaction have been developed from the fact that excluding water from mating surfaces can serve as a driving force in molecular association (Connelly et al. 1994; Ladbury 1996). Nevertheless, in some systems such as antibody–antigen interaction (Bhat et al. 1994), protein–carbohydrate interaction (Quiocho et al. 1989) and protein–peptide interaction (Tame et al. 1994), the inclusion of water appears to confer unusual specificity and enhanced affinity on the intermolecular interaction.
The source of heat capacity changes observed in protein–protein interactions is now becoming a subject of controversy. Various lines of evidence suggest that in an interaction mediated by a large number of water molecules, the retention of water molecules in a binding site can cause a significant contribution to the change in heat capacity upon binding (Morton and Ladbury 1996). There are also reports speculating that the burial of water in protein–protein interfaces is responsible for some of the negative changes in heat capacity (Bhat et al. 1994; Guinto and di Cera 1996; Stites 1997). A poor correlation between the measured and the calculated values for ΔCp in E3-PSBD complex formation casts doubt on the universality of the empirical relationship in equation (4) above and supports the suggestion that although the ΔCp for protein folding often correlates well with the burial of nonpolar surface, a similar correlation may not apply to protein–protein interactions (Raman et al. 1995).
The association of E3 with the PSBD would have been wrongly inferred as being governed by hydrophobic interactions, on the basis that it is entropy-driven. The B. stearothermophilus E3-PSBD complex must be added to the growing list of protein–protein interactions that show little, if any, correlation between ΔCp and the nonpolar surface area buried on binding (Raman et al. 1995; Pearce et al. 1996; Frisch et al. 1997). The large favorable enthalpy and small favorable entropy changes observed in the E1-PSBD interaction would normally be expected to imply more secondary forces, for example hydrogen bonds, as being involved, and less in the way of hydrophobic interactions between the two molecules. However, the strong component of electrostatic interaction in the E3-PSBD complex that we have seen to be associated with very different thermodynamic parameters makes it difficult to predict much with certainty about the interface in the E1-PSBD complex. A more detailed analysis of that binding interface will have to await a three-dimensional structure of the PSBD-complexed form of E1.
Materials and methods
Materials
All reagents used were of analytical grade. Unless otherwise indicated, buffers and chemicals were purchased from Sigma Chemicals. Bacteriological media were from Difco Laboratories. Ampicillin was purchased from Beecham Research. Restriction endonucleases BamHI and NcoI were supplied by Pharmacia and New England Biolabs, respectively. The expression vector pET11d was supplied by Novagene. Pfu DNA polymerase and T4 DNA ligase were from Stratagene and Promega, respectively. Isopropyl-1-thio-β-D-galactopyranoside (IPTG) was supplied by Melford Laboratory. E.coli strain JM 109 (endA1, recA1, gyrA96, thi, hsdR17 (rk−,mk+), relA1, supE44,D(lac-proAB), [F`, traD36, proAB, lacIqZDM15] was used for genetic manipulations, and the lambda lysogen strain BL21(DE3), which carries the T7RNA polymerase gene under the control of the lacUV5 promoter, was used for gene expression (Studier et al. 1990). HBS buffer (pH 7.4) is composed of 10 mM Hepes, 150 mM NaCl, and 3.4 mM EDTA.
Construction of the plasmid pET11DD
The subgene encoding the DD (residues 1–170) of B. stearothermophilus E2 in the plasmid NAVDD (Hipps and Perham 1992) was amplified by polymerase chain reaction (PCR). The 21bp forward primer incorporated two mismatches to create an NcoI restriction site and to alter the translation initiation codon from GTG to ATG. Primer sequences were as follows: 5`-TAGAC AGACCATGGCTTTTGA-3` (forward, mismatches underlined) and 5`-ACCCGGGGATCCGTCGCCGC-3` (reverse). The resulting 573bp DNA fragment was treated with NcoI and BamHI and purified by the "crush and soak" method (Maniatis et al. 1982) after electrophoresis in a 5% polyacrylamide gel. This fragment was then ligated into NcoI/BamHI-cut, calf intestine alkaline phosphatase-treated pET11d. The resulting plasmid, pET11DD, was transformed into the E. coli strain JM 109 cells. The insert DNA was completely sequenced using the TagTrack™ Sequencing System (Promega) to check the fidelity of the PCR.
