

Abstract
The F1F0 ATP synthase is a reversible molecular motor that employs a rotary catalytic cycle to couple a chemiosmotic membrane potential to the formation/hydrolysis of ATP. The multisubunit enzyme contains two copies of the b subunit that form a homodimer as part of a narrow, peripheral stalk structure that connects the membrane (F0) and soluble (F1) sectors. The three-dimensional structure of the b subunit is unknown making the nature of any interactions or conformational changes within the F1F0 complex difficult to interpret. We have used circular dichroism and analytical ultracentrifugation analyses of a series of N- and C-terminal truncated b proteins to investigate its stability and structure. Thermal denaturation of the b constructs exhibited distinct two-state, cooperative unfolding with Tm values between 30 and 40°C. CD spectra for the region comprising residues 53–122 (b53–122) showed θ222/θ208 = 0.99, which reduced to 0.92 in the presence of the hydrophobic solvent trifluoroethanol. Thermodynamic parameters for b53–122 (ΔG, ΔH and ΔCp) were similar to those reported for several nonideal, coiled-coil proteins. Together these results are most consistent with a noncanonical and unstable parallel coiled-coil at the interface of the b dimer.
Keywords: Protein domains, coiled-coil, thermal denaturation, ultracentrifugation, circular dichroism
The F1F0 ATP synthase catalyzes the formation of ATP from ADP and inorganic phosphate using a transmembrane proton gradient as an energy source. This enzyme, found in similar form in the membranes of mitochondria, bacteria, and chloroplasts, can be divided into the peripheral F1 sector that houses the sites of ATP synthesis/hydrolysis, and the integral F0 sector that conducts protons across the membrane. In the prototypical complex from Escherichia coli, the F1 sector contains five types of subunits in a stoichiometry α3β3γδɛ, and the F0 sector contains three types of subunits in a stoichiometry of ab2c10. It is believed that the catalytic activities of F1 and F0 are coupled through the rotary motion of a c10γɛ subcomplex (the "rotor") relative to the remainder of the enzyme. In this mechanism, the movement of a ring of c subunits past ab2 in the F0 sector is linked to the passage of protons across the membrane, while rotation of the centrally located γɛ drives conformational changes in the catalytic sites of the α3β3 hexamer resulting in substrate binding, catalysis, and product release. The remaining subunits, b and δ, form a second, peripheral stalk that maintains an apparently static link between the a subunit and the catalytic hexamer. For recent reviews of the structure and mechanism of ATP synthase, see Leslie and Walker (2000), Noji and Yoshida (2001), and Dunn et al. (2000).
The b subunit of E. coli has a hydrophobic N-terminal 24 amino-acid sequence that spans the cytoplasmic membrane followed by a polar 132-residue C-terminal region that extends into the cytoplasm where it binds F1. The transmembrane region interacts with both the a and c subunits in the proton-translocating F0 sector (Kumamoto and Simoni 1986; Jones et al. 2000) while the polar domain contacts the α, β and δ subunits of the F1 domain (Ogilvie et al. 1998; McLachlin et al. 2000). Besides being essential for assembling the enzyme and holding F1 and F0 together, the b subunit has been suggested to store elastic energy transiently during the catalytic cycle by bending or stretching in response to the torque generated by rotation (Cherepanov et al. 1999).
A nuclear magnetic resonance (NMR) structure of the first 34 residues of the b subunit has shown a helical transmembrane segment followed by a bend at residues 23–26 in the membrane-proximal region (Dmitriev et al. 1999). The remainder of the subunit has not been defined at high resolution, but electron microscopic images show the peripheral stalk as a narrow structure rising from the membrane and running up the side of F1 (Wilkens et al. 2000). The electron microscopic and cross-linking studies (Ogilvie et al. 1998; McLachlin et al. 1998, 2000) of b have indicated that the polar domain covers most of the ∼130 Å from the membrane to the top of F1 where δ has been localized. Sequence analysis of bacterial b subunits reveals no strong homology to proteins of known high-resolution structure, but predictive algorithms support a predominantly helical protein (Fig. 1A ▶).
Fig. 1.

Proposed secondary structures and domains of the b subunit and truncations. (A) The predicted secondary structure for the b subunit is indicated as α-helix, bend, or turn and the solution structure of the b2–34 construct (Dmitriev et al. 1999). (B) The domain structure proposed from deletion analysis (Revington et al. 1999) includes the transmembrane, tether, dimerization, and δ-binding domains as shown. (C) The constructs of the b subunit used in this study are illustrated, showing their sequence relationship to the domains outlined in panel B.
Experimentally, the expressed cytoplasmic region of b exists in solution as a highly helical, extended dimer capable of binding F1 (Dunn 1992). More recently, cross-linking studies and hydrodynamic analyses of truncation mutants (Revington et al. 1999; McLachlin and Dunn 2000) have allowed division of the cytoplasmic region into three domains as shown in Figure 1B ▶. Residues 25–52 form the "tether domain", predicted to include an amphipathic helix and known to contain residues that interact with cytoplasmic loops of the a subunit (McLachlin et al. 2000). The 53–122 segment of b, when expressed separately (b53–122), has been shown to form a dimer with an affinity similar to that of the entire soluble domain, b24–156. This section of the protein contains a heptad repeat motif (McCormick et al. 1993) and behaves hydrodynamically like a pair of straight parallel helices (Revington et al. 1999) suggesting that the dimer may exist as a left-handed coiled-coil. Residues in this region can be cross-linked to the α and β subunits of F1 (McLachlin et al. 2000).The sequence from 123 to the C terminus at residue 156 contains the key residues for interaction with the δ subunit (McLachlin et al. 1998) and has been suggested to fold into a structure more globular than the rest of the protein (Revington et al. 1999; Dunn et al. 2000).
In the absence of high-resolution structural data for the b subunit, we have turned to alternative methods to provide evidence for the coiled-coil and globular substructures in this protein. We have used circular dichroism spectroscopy and analytical ultracentrifugation to determine the secondary structure and the thermodynamic properties of the soluble region of the b subunit and several truncated constructs. The extended structure of the polar region of the b subunit with autonomous domains provided a unique opportunity to use a subtractive approach to understand the contributions of each region to the overall structure of the protein and several truncated variations. Because the role of the b subunit in the rotary catalytic mechanism is believed to be primarily structural, this analysis also provided a first detailed look at the strength and nature of the interactions that stabilize its conformation.
Results
Relationship of b constructs with predicted secondary structure and domains of the b subunit
As shown in Figure 1A ▶, the secondary structure of the cytoplasmic region of the b subunit of E. coli ATP synthase is predicted to consist of a bend sequence (residues 23–26) and two long α-helical regions (residues 34–78, 85–156), separated only by a turn sequence (79–84). The bend at positions 23–26 has been observed in the NMR structure of the N-terminal 34-residue fragment (Dmitriev et al. 1999), and the helicity of the expressed cytoplasmic region has been determined from CD spectra (Dunn 1992). The domain model described above (i.e., the transmembrane domain and the three sections of the cytoplasmic region) is presented in Figure 1B ▶. Truncations of the soluble region of the b subunit were selected in this study (Fig. 1C ▶) to explore differences in the secondary structure and thermodynamic stability of the various segments in the absence of a three-dimensional structure. Comparison of the thermodynamic properties, ΔG, ΔH and ΔCp with those for proteins of known three-dimensional structure should provide insight into the tertiary structure of the b subunit. The parental protein, b24–156 (McLachlin and Dunn 1997), represented the entire soluble region of the b subunit lacking only the leading 23 residues proposed to span the membrane. The structure of the tether domain was studied by deletion of the first, hydrophobic third of the domain resulting in b34–156. The construct produced by deletion of the rest of the tether domain, b53–156, was found to aggregate and therefore could not be analyzed usefully. The four-residue, C-terminal truncation producing b24–152 resulted in a molecule with properties dramatically different from those of b24–156 (McLachlin et al. 1998) and therefore was selected for this study as well as b24–134, a construct lacking two thirds of the δ-binding domain. Lastly, the b53–122 protein, the isolated dimerization sequence (Revington et al. 1999), was examined.