Protein purification
E. coli BL21(DE3) cells transformed with pET11DD were grown at 37°C in 2 L of LB medium (Maniatis et al. 1982) supplemented with 50 μg/mL ampicillin until the A600 was between 0.7 and 1.0. The cells were then induced with IPTG (final concentration of 1mM) and incubated for a further 2 h. The cells were harvested by centrifugation at 4,500 × g for 40 min, resuspended in 100 mM Tris-HCl buffer (pH 7.5) containing 1 mM EDTA, 0.02% sodium azide, 0.1 mM PMSF and disrupted in a French press at 4°C with cell pressure of 140 MPa. Cell debris was removed by ultracentrifugation at 20,000 × g for 20 min, and the supernatant was fractionally precipitated with solid ammonium sulphate (35%–70%). The protein in this fraction was subsequently dissolved in 10 mL of buffer A (20 mM potassium phosphate, pH 7.0, 0.02% sodium azide) and dialyzed overnight at 4°C against 2 L of the same buffer. The solution was then applied to a Pharmacia Hi-load™ S Sepharose column preequilibrated with buffer A and eluted with buffer B (20 mM potassium phosphate, pH 7.0, 0.02% sodium azide, 1M NaCl), applying a 15%–75% gradient over 4 column volumes at a flow rate of 1 mL/min. The fractions containing the DD (as judged by SDS-PAGE, apparent molecular mass 25 kD) were pooled, dialyzed again and loaded onto a Pharmacia Mono™ Q high-performance anion-exchange column preequilibrated with buffer A. The protein was eluted with buffer B, again applying a 15%–75% gradient over 4 column volumes at a flow rate of 2 mL/min. Fractions containing the desired protein were pooled and concentrated by using Centriprep™ filtration. The recombinant E1 and E3 components were purified as described previously (Lessard and Perham 1994; Lessard et al. 1998). The purity of a protein during the purification process was monitored by means of SDS-PAGE using the Pharmacia PhastSystem™. Protein solutions were prepared by exhaustive dialysis at 4°C against a large excess of deionized water and then concentrated to approximately 4.1 mM for DD, 1.5 mM for E3, and 0.28 mM for E1.
Isothermal titration calorimetry
ITC measurements were carried out over different temperature ranges (10–37°C) using MCS-ITC and VP-ITC titration calorimeters (MicroCal, Northampton, MA) to obtain enthalpy and heat capacity changes (Wiseman et al. 1989). All of the protein samples for microcalorimetry were highly concentrated and then exhaustively dialyzed against deionized water. They were diluted into the various different buffers before the titration experiments, to minimize mixing heat effects caused by differences in solution composition. Diluted protein samples were then briefly but gently degassed before being added to the calorimeter cell. The DD, usually at a concentration of approximately 200 μM, was injected in 10-μL increments into the reaction cell (cell volume 1.31–1.41 mL) containing E3 at a concentration of around 7 μM (or E1 at around 9 μM) until complete saturation. A 250-μL injection syringe with 310–400 rpm stirring was used to give a series of 10 μL injections at 3-min intervals. Control experiments for heats of mixing and dilution were performed under identical conditions and used for data correction in subsequent analysis. Data acquisition and subsequent nonlinear regression analysis were done in terms of a simple binding model, using the Microcal ORIGIN software package.
Differential scanning calorimetry
DSC measurements were carried out using a VP-DSC differential scanning calorimeter (Microcal) at a scan rate of 60°C/h in HBS buffer (10 mM Hepes, 150 mM NaCl, 3.4 mM EDTA, pH 7.4) (Cooper and Johnson 1994). Sample and reference solutions were gently degassed for approximately 3 min before loading into the cell. Samples were scanned from 20° to 110°C, but no re-scan was possible owing to the precipitation of denatured E3 and E1. DSC scans were corrected by subtraction of the data from suitable controls, and concentrations were normalized to determine the midpoint melting temperature (Tm). The following concentrations were used for DSC experiments: approximately 50 μM for DD, 10 μM for PSBD, 340 μM for lipoyl domain (LD), 3.0 μM for E3, and 4.0 μM for E1.
General protein methods
Concentrations of protein samples were estimated from both amino acid analysis and UV absorbance measurements assuming A2800.1% = 1.31 (E3, Mr = 100,045), 0.794 (E1, Mr = 153,333), and 0.48 (DD, Mr = 18,383) as reported (Hipps and Perham 1992; Lessard 1995). SDS-PAGE and amino acid analysis were carried out as described (Lessard 1995).
Acknowledgments
This work was supported in part by a research grant (to R.N.P.) from the Biotechnology and Biological Sciences Research Council (BBSRC). We thank the BBSRC and the Wellcome Trust for their support of the core facilities in the Cambridge Centre for Molecular Recognition, and the BBSRC and Engineering and Physical Sciences Research Council for funding the biological microcalorimetry facilities in the University of Glasgow. We thank the Cambridge Overseas Trust and St John's College, Cambridge for financial support to H.I.J. We also thank C. Fuller, D. Pate, and M. Nutley for skilled technical assistance and Drs. G.J. Domingo and H.J. Chauhan for help with protein purification.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
PDH, pyruvate dehydrogenase
E1, pyruvate decarboxylase (EC 1.2.4.1)
E2, dihydrolipoyl acetyltransferase (EC 2.3.1.12)
E3, dihydrolipoyl dehydrogenase (EC 1.8.1.4)
PSBD, peripheral subunit-binding domain
LD, lipoyl domain
CD, catalytic domain
DD, di-domain
SPR, surface plasmon resonance
ITC, isothermal titration microcalorimetry
DSC, differential scanning calorimetry
HBS, Hepes buffered saline
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.4970102.
References
- Ævarsson, A., Seger, K., Turley, S., Sokatch, J. R., and Hol, W.G.J. 1999a. Crystal structure of 2-oxoisovalerate dehydrogenase and the architecture of 2-oxo acid dehydrogenase multienzyme complexes. Nat. Struct. Biol. 6 785–792. [DOI] [PubMed] [Google Scholar]
- Ævarsson, A., Chuang, J.L., Wynn, R.M. Turley, S., Chuang, D.T., and Hol, W.G.J. 1999b. Crystal structure of human branched-chain α-ketoacid dehydrogenase and the molecular basis of mutienzyme complex deficiency in maple syrup urine disease. Structure 8 277–291. [DOI] [PubMed] [Google Scholar]
- Bhat, T.N., Bentley, G.A., Boulot, G., Greene, M.I., Tello, D., Dall'Acqua, W., Souchon, H., Schwarz, F.P., Mariuzza, R.A., and Poljak, R.J. 1994. Bound water molecules and conformational stabilization help mediate an antigen-antibody association. Proc. Natl. Acad. Sci.. 91 1089–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradshaw, J.M. and Waksman, G. 1998. Calorimetric investigation of proton linkage by monitoring both the enthalpy and association constant of binding: Application to the interaction of the Src SH2 domain with a high-affinity tyrosyl phosphopeptide. Biochemistry 37 15400–15407. [DOI] [PubMed] [Google Scholar]
- Connelly, P.R., Aldape, R.A., Bruzzese, F.J., Chambers, S.P., Fitzgibbon, M.J., Fleming, M.A., Itoh, S., Livingston, D.J., Navia, M.A., Thomson, J.A., and Wilson, K.P. 1994. Enthalpy of hydrogen bond formation in a protein-ligand binding reaction. Proc. Natl. Acad. Sci. 91 1964–1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper, A. 1999. Thermodynamic analysis of biomolecular interactions. Curr. Opin. Chem. Biol. 3 557–563. [DOI] [PubMed] [Google Scholar]
- Cooper, A. and Johnson, C.M. 1994. Differential scanning calorimetry. In Methods in molecular biology, Vol 22: Microscopy, optical spectroscopy and macroscopic techniques. (eds. C. Jones, B. Mulloy, and A.H. Thomas A.H.), pp. 125–136. Humana Press; Totowa, NJ. [DOI] [PubMed]
- Cooper, A., Johnson, C.M., Lakey, J.H., and Nöllmann, M. 2001. Heat does not come in different colours: Entropy–enthalpy compensation, free energy windows, quantum confinement, pressure perturbation calorimetry, solvation and the multiple causes of heat capacity effects in biomolecular interactions. Biophys. Chem. 28 215–230. [DOI] [PubMed] [Google Scholar]
- de Kok, A., Hengeveld, A.F., Martin, A., and Westphal, A.H. 1998. The pyruvate dehydrogenase multienzyme complex from Gram-negative bacteria. Biochim. Biophys. Acta 1385 353–366. [DOI] [PubMed] [Google Scholar]