Thermal stability of the dimer of b subunit
Sedimentation equilibrium analytical ultracentrifugation allows determination of the molecular weights of proteins in solution under native conditions. Previously, this technique has been used to define the regions of the b subunit necessary for dimer formation (Revington et al. 1999). The thermal stability of the dimer was examined by measuring the molecular weight of the species over a temperature range from 5–40°C (Fig. 2 ▶). A single-species model was used to estimate the single molecular weight that best fit the concentration gradient observed in the ultracentrifuge cell after equilibration at each temperature. The molecular weight value calculated in this manner reflected the average molecular weight of the solution species (Mobs). The observed molecular weights of all truncations of the b subunit studied were close to values expected for dimers (Mobs/ M1 = 2) over the 5–25°C range and then showed a sharp decline in the 30–40°C range reflecting an increase in the monomeric species. Full conversion to the monomeric state could not be observed by ultracentrifugation because of an upper operating limit (40°C) of the instrument. The dissociation constants (Kd) of this transition were determined by the fitting of multiple data sets to a monomer-dimer equilibrium at each temperature. The resulting Kd values at 5°C ranged from 1–4 μM for all of the constructs studied (Table 1) indicating that at low temperatures and at concentrations >∼10–40 μM the dimer is the predominant species. At higher temperatures, calculation of the Kd was complicated by thermal unfolding of the proteins (vide infra).
Fig. 2.

Sedimentation equilibrium analysis of the conversion of b24–156 (▪) and the dimerization domain construct b53–122 (□) from dimer to monomer over the temperature range of 5–40°C. The ratio of the average molecular weight observed by equilibrium sedimentation (Mobs) divided by the molecular weight of the monomer (M1) calculated from the amino-acid sequence is plotted as a function of temperature. Error bars indicate the 95% confidence interval from the simultaneous fit of three data sets. Values for Kd were calculated at each data point as described in Materials and Methods. The reported Tm was the temperature at which the Kd was equal to the protein concentration (Pt) used in that experiment. Linear interpolation of the two nearest Kd values was used to estimate the reported Tm values (Table 1).
Table 1.
Thermodynamic parameters derived from sedimentation equilibrium ultracentifugation and circular dichroic analysis of the thermal denaturation of the b subunit truncations
| Sedimentation analysis | Circular dichroism analysis | |||||
| Construct | Tm(UC) (°C) | Kd(5°) (μM) | Tm(CD) (°C) | ΔGu(20°C) (kcal mol−1) | ΔHu(Tm) (kcal mol−1) | ΔCρ(Tm) (kcal mol−1 K−1) |
| b24–156 | 37 ± 3 | 1.0 ± 0.6 | 39.4 ± 1.5 | 1.7 | 51.2 ± 8.7 | 2.4 ± .3 |
| b34–156 | 37 ± 3 | 1.6 ± 0.7 | 40.7 ± 1.3 | 1.8 | 49.6 ± 6.4 | 2.1 ± .2 |
| b24–152 | —a | 3.5 ± 1.1 | 40.5 ± 1.6 | 1.8 | 49.6 ± 8.3 | 2.2 ± .2 |
| b24–134 | 36 ± 3 | 1.6 ± 0.5 | 40.5 ± 1.5 | 1.8 | 49.3 ± 7.4 | 2.1 ± .2 |
| b53–122 | 32 ± 3 | 1.6 ± 1.2 | 30.2 ± 1.7 | 1.0 | 30.9 ± 3.4 | 0.4 ± .1 |
a Not available because of degradation of sample during extended high-temperature sedimentation equilibrium runs.
Characterization of the b subunits by circular dichroism
CD spectra of the b protein and its truncation mutants (Fig. 3 ▶) revealed large negative ellipticities at θ208 and θ222, indicative of significant α-helical content in the proteins. The spectrum of b24–156 exhibited the most intense profile and was very similar in magnitude to previous spectra of the soluble region of b (Dunn 1992; Greie et al. 2000). The CD spectra of the truncated b proteins exhibited intensity differences from this parent protein at both 208 and 222 nm. Because these mutant proteins comprised truncations at both the N and C termini that retained a dimeric structure, it was expected that the CD spectra might provide evidence for regions of greater helical content or the nature of helical interactions at the dimer interface.
Fig. 3.
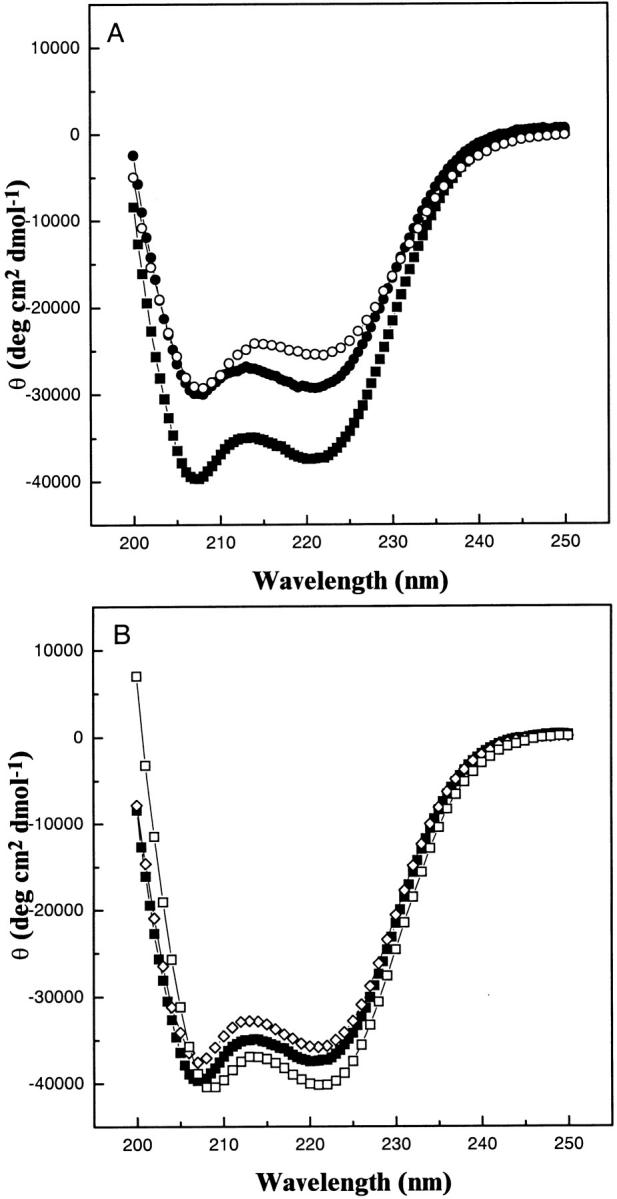
CD spectra of the b subunit proteins in 25 mM Na2HPO4, pH 7.5 at 20°C. (A) Comparison of the C-terminal truncations, b24–152 (○) and b24–134 (•) with the full-length cytoplasmic region, b24–156 (▪). (B) The N-terminal truncation b34–156 (⋄), and the dimerization domain, b53–122 (□), plotted with the parental b24–156 (▪).