- Dunitz, J.D. 1995. Win some, lose some—Enthalpy–entropy compensation in weak intermolecular interactions. Chem. Biol. 2 709–712. [DOI] [PubMed] [Google Scholar]
- Frisch, C., Schreiber G., Johnson, C.M., and Fersht A.R. 1997. Thermodynamics of the interaction of barnase and barstar: Changes in free energy versus changes in enthalpy on mutation. J. Mol. Biol. 267 696–706. [DOI] [PubMed] [Google Scholar]
- Gómez, J. and Freire, E. 1995. Thermodynamic mapping of the inhibitor site of the aspartic protease endothiapepsin. J. Mol. Biol. 252 337–350. [DOI] [PubMed] [Google Scholar]
- Guex, N. and Peitsch, M.C. 1997. SWISS-MODEL amd the Swiss-PdbViewer: An environment for comparative protein modelling. Electrophoresis 18 2714–2723. [DOI] [PubMed] [Google Scholar]
- Guinto E.R. and di Cera, E. 1996. Large heat capacity change in a protein-monovalent cation interaction. Biochemistry 35 8800–8804. [DOI] [PubMed] [Google Scholar]
- Hipps, D.S., Packman, L.C., Allen, M.D., Fuller, C., Sakaguchi, K., Appella, E., and Perham, R.N. 1994. The peripheral subunit-binding domain of the dihydrolipoyl acetyltransferase component of the pyruvate dehydrogenase complex of Bacillus stearothermophilus: Preparation and characterization of its binding to the dihydrolipoyl dehydrogenase component. Biochem J. 297 137–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hipps, D.S. and Perham, R.N. 1992. Expression in Escherichia coli of a sub-gene encoding the lipoyl and peripheral subunit-binding domains of the dihydrolipoyl acetyltransferase component of the pyruvate dehydrogenase complex of Bacillus stearothermophilus. Biochem. J. 283 665–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard, S.J. and Thornton, J.M. 1993. `NACCESS', Computer Program, Department of Biochemistry and Molecular Biology, University College London. Further information can be found at http://sjh.bi.umist.ac.uk/naccess.html.
- Izard, T., Ævarsson, A., Allen M.D., Westphal, A.H., Perham, R.N., de Kok, A., and Hol, W.G.J. 1999. Principles of quasi-equivalence and Euclidean geometry govern the assembly of cubic and dodecahedral cores of pyruvate dehydrogenase complexes. Proc. Natl. Acad. Sci.,96 1240–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janin J. 1999. Wet and dry interfaces: The role of solvent in protein–protein and protein–DNA recognition. Structure 7 R277–R279. [DOI] [PubMed] [Google Scholar]
- Jones, D.D., Stott, K.M., Howard, M.J., and Perham, R.N. 2000. Restricted motion of the lipoyl-lysine swinging arm in the pyruvate dehydrogenase complex of Escherichia coli. Biochemistry 39 8448–8459. [DOI] [PubMed] [Google Scholar]
- Kalia, Y.N., Brocklehurst, S.M., Hipps, D.S., Appella, E., Sakaguchi, K., and Perham, R.N. 1993. The high-resolution structure of the peripheral subunit-binding domain of dihydrolipoamide acetyltransferase from the pyruvate dehydrogenase multienzyme complex of Bacillus stearothermophilus. J. Mol. Biol. 230 323–341. [DOI] [PubMed] [Google Scholar]
- Kauzmann, W. 1959. Some factors in the interpretation of protein denaturation. Adv. Protein Chem. 14 1–63. [DOI] [PubMed] [Google Scholar]
- Kraulis, P. 1991. MOLSCRIPT, a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallog. 24 946–950. [Google Scholar]
- Ladbury, J.E. 1996. Just add water! The effect of water on the specificity of protein-ligand binding sites and its potential application to drug design. Chem. Biol. 3 973–980. [DOI] [PubMed] [Google Scholar]
- Lawson, J.E., Niu, X.-D., and Reed, L.J. 1991. Functional analysis of the domains of dihydrolipoamide acetyltransferase from Saccharomyces cerevisiae. Biochemistry 30 11249–11254. [DOI] [PubMed] [Google Scholar]
- Lee, B.K. and Richards, F.M. 1971. Solvent accessibility of groups in proteins. J. Mol. Biol. 55 379–400. [DOI] [PubMed] [Google Scholar]
- Lessard, I.A.D. 1995. "Protein–protein Interaction and Molecular Self-assembly of the Pyruvate Dehydrogenase Multienzyme Complex," PhD thesis, Department of Biochemistry, University of Cambridge, Cambridge, UK.