Comparison of the CD spectra for b24–152, b24–134, and b24–156 in Figure 3A ▶ showed the structural effects of deleting portions of the δ-binding region of the proteins. Removal of the four C-terminal residues of b24–156 to produce b24–152 resulted in a 32% decrease in mean residue ellipticity at 222 nm (θ222), suggesting a large decrease of the fraction of residues in helical conformation. Removal of a further 18 residues from b24–152 to give b24–134 resulted in a more negative θ222 value and a θ208 value almost identical to that of the b24–152 construct, although both minima were still substantially less intense than those of the b24–156 construct. These spectral changes suggest that removal of the C-terminal four residues caused a loss of helical structure in a considerable portion of the C-terminal region and that further truncation back to residue 134 removed some of these nonhelical residues. The ratio of the magnitudes of the 222 and 208 nm minima (θ222/θ208) for b24–156 , and b24–152 were near 0.88, which is intermediate for that commonly observed for regular α-helix (≈ 0.83) and coiled-coil structures (≥1.0) (Lau et al. 1984), while the value for b24–134, was 0.97. A comparison of the θ222/θ208 values for b24–156, b24–152 and b24–134 indicate that the helical structure present in the 24–134 region is largely in a coiled-coil conformation while the remaining C-terminal region (residues 134–156) is a mix of α-helical and nonhelical structure.
Figure 3B ▶ shows that the CD spectrum of the N-terminal truncation protein, b34–156, was almost identical to that of the full-length soluble region of the b subunit (b24–156), suggesting that the shortened protein retained a near identical fraction of helical structure as the parent. The dimerization domain construct, b53–122, had a θ222 value 10% greater and a θ222/θ208 ratio 14% higher (0.99) than the full soluble domain, signifying that the removed regions were of less helical character. The large increases in θ208 and θ222 of b53–122 over b24–134 (Fig. 3A ▶) suggest that most of the helical content in the latter construct was in the dimerization sequence and that the tether region, residues 24–53, adopts a less helical structure. The observation that b53–122 had a θ222/θ208 ∼1.0 and had a red-shifted minimum compared to the full-length protein is consistent with this region forming a coiled-coil structure (Lau et al. 1984).
Interactions within the b dimerization domain
The dimerization region, b53–122, was further characterized by CD spectroscopy to probe the nature of hydrophobic and ionic interactions within this region of the b protein. Figure 4A ▶ shows the CD spectrum of the b53–122 protein in aqueous buffer and in the presence of 50% trifluoroethanol (TFE). This latter solvent has been used as a successful probe for helix interactions observed in coiled-coil proteins. In aqueous buffer the CD spectrum of b53–122 was similar to that obtained in Figure 3 ▶ having θ222/θ208 = 0.99 and indicative of a coiled-coil structure. In the presence of 50% TFE the CD spectrum of b53–122 had an increased magnitude for θ208 and a negligible change at θ222 resulting in a decrease in θ222/θ208 to 0.92. This observation indicated that TFE interrupted the interactions between helical segments in b53–122. Similar observations have been noted for the disruption of the synthetic coiled-coil proteins c-Myc and Max (Lavigne et al. 1995) and a series of hybrid coiled-coils from cortexillin and GCN4 (Lee et al 2001).
Fig. 4.
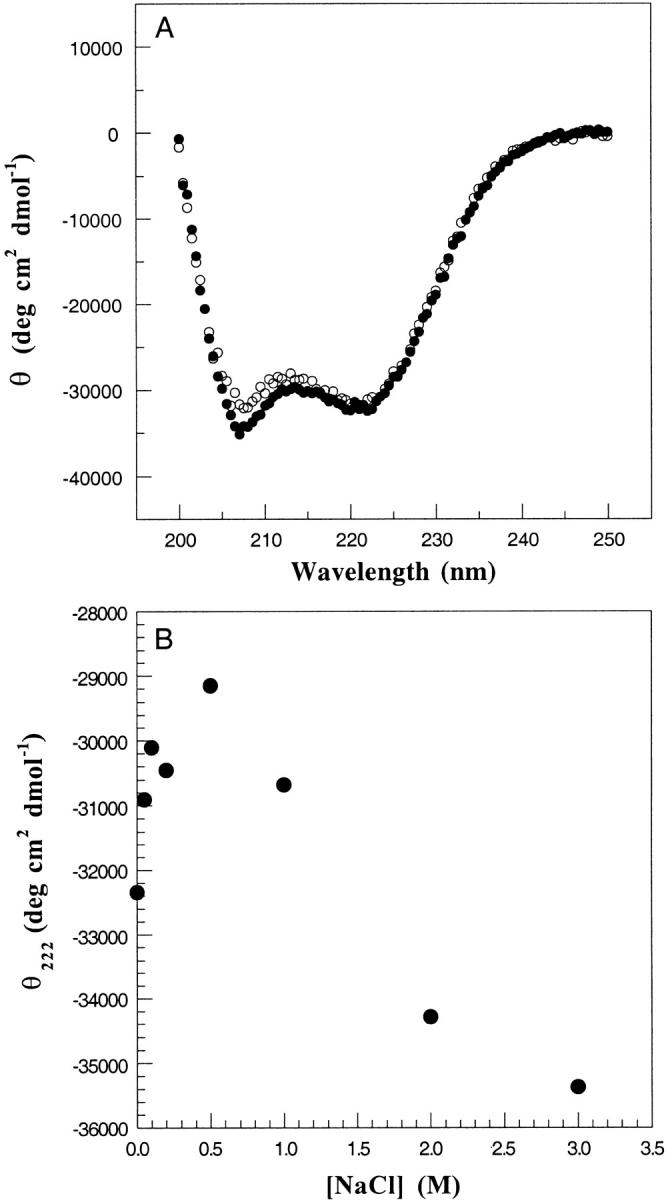
Effect of trifluoroethanol and NaCl on the structure of the b53–122 protein. (A) CD spectra of b53–122 protein recorded in 25 mM Na2HPO4, pH 7.5 (•) and with the addition of 50% trifluoroethanol (○) at 5°C. (B) Dependance of θ222 on increasing NaCl concentration for spectra of b53–122 protein in 25 mM Na2HPO4, pH 7.5 at 5°C and at the NaCl concentrations indicated.
Helical proteins are stabilized by i+3 and i+4 ionic interactions between arginine/lysine and aspartate/glutamate side-chain pairs. In the case of coiled-coil proteins, further interactions may occur between the helices. To determine whether ionic interactions were important for the stability of b53–122, CD spectra were recorded as a function of increasing ionic strength. Figure 4B ▶ shows a graph of θ222 at NaCl concentrations ranging from 0–3 M. The graph shows there was a steady decrease in the magnitude of θ222 as the salt concentration increased to 500 mM. This observation is consistent with the disruption of ionic interactions that stabilize helical structure. At NaCl concentrations >500 mM this trend was reversed and an increase in magnitude for θ222 was observed. This result is typical of a greater hydrophobic interaction at higher ionic strength. This pattern was nearly identical to that observed for the coiled-coil protein GCN4 (Kenar et al. 1995).
Mechanism of unfolding for the dimeric b subunit
Analytical ultracentrifugation experiments in this work and previous studies (McLachlin et al. 1998; Revington et al. 1999) show that the truncated b proteins that include the 53–122 sequence exist in temperature-sensitive, reversible dimer-monomer equilibria. However, these experiments did not give an indication of the structural changes that occurred in this transition or whether the monomer retained a folded state. It was further recognized that the broad range of truncated constructs here could provide important information regarding region-specific contributions towards overall stability of the proteins and details about the cooperativity of folding of the b subunit. These details were probed by a series of dilution and thermal unfolding experiments.