- Lessard, I.A.D., Fuller, C., and Perham, R.N. 1996. Competitive interaction of component enzymes with the peripheral subunit-binding domain of the pyruvate dehydrogenase multienzyme complex of Bacillus stearothermophilus: Kinetic analysis using surface plasmon resonance detection. Biochemistry 35 16863–16870. [DOI] [PubMed] [Google Scholar]
- Lessard, I.A.D., Domingo, G.J., Borges, A., and Perham, R N. 1998. Expression of genes encoding the E2 and E3 components of the Bacillus stearothermophilus pyruvate dehydrogenase complex and the stoichiometry of subunit interaction in assembly in vitro. Eur. J. Biochem. 258 491–501. [DOI] [PubMed] [Google Scholar]
- Lessard, I.A.D. and Perham, R.N. 1994. Expression in Escherichia coli of genes encoding the E1α and E1β subunits of the pyruvate dehydrogenase complex of Bacillus stearothermophilus and assembly of a functional E1 component (α2β2) in vitro. J. Biol. Chem. 269 10378–10383. [PubMed] [Google Scholar]
- Lessard, I.A.D. and Perham, R.N. 1995. Interaction of component enzymes with the peripheral subunit-binding domain of the pyruvate dehydrogenase multienzyme complex of Bacillus stearothermophilus: Stoichiometry and specificity in self-assembly. Biochem. J. 306 727–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mande, S. S., Sarfaty, S., Allen, M.D., Perham, R. N., and Hol, W.G.J. 1996. Protein–protein interactions in the pyruvate dehydrogenase multienzyme complex: Dihydrolipoamide dehydrogenase complexed with the binding domain of dihydrolipoamide acetyltransferase. Structure 4 277–286. [DOI] [PubMed] [Google Scholar]
- Maniatis, T., Fritsch, E. F., and Sambrook, J. 1982. Molecular cloning: A laboratory manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Mattevi, A, Obmolova, G, Kalk, K.H., Westphal, A.H., de Kok, A., and Hol, W.G.J. 1993. Refined crystal structure of the catalytic domain of dihydrolipoyl transacetylase (E2p) from Azotobacter vinelandii at 2.6Å resolution. J. Mol. Biol. 230 1183–1199. [DOI] [PubMed] [Google Scholar]
- Morton, C.J. and Ladbury, J.E. 1996. Water-mediated protein–DNA interactions: The relationship of thermodynamics to structural detail. Protein Sci. 5 2115–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy, K.P. and Gill, S.J. 1991. Solid model compounds and the thermodynamics of protein unfolding. J. Mol. Biol. 222 699–709. [DOI] [PubMed] [Google Scholar]
- Murphy, K.P., Xie, D., Garcia, K.C., Amzel, L.M., and Freire, E. 1993. Structural energetics of peptide recognition: Angiotensin II/antibody binding. Proteins 15 113–120. [DOI] [PubMed] [Google Scholar]
- Ortiz-Salmerón, E., Barón, C., and García-Fuentes, L. 1998. Enthalpy of captopril-angiotensin I-converting enzyme binding. FEBS Lett. 435 219–224. [DOI] [PubMed] [Google Scholar]
- Patel, M.S. and Roche, T.E. 1990. Molecular biology and biochemistry of pyruvate dehydrogenase complexes. FASEB J. 4 3224–3233. [DOI] [PubMed] [Google Scholar]