For dimeric proteins the generally accepted folding/unfolding pathway can be described as
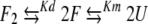 |
(1) |
where F2 is the folded dimer, F is a folded monomer, U is unfolded monomer, and Kd and Km are the equilibrium constants for dimer dissociation and monomer unfolding, respectively. The first portion of this pathway is bimolecular. Under conditions such as low temperature, where the folded monomer (F) is sufficiently stable, Kd can be measured by examining concentration-dependent spectral changes reflective of the dimer to monomer transition. At higher temperatures, the unfolded species is favored such that a concentration-dependent spectral change is a product of Kd and the second unimolecular equilibrium (Km). To probe the dimer to monomer equilibrium (Kd) for b53–122 CD spectra were collected for a range of concentrations at 5°C (Fig. 5 ▶). This temperature was chosen because sedimentation equilibrium indicated a Mobs/M1 ≈ 2 (dimer) and maximum θ208 and θ222 values (see Fig. 6 ▶). The plot shown in Figure 5 ▶ shows the change in θ222 of b53–122 as the concentration was decreased from about 50 to 0.5 μM at 5°C. The data shows that θ222 leveled off near −43,000 deg cm2 dmol−1 at the highest concentrations studied and decreased to about −20,000 deg cm2 dmol−1 although this lower plateau could not be clearly determined because of instrument sensitivity. The magnitude of these spectral changes and their concentration-dependent nature are in excellent agreement with other studies showing the concentration-dependent dissociation of oligomeric α-helical proteins (Ho and DeGrado 1987; Donaldson et al. 1995; Lee et al. 2001). The decrease in magnitude of θ222 with decreasing concentration likely arose from a combination of a decrease in helical structure and removal of helix-helix interactions at the subunit interface as the dimer (F2) dissociated to form monomer (F). The dilutions of b53–122 covered a range where the protein was >90% dimer to concentrations where the population had shifted to 30% dimer, based on dissociation constants derived from sedimentation equilibrium analysis. Analysis of the data in Figure 5 ▶ was done using equation 2 for a dimer to monomer equilibrium having a dissociation constant Kd.
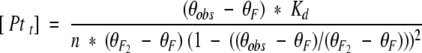 |
(2) |
Fig. 5.

Concentration dependence of the ellipticity for the b53–122 protein. The plot shows the mean residue ellipticity at 222 nm (θ222) vs the protein concentration. The concentrations ranged from 0.5–50 μM in 25 mM Na2HPO4, pH 7.5 and 5°C. The curve shown is the best fit line according to equation 2 for a folded dimer (F2) to folded monomer (F) equilibrium yielding a Kd = 0.6 ± 0.2 μM.
Fig. 6.
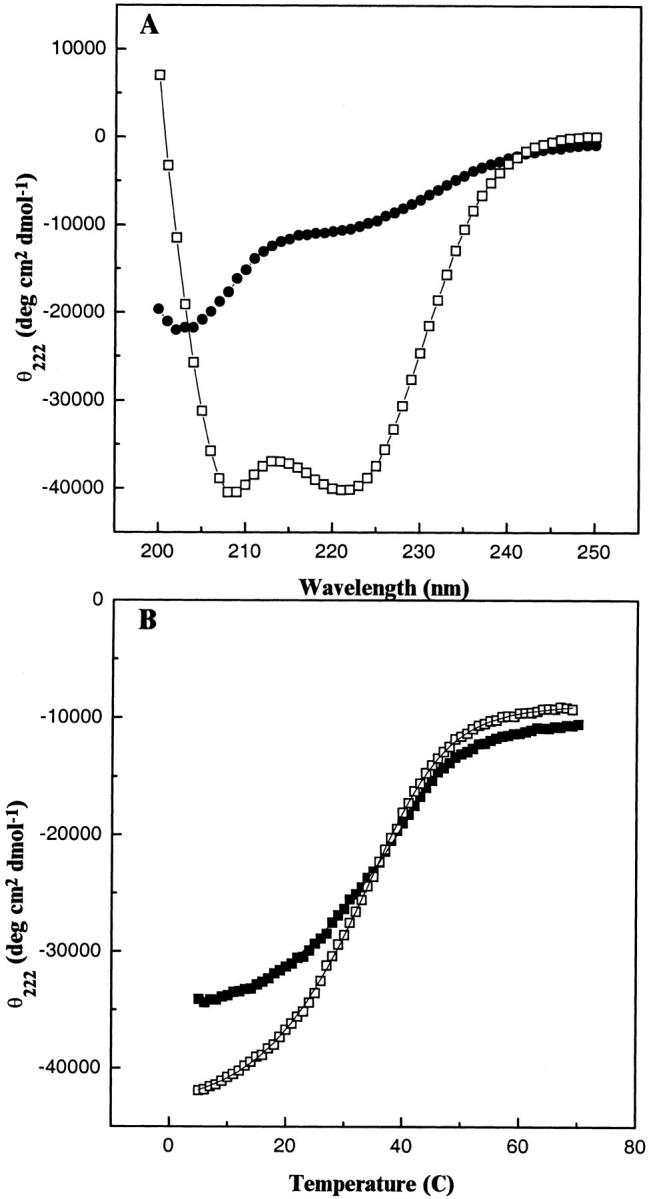
CD analysis of the temperature-dependent denaturation of b53–122. (A) Spectra of the folded dimeric and unfolded monomeric forms of b53–122. The spectra show 100 μM b53–122 at 5°C (□) and after thermal unfolding at 70°C (•). (B) Thermal unfolding profiles of b subunit proteins monitored by CD spectroscopy. The mean residue ellipticity at 222 nm (θ222) for b53–122 (□) and b24–156 (▪) were monitored as a function of temperature. The θ222 values were acquired at 1°C intervals using a 1-min equilibrium time at each temperature. The curves drawn through the data indicate the best fit lines for the thermal unfolding based on equation 3.
In this equation, the mean residue ellipticities of the folded dimer and folded monomer are expressed as θF2 and θF, respectively, and θobs was the experimental ellipticity at a given total protein concentration, Pt. An acceptable fit was found for θF2 = −43,700 deg cm2 dmol−1 and θF = −15,000 ± 5000 deg cm2 dmol−1 yielding a Kd of 0.6 ± 0.2 μM, in good agreement with that observed in analytical ultracentrifugation studies (1.6 μM, Table 1). The magnitude of the ellipticity for the monomer (−15,000 deg cm2 dmol−1) indicated that it had a significant amount of residual α-helical secondary structure.
Thermal unfolding
To assess the unfolding process, a series of CD spectra of the truncated b proteins were collected as a function of increasing temperature. Figure 6A ▶ shows a comparison of CD spectra for 100 μM b53–122 acquired at 5 and 70°C. The spectrum at 5°C represented the folded dimeric protein (F2) comprised largely of α-helical structure. Based on the results of Figure 5 ▶, the population of folded monomeric (F) protein should be negligible at this temperature and high concentration. At 70°C, the magnitude of the CD signals at 222 and 208 nm were significantly reduced, consistent with the loss of most secondary structure. The magnitude of the residual ellipticity at 222 nm and 70°C was similar to that observed for other thermal denaturation studies where a substantial negative ellipticity often is observed (Hackel et al. 2000). The spectrum at 70°C also contained a minimum near 200 nm and a positive slope between 200 and 250 nm indicative of the unfolded form of b53–122 (U) and was similar to spectra obtained for the unfolded forms of other helical proteins at high temperature (O'Shea et al. 1989 ; De Francesco et al. 1991). Spectra of b53–122 collected at 5, 20, 40, and 70°C (data not shown) also revealed an isodichroic point at 203 nm, consistent with a two-state helix-to-random coil transition (Greenfield et al. 1967). These observations indicated that at protein concentrations significantly higher than Kd and temperatures higher than 5°C, the folded monomer species (F) is not significantly populated. Therefore, unfolding of b53–122 could be modeled as a single cooperative transition from the folded, dimeric state (F2) to the unfolded monomer (U). This type of transition, which has been observed for a variety of other dimeric, helical proteins including coiled-coils can be analyzed to determine the thermodynamics of the unfolding process (De Francesco et al. 1991; Lee et al. 2001).
The thermal unfolding of the complete series of truncated b proteins was examined by measuring the change in ellipticity as a function of temperature (Fig. 6B ▶). Each protein showed a smooth sigmoidal curve suggestive of a cooperative unfolding transition from folded dimer (F2) at low temperatures to U at higher temperatures. The unfolding data for each b protein was fit using the approach of Lavigne et al. (1998) according to equation 3.
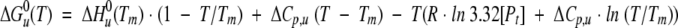 |
(3) |
The results of the curve fitting yielded thermodynamic parameters for the enthalpy of unfolding at the transition midpoint [ΔHu(Tm)], the heat capacity (ΔCp) and the unfolding energy (ΔGu). Examination of the unfolding midpoints (Tm) showed a surprisingly narrow range of temperatures (Table 1). As all experiments were done at similar concentrations (16–27 μM), these measured values for Tm were used as an indicator of overall protein stability. The results show that all the b proteins except b53–122 have a midpoint near 40°C. The latter had a depressed Tm indicating it was the least stable of the proteins. However, the range observed here was much smaller than that typically observed for other proteins. Jelesarov and Bosshard (1996) observed that a series of alanine substitutions for leucines at the interface of model coiled-coil peptides varied the Tm over a range of 27°C while changing the ΔGu of the denaturation process by only 2.7 kcal mol−1. Therefore, relatively minor differences in stability of the b proteins studied here could result in significantly different Tm. The midpoints of the melting curves were comparable to the midpoints of melting determined from the sedimentation analysis (Table 1), supporting the idea that the temperature-dependent changes observed in the CD reflected a dimer to monomer transition.
The enthalpies of unfolding [ΔHu(Tm)] and heat capacities (ΔCp) also are shown in Table 1. Comparison of ΔHu for all protein constructs revealed there was little change between the parent protein (51.2 kcal mol−1), mutants (b24–152, b24–134) where residues were deleted at the C terminus (49.6; 49.3 kcal mol−1) and the mutant (b34–156) where residues were removed from the N terminus (49.6 kcal mol−1) of the protein. However, a decrease of about 20 kcal mol−1 was noted when residues from both N and C termini were deleted (b53–122). The magnitude of heat capacity, ΔCp, is proportional to the change in exposed hydrophobic surface area between the folded and unfolded states (Makhatadze and Privalov 1995). Therefore, a larger value of ΔCp usually is attributed to the exposure of more nonpolar side chains during the unfolding process. Although the errors on the values for ΔCp average 15%, some important trends were observed with respect to the primary sequence of the b proteins. Comparison of the proteins b24–156 and b24–152 indicated a 10% decrease in ΔCp, while a further truncation back to position V134 made little if any difference. These changes are consistent with the effects of these truncations previously observed in hydrodynamic analyses and will be dealt with in detail in the Discussion. Removal of residues at the N terminus of the protein (b34–156) resulted in a similar decrease in ΔCp, likely a result of the removal of a stretch of hydrophobic residues (VWPPLMAAI) just C-terminal to the membrane spanning region. While these changes were relatively small a much larger change was noted when further truncations were done to both the N and C termini. The heat capacity for b53–122, (0.39 kcal mol−1 K−1) was nearly fivefold smaller than those of the longer b proteins, which averaged 2.19 kcal mol−1 K−1. Most of the decrease in ΔCp in this construct was likely the result of removal of the hydrophobic sequence between residues 124 and 132, which is the only significant hydrophobic stretch in the soluble domain and which must have sizable buried nonpolar surface area in the folded form of the protein.
Discussion
In the rotary mechanism of ATP synthase, the function of the second stalk is to maintain an essentially static relationship between the a subunit of F0 and the α3β3 hexamer of F1. As a result of this stator function, rotation of the asymmetric antiparallel coiled-coil section of γ within α3β3 drives conformational changes in those subunits, resulting in catalysis. In addition, the second stalk may serve to store energy through elastic deformation. Understanding how the b subunit dimer, which makes up most of the second stalk, fulfills its function will require detailed knowledge of its structure and of any conformational changes that it undergoes. In the absence of a high-resolution structure of the b subunit, we have used a spectroscopic and thermodynamic approach toward understanding the structure and stability of the b subunit dimer and its constituent domains.
Domain model of the b subunit
The CD spectrum of the cytoplasmic region of the b subunit, b24–156, shows a predominantly helical character in agreement with previously published spectra of b constructs containing essentially the entire cytoplasmic region (Dunn 1992; Rodgers et al. 1997; Greie et al. 2000). The thermodynamic values of ΔCp (2.4 kcal mol−1 K−1) measured for b24–156 are consistent with literature values observed for elongated proteins such as tropomyosin fragments in the same size range (1.5–2.6 kcal mol−1 K−1) (Privalov 1982) that have limited hydrophobic interfaces rather than those of globular proteins such as myoglobin (3.4 kcal mol−1 K−1). These observations support the highly extended nature of the b subunit as judged from electron microscopy images (Wilkens et al. 2000) and hydrodynamic data (Revington et al. 1999).
The truncated versions of the b subunit produced CD spectra indicative of highly helical, folded proteins supporting their use for studies of the putative domains (Fig. 1 ▶), as in the absence of a globular fold those domains should be effectively independent. In particular, the shortest construct, b53–122, retained a highly helical structure even though a total of 63 residues had been removed from the N and C termini of the soluble region. This indicates the deleted regions (24–52, 123–156) are not essential for the folding of the 53–122 domain in the b subunit. Examination of the spectra of the C-terminal truncations (b24–152, b24–134) revealed significant differences compared to b24–156. At first glance, the 32% decrease in ellipticity in b24–152 compared to b24–156 appears too large for a simple deletion of the four C-terminal residues. However, this change is consistent with ultracentrifugation data showing a significant change in shape and hydrodynamic properties when the four C-terminal residues were removed (McLachlin et al. 1998). The centrifugation data support a model where the extreme C terminus of the δ-binding region, which had been suggested to form an amphipathic helix, folds back and interacts with another section between residues 122 and 152. Comparison of the CD spectra suggests that this folded C-terminal conformation in b24–156 stabilizes α-helical structure in the remainder of the δ-binding region resulting in the significantly higher θ222 observed for b24–156 relative to b24–152. The disruption of this C-terminal structure, whether by truncation (McLachlin et al. 1998), mutation (Dunn et al. 2000), or temperature (M. Revington and S. Dunn, unpubl.) eliminates the b2–δ interaction that is critical to formation of the second stalk. The similarity of the values for Tm, ΔGu and ΔHu, indicate the C-terminal region contributes little towards the overall stability of the b subunit.
The CD spectra of the N-terminal truncations imply that the tether domain (residues 24–53) has little helical character. While the current studies do not allow a complete thermodynamic characterization for this entire region, comparison of the proteins b24–156 and b34–156 shows only a moderate decrease in ΔCp (0.32 kcal mol−1 K−1) is noted upon deletion of the first 10 residues. Although this deleted sequence contains several nonpolar side chains (VWPPL MAAI), the small difference in ΔCp indicates these must be significantly exposed to solvent in the folded form and likely contribute little to the stability of the b subunit. This is further reflected in the small difference in stability between b24–156 and b34–156 (ΔΔGu 0.12 kcal mol−1) and their similar hydrodynamic properties (Revington et al. 1999). The limited structure in the tether domain also is supported by mutational studies where insertion or deletion of several residues in this domain, or near its boundary with the dimerization domain, does not disrupt ATP synthase function in vivo (Sorgen et al. 1998, 1999).