- Pearce, K.H., Jr, Ultsch, M.H., Kelly, R.F., de Vos, A.M., and Wells, J.A. 1996. Structural and mutational analysis of affinity-inert contact residues at the growth hormone-receptor interface. Biochemistry 35 10300–10307. [DOI] [PubMed] [Google Scholar]
- Perham, R.N. 2000. Swinging arms and swinging domains in multifunctional enzymes: Catalytic machines for multistep reactions. Annu. Rev. Biochem. 69 961–1004. [DOI] [PubMed] [Google Scholar]
- Quiocho, F.A., Wilson, D.K., and Vyas, N.K. 1989. Substrate specificity and affinity of a protein modulated by bound water molecules. Nature 340 404–407. [DOI] [PubMed] [Google Scholar]
- Raman, C.S., Allen, M.J., and Nall, B.T. 1995. Enthalpy of antibody–cytochrome c binding. Biochemistry 34 5831–5838. [DOI] [PubMed] [Google Scholar]
- Reed, L.J. and Hackert, M.L. 1990. Structure–function relationships in dihydrolipoamide acyltransferases. J. Biol. Chem. 265 8971–8974. [PubMed] [Google Scholar]
- Robien, M.A., Clore, G.M., Omichinski, J.G., Perham, R.N., Appella, E., Sakaguchi, K., and Gronenborn, A.M. 1992. Structure of the E3-binding domain of the dihydrolipoamide succinyltransferase core from the 2-oxoglutarate dehydrogenase multienzyme complex of Escherichia coli. Biochemistry 31 3463–3471. [DOI] [PubMed] [Google Scholar]
- Sanderson, S.J., Miller, C., and Lindsay, J.G. 1996. Stoichiometry, organization and catalytic function of protein-X of the pyruvate dehydrogenase complex from bovine heart. Eur. J. Biochem. 236 68–77. [DOI] [PubMed] [Google Scholar]
- Spolar, R.S., Livingstone, J.R., and Record, M.T. 1992. Use of liquid-hydrocarbon and amide transfer data to estimate contributions to thermodynamic functions of protein folding from the removal of nonpolar and polar surface from water. Biochemistry 31 3947–3955. [DOI] [PubMed] [Google Scholar]
- Stepp, L.R. and Reed, L. J. 1985. Active-site modification of mammalian pyruvate dehydrogenase by pyridoxal 5`-phosphate. Biochemistry 24 7187–7191. [DOI] [PubMed] [Google Scholar]
- Stites W.E. 1997. Protein–protein interactions: Interface structure, binding thermodynamics, and mutational anlaysis. Chem. Rev. 97 1233–1250. [DOI] [PubMed] [Google Scholar]
- Studier, F.W., Rosenberg, A.H., Dunn, J.J., and Dubendorff, J.W. 1990. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 185 60–89. [DOI] [PubMed] [Google Scholar]
- Tame, J.R.H., Murshudov, G.N., Dodson, E.J., Neil, T.K., Dodson, G.G., Higgins, C.F., and Wilkinson, A.J. 1994. The structural basis of sequence-independent peptide binding by OppA. Science 264 1578–1581. [DOI] [PubMed] [Google Scholar]
- Velazquez-Campoy, A., Todd, M. J., and Freire, E. 2000. HIV-1 protease inhibitors: Enthalpic versus entropic optimization of the binding affinity. Biochemistry 39 2201–2207. [DOI] [PubMed] [Google Scholar]
- Wiseman, T, Williston, S., Brandts, J.F., and Nin, L-N. 1989. Rapid measurement of binding constants and heat of binding using a new titration calorimeter. Anal. Biochem. 179 131–137. [DOI] [PubMed] [Google Scholar]