Evidence for a coiled-coil dimerization domain
The ΔCp data for the cytoplasmic region of the b subunit support an elongated structure for this protein. This conclusion is supported by previous hydrodynamic and NMR experiments (Revington et al. 1999) that both showed the b53–122 protein adopts a highly extended shape. One tertiary structure consistent with this observation would place a parallel coiled-coil in the dimerization domain (residues 53–122) of the b subunit. This structure has been suggested previously based on a weak heptad repeat found in the region (Dunn 1992; McCormick et al. 1993) and a requirement to span the membrane-F1 sector apex (∼130Å). The b53–122 sequence from E. coli (residues 63–73 and 107–121) and the corresponding regions from other bacteria, (Fig. 7 ▶) display two regions predicted to have a left-handed coiled-coil structure (Lupas et al. 1991). In general, several of the a and d positions of the heptad repeats in b53–122 contain alanine or larger hydrophobic residues (Fig. 7 ▶) typical of coiled-coil sequences.
Fig. 7.

Alignment of the dimerization region of the b subunits of Escherichia coli and other bacterial species using the Clustal W alignment program (Thompson et al. 1994). The arrangement of the heptad repeat identified in the E. coli b subunit according to the COILS program (Lupas 1996) is shown. The last line shows a consensus sequence for this alignment where an upper-case letter indicates that amino acid is present in at least 70% of the 10 sequences (X = I, L, V).
Circular dichroism experiments are most consistent with the dimerization domain forming a coiled-coil. In the b53–122 protein, θ222/θ208 reached a value of 0.99, approaching that observed for a variety of ideal coiled-coil peptide systems (Lavigne et al. 1995), where θ222/θ208 typically ranges from 1.0–1.05. This shows that b53–122 may contain a small amount of either unstructured polypeptide or α-helical structure which yields a lower θ222/θ208. Further, concentration dependence studies show a decrease in ellipticity that could be fitted to a dimer to monomer transition similar to that observed for designed coiled-coil systems (Ho and DeGrado, 1987; Lee et al. 2001). The b53–122 protein also shows sensitivity to hydrophobic solvents, a characteristic of many coiled-coils. In the presence of trifluoroethanol θ222/θ208 decreased from 0.99 to 0.92, a trend observed for the c-Myc and Max coiled-coils (Lavigne et al. 1998) and hybrid cortexillin/GCN4 coiled coils (Lee et al. 2001).
While CD spectra support a coiled-coil structure for b53–122 some question arises regarding the stability of this protein. The unfolding temperature for b53–122 (∼30°C) falls well below that expected for stable, near-ideal coiled-coils such as those derived from tropomyosin, GCN4, or fos/jun, which typically display a Tm near 60°C (O'Shea et al. 1989). Further, physical characteristics typically used to measure the stability such as the Kd and ΔGu of b53–122, are near 1 μM at 5°C and 1 kcal mol−1 respectively, significantly weaker than reported for these other coiled-coils (De Francesco et al. 1991). However, one excellent measure for protein structure is the difference in heat capacity between the folded and unfolded structure which ranges from 0.4–1.3 kcal mol−1 K−1 for ideal coiled-coils to 1.3–3.0 kcal mol−1 K−1 for more globular proteins ( Privalov 1979, 1982; Lee et al. 2001). The ΔCp value for b53–122 (0.39 kcal mol−1 K−1), falls on the low end of the range commonly observed for coiled-coils and more importantly, is very distinct from that observed for globular proteins.
Several examples of less-stable coiled-coil proteins have been characterized recently both thermodynamically and structurally based on the coiled-coil homo- and heterodimers formed by c-Myc and Max (Lavigne et al. 1998) and a series of hybrid coiled-coils from cortexillin and GCN4 (Lee et al 2001). In nearly all of these cases, there is a striking similarity between ΔCp, ΔHu, ΔGu, and Tm for these proteins and b53–122. For example, the Tm for b53–122 (32°C) is very similar to that determined for the c-Myc-Max heterodimer (38°C), the Max2 homodimer (41°C) and several of the cortexillin and GCN4 hybrid coiled coils which exhibit Tm values of 30–40°C. Further the heat capacity for unfolding for b53–122 (ΔCp 0.39 kcal mol−1 K−1) is close to the range observed for these coiled-coil proteins (ΔCp 0.4–0.5 kcal mol−1 K−1). Like b53–122 several of the hybrid GCN4 and cortexillin coiled-coils fall below the traditional stabilities of some synthetic, ideal coiled-coils such as those derived from GCN4 (O'Shea et al. 1989) or the transcription factor LFB1 (De Francesco et al. 1991). It has been suggested that suboptimal residues in the a and d heptad positions of these proteins leads to decreases in their overall stabilities. For example, the c-Myc and Max proteins utilize several glutamate, histidine and asparagine residues at the a and d positions (Lavigne et al. 1995). while several of the hybrid cortexillin sequences exhibit an asparagine residue at one a position (Lee et al. 2001). Both these positions typically are occupied with large aliphatic side-chain residues in order to maximize stability (Zhu et al. 1993). In the b53–122 sequence, two a positions are occupied by unusual residues (K58, R113) which could confer a decreased stability. Interestingly both these residues are positioned to allow i+3 charged interactions (D55-K58 and E110-R113) an arrangement that would allow the hydrophobic methylene groups of the a side chains to interact at the dimer interface. An extensive network of similar ionic interactions have been noted from crystallographic studies of the rod domain of the coiled-coil cortexillin I (Burkhard et al. 2000). A characteristic i+5 interaction between E112-R117` across the coiled-coil interface of b53–122 would also be possible. These types of ionic interactions in b53–122 are supported by a decrease in α-helical structure with increasing ionic strength (to 500 mM NaCl) and a subsequent increase in helical structure at higher salt concentrations. A near-identical trend in stability has been previously observed for the coiled-coil GCN4.
Some observations point to a noncanonical, coiled-coil structure of the b dimer. In particular, b53–122 contains a high frequency of alanine residues at the d positions apparent in phylogenetic analyses (Fig. 7 ▶). Similar observations in tropomyosin (Brown et al. 2001) have been shown to give rise to bends in a left-handed coiled-coil. Further, the predicted coiled-coil regions for the b53–122 protein also contain a discontinuity between residues 80–106, perhaps indicating a unique helical alignment of the subunits or the use of imperfect heptads providing a less closely packed interface (Brown et al. 1996). Both cases would still satisfy the elongated structure observed in sedimentation velocity (Revington et al. 1999) or small-angle X-ray scattering experiments (B. Shilton, M. Revington, and S. Dunn, unpubl.). Further, a coiled-coil structure for the 53–122 region of the b subunit would span ∼105Å or 75% of the required distance (130 Å) from the membrane surface to the apex of the α3β3 hexamer in the F1F0 ATP synthase. The instability of this dimeric protein observed in this work would be consistent with its role as a flexible linker that stores elastic energy and responds to the torque of rotational catalysis (Cherepanov et al. 1999).
Materials and Methods
Protein expression, purification, and quantification
Protein constructs were expressed and purified as previously described (Revington et al. 1999). Protein concentrations for sedimentation equilibrium studies were determined from extinction coefficients (ɛ280) based on quantitative amino acid analysis using samples of known A280.
Protein concentrations of circular dichroism samples were determined from the isodichroic point at 203 nm using the mean residue ellipticity (θmre) (Holtzer and Holtzer 1992). b53–122 was used as a reference protein where its concentration was determined using quantitative amino-acid analysis, ellipticity at 203 nm was measured, and a θmre was calculated. Because the magnitude of θmre is independent of secondary structure at the isodichroic point, this was used to calculate the concentrations of the other b constructs using the equation [P] = θ203/n(θmre) where [P] is the protein concentration, θ203 is the observed ellipticity at 203 nm and n is the number of residues in the construct.
Analytical ultracentrifugation
Sedimentation equilibrium data were collected with a Beckman Optima XL-A Analytical Ultracentrifuge using an An-60 Ti rotor and 6-channel cells. The samples were studied in a buffer consisting of 50 mM Tris- HCl, pH 7.5, 100 mM NaCl, and 1 mM EDTA. The proteins were monitored at 280 nm and data collected in 0.002-cm radial increments with the absorbance value at each point being the average of 10 measurements. Samples were initially cooled to 5°C, allowed to equilibrate for ∼24 h, and then scanned. The temperature then was raised in 5°C increments to 40°C. The samples were scanned at each temperature increment after equilibration. After the 40°C scans, the temperature was returned to 10 or 20°C and allowed to reequilibrate. The samples then were scanned to compare values to those already measured to determine the extent of protein degradation or irreversibility of the thermal denaturation process. Complete temperature-denaturation studies were performed on three samples of each construct.
The data sets were analyzed by Beckman XLA software and Beckman XLA macros used in conjunction with ORIGIN (Microcal) software. The three data sets collected on each construct were then simultaneously fitted using the Multifit macro. Initially, the data were fit assuming a single-solution species that results in determination of the average molecular weight of the solution species present, Mobs. The data sets also were fit for a monomer-dimer equilibrium with the association constant, Ka, as the variable with the molecular weight of the monomer inferred from the amino-acid sequence, M1, as a constant. The partial specific volumes of proteins were calculated from the amino-acid sequences by the method of Cohn and Edsall (1943). The density of the solvent was measured using a pycnometer.
Circular dichroism spectra
Protein samples for circular dichroism analysis were prepared in 25 mM Na2HPO4, pH 7.5. Circular dichroism spectra were collected on Aviv 62A DS and Jasco 810 spectropolarimeters. The Aviv spectropolarimeter used a thermoelectric cell holder to control temperature while the Jasco instrument used jacketed cells connected to an external water bath. Spectra were recorded in cells of path lengths ranging from 0.1 mm to 1 cm. Far ultraviolet (UV) wavelength scans were collected from 200–250 nm in 1 nm steps. The readings were the average of 3 sec at each wavelength and the reported ellipticity values were the average of three determinations for each sample. Temperature scans were collected at 222 nm for the range of 5–70°C in 1°C steps with 1 min equilibration time between readings. The observed ellipticity (θobs) was converted to molar mean residue ellipticity (θmre) in units of degrees cm−2 decimole−1 using θmre = (θobs•MRW) / (10•l•c) where MRW is the mean residue weight for the polypeptide in g dmol−1 residue−1, one is the pathlength in centimeters and c is the polypeptide concentration in g mL−1.
Data analysis
Calculations of Tm from the sedimentation equilibrium were calculated at 5°C intervals from the determined Kd values for the monomer-dimer equilibria between 5–40°C as described above. The Tm was the temperature at which the Kd value was equal to the protein concentration (Pt) used in that experiment. Linear interpolation of the two nearest Kd values was used to estimate the reported Tm values.
Thermal denaturation data were analyzed using the program xcrvfit (Boyko and Sykes, University of Alberta) according to the fitting method of Lavigne et al. (1998). Multiple iterations were used to determine the Gibbs free energy of unfolding at temperature T [ΔGu(T)], the change in molar enthalpy [ΔHu(Tm)] at the transition midpoint (Tm), and the heat capacity ΔCp based on equation 3. The fitting algorithm included corrections for the linear dependence of θN and θF in the pre- and posttransition regions. Concentration-dependent data were fitted using the program Kaleidagraph according to equation 2 (Ho and DeGrado 1987).
Acknowledgments
The authors thank Yumin Bi for preparation of some of the b53–122 protein used in these experiments and Kathy Barber for collection of some of the CD spectra. We are grateful to Dr. L. Lee of the Department of Chemistry and Biochemistry of the University of Windsor for use of the AVIV CD spectropolarimeter. This research was supported by operating grants MT-10237 (SDD) and MT-13105 (GSS) from the Canadian Institutes for Health Research and by an institutional grant from the Canada Foundation for Innovation for the Jasco 810 spectropolarimeter.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.3200102.
References
- Brown, J.H., Cohen C., and Parry, D.A. 1996. Heptad breaks in α-helical coiled coils: Stutters and stammers. Proteins 26 134–145. [DOI] [PubMed] [Google Scholar]
- Brown, J.H., Kim, K-H., Jun, G., Greenfield, N.J., Dominguez, R., Volkmann, N., Hitchcock-DeGregori, S.E., and Cohen, C. 2001. Proc. Nat. Acad. Sci. 98 8496–8501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhard, P., Kammerer, R.A., Steinmetz, M.O., Bourenkov, G.P., and Aebi, U. 2000. The coiled-coil trigger site of the rod domain of cortexillin I unveils a distinct network of interhelical and intrahelical salt bridges. Structure 8 223–230. [DOI] [PubMed] [Google Scholar]
- Cherepanov, D.A., Mulkidjanian, A.Y., and Junge, W. 1999. Transient accumulation of elastic energy in proton translocating ATP synthase. FEBS Lett. 449 1–6. [DOI] [PubMed] [Google Scholar]
- Cohn, E.J. and Edsall, J.T. 1943. In Proteins, amino acids, and peptides, pp. 157–161. Rheinhold, New York.
- De Francesco, R., Pastore, A., Vecchio, G., and Cortese, R. 1991. Circular dichroism study on the conformational stability of the dimerization domain of transcription factor LFB1. Biochemistry 30 143–147. [DOI] [PubMed] [Google Scholar]
- Dmitriev, O., Jones, P.C., Jiang, W., and Fillingame, R.H. 1999. Structure of the membrane domain of subunit b of the Escherichia coli F0F1 ATP synthase. J. Biol. Chem. 274 15598–15604. [DOI] [PubMed] [Google Scholar]
- Donaldson, C., Barber, K.R., Kay, C.M., and Shaw, G.S. 1995. Human S100b protein: Formation of a tetramer from synthesic calcium-binding site peptides. Protein Sci. 4 765–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn, S.D. 1992. The polar domain of the b subunit of Escherichia coli F0F1-ATPase forms an elongated dimer that interacts with the F1 sector. J. Biol. Chem. 267 7630–7636. [PubMed] [Google Scholar]
- Dunn, S.D., Bi, Y., and Revington, M. 2000. A reexamination of the structural and functional consequences of mutation of alanine-128 of the b subunit of Escherichia coli ATP synthase to aspartic acid. Biochim. Biophys. Acta. 1459 521–527. [DOI] [PubMed] [Google Scholar]
- Dunn, S.D., McLachlin, D.T., and Revington, M. 2000. The second stalk of Escherichia coli ATP synthase. Biochim Biophys Acta 1458 356–363. [DOI] [PubMed] [Google Scholar]
- Greenfield, N., Davidson, B., and Fasman, G.D. 1967. The use of computed optical rotatory dispersion curves for the evaluation of protein conformation. Biochemistry 6 1630–1637. [DOI] [PubMed] [Google Scholar]
- Greie, J.C., Deckers-Hebestreit, G., and Altendorf, K. 2000. Secondary structure composition of reconstituted subunit b of the Escherichia coli ATP synthase. Eur. J. Biochem. 267 3040–3048. [DOI] [PubMed] [Google Scholar]
- Hackel, M., Konno, T., and Hinz, M. 2000. A new alternative method to quantify residual structure in `unfolded' proteins. Biochim. Biophys. Acta. 1479 155–165. [DOI] [PubMed] [Google Scholar]
- Ho., S.P. and DeGrado, W.F. 1987. Design of a 4-helix bundle protein: Synthesis of peptides which self-associate into a helical protein. J. Am. Chem. Soc. 109 6751–6758. [Google Scholar]
- Holtzer, M.E. and Holtzer, A. 1992. α-Helix to random coil transitions: Determination of peptide concentration from the CD at the isodichroic point. Biopolymers 32 1675–1677. [DOI] [PubMed] [Google Scholar]
- Jelesarov, I. and Bosshard, H.R. 1996. Thermodynamic characterization of the coupled folding and association of heterodimeric coiled coils (leucine zippers). J. Mol. Biol. 263 344–358. [DOI] [PubMed] [Google Scholar]
- Jones, P.C., Hermolin, J., Jiang, W., and Fillingame, R.H. 2000. Insights into the rotary catalytic mechanism of F0F1 ATP synthase from the cross-linking of subunits b and c in the Escherichia coli enzyme. J. Biol. Chem. 275 31340–31346. [DOI] [PubMed] [Google Scholar]
- Kenar K.T., Garcia-Moreno, B., and Friere, E. 1995. A calorimetric characterization of the salt dependence of the stability of the GCN4 leucine zipper. Protein Sci. 41934–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumamoto, C.A. and Simoni, R.D. 1986. Genetic evidence for interaction between the a and b subunits of the F0 portion of the Escherichia coli proton translocating ATPase. J. Biol. Chem. 261 10037–10042. [PubMed] [Google Scholar]
- Lau, S.Y., Taneja, A.K., and R.S. Hodges. 1984. Synthesis of a model protein of defined secondary and quaternary structure. Effect of chain length on the stabilization and formation of two-stranded α-helical coiled-coils. J. Biol. Chem. 259 13253–13261. [PubMed] [Google Scholar]
- Lavigne, P., Crump, M.P., Gagne, S.M., Hodges, R.S., Kay, C.M., and Sykes, B.D. 1998. Insights into the mechanism of heterodimerization from the 1H-NMR solution structure of the c-Myc-Max heterodimeric leucine zipper. J. Mol. Biol. 281 165–181. [DOI] [PubMed] [Google Scholar]
- Lavigne, P., Kondejewski, L.H., Houston, M.E.J., Sonnichsen, F.D., Lix, B., Sykes, B.D., Hodges, R.S., and Kay, C.M. 1995. Preferential heterodimeric parallel coiled-coil formation by synthetic Max and c-Myc leucine zippers: A description of putative electrostatic interactions responsible for the specificity of heterodimerization. J. Mol. Biol. 254 505–520. [DOI] [PubMed] [Google Scholar]
- Lee, D.L., Lavigne, P., and Hodges, R.S. 2001. Are trigger sequences essential in the folding of two-stranded a-helical coiled coils. J. Mol. Biol. 306 539–553. [DOI] [PubMed] [Google Scholar]
- Leslie, A.G. and Walker, J.E. 2000. Structural model of F1-ATPase and the implications for rotary catalysis. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 355 465–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupas, A. 1996. Coiled coils: New structures and new functions. Trends Biochem. Sci. 21 375–382. [PubMed] [Google Scholar]
- Lupas, A., Van Dyke, M., and Stock, J. 1991. Predicting coiled coils from protein sequences. Science 252 1162–1164. [DOI] [PubMed] [Google Scholar]
- Makhatadze, G.I. and Privalov, P.L. 1995. Energetics of protein structure. Adv. Protein Chem. 47 307–425. [DOI] [PubMed] [Google Scholar]
- McCormick, K.A., Deckers-Hebestreit, G., Altendorf, K., and Cain, B.D. 1993. Characterization of mutations in the b subunit of F1F0 ATP synthase in Escherichia coli. J. Biol. Chem. 268 24683–24691. [PubMed] [Google Scholar]
- McLachlin, D.T., Bestard, J.A., and Dunn, S.D. 1998. The b and δ subunits of the Escherichia coli ATP synthase interact via residues in their C-terminal regions. J. Biol. Chem. 273 15162–15168. [DOI] [PubMed] [Google Scholar]
- McLachlin, D.T., Coveny, A.M., Clark, S.M., and Dunn, S.D. 2000. Site-directed cross-linking of b to the α, β, and a subunits of the Escherichia coli ATP synthase. J. Biol. Chem. 275 17571–17577. [DOI] [PubMed] [Google Scholar]
- McLachlin, D.T. and Dunn, S.D. 1997. Dimerization interactions of the b subunit of the Escherichia coli F1F0-ATPase. J. Biol. Chem. 272 21233–21239. [DOI] [PubMed] [Google Scholar]
- McLachlin, D.T. and Dunn, S.D. 2000. Disulfide linkage of the b and δ subunits does not affect the function of the Escherichia coli ATP synthase. Biochemistry 39 3486–3490. [DOI] [PubMed] [Google Scholar]
- Noji, H. and Yoshida, M. 2001. The rotary machine in the cell, ATP synthase. J. Biol. Chem. 276 1665–1668. [DOI] [PubMed] [Google Scholar]
- Ogilvie, I., Wilkens, S., Rodgers, A.J., Aggeler, R., and Capaldi, R.A. 1998. The second stalk: The δ-b subunit connection in ECF1F0. Acta. Physiol. Scand. Suppl. 643 169–175. [PubMed] [Google Scholar]
- O-Shea, E.K., Rutkowski, R., and Kim, P.S. 1989. Evidence that the leucine zipper is a coiled coil. Science 243 538–542. [DOI] [PubMed] [Google Scholar]
- Privalov, P.L. 1979. Stability of proteins. Small globular proteins. Adv. Protein Chem. 33 167–242. [DOI] [PubMed] [Google Scholar]
- Privalov, P.L. 1982. Stability of proteins. Proteins which do not present a single cooperative system. Adv. Protein Chem. 35 1–104. [PubMed] [Google Scholar]
- Revington, M., McLachlin, D.T., Shaw, G.S., and Dunn, S.D. 1999. The dimerization domain of the b subunit of the Escherichia coli F(1)F(0)-ATPase. J. Biol. Chem. 274 31094–31101. [DOI] [PubMed] [Google Scholar]
- Rodgers, A.J. and Capaldi, R.A. 1998. The second stalk composed of the b- and δ-subunits connects F0 to F1 via an α-subunit in the Escherichia coli ATP synthase. J. Biol. Chem. 273 29406–29410. [DOI] [PubMed] [Google Scholar]
- Rodgers, A.J., Wilkens, S., Aggeler, R., Morris, M.B., Howitt, S.M., and Capaldi, R.A. 1997. The subunit δ-subunit b domain of the Escherichia coli F1F0 ATPase. The b subunits interact with F1 as a dimer and through the δ subunit. J. Biol. Chem. 272 31058–31064. [DOI] [PubMed] [Google Scholar]
- Sorgen, P.L., Bubb, M.R., and Cain, B.D. 1999. Lengthening the second stalk of F(1)F(0) ATP synthase in Escherichia coli. J. Biol. Chem. 274 36261–36266. [DOI] [PubMed] [Google Scholar]
- Sorgen, P.L., Caviston, T.L., Perry, R.C., and Cain, B.D. 1998. Deletions in the second stalk of F1F0-ATP synthase in Escherichia coli. J. Biol. Chem. 273 27873–27878. [DOI] [PubMed] [Google Scholar]
- Thompson, J.D., Higgins, D.G., and Gibson, T.J. 1994. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkens, S., Zhou, J., Nakayama, R., Dunn, S.D., and Capaldi, R.A. 2000. Localization of the delta subunit in the Escherichia coli F(1)F(0)-ATPsynthase by immuno electron microscopy: The delta subunit binds on top of the F(1). J. Mol. Biol. 295 387–391. [DOI] [PubMed] [Google Scholar]
- Zhu, B.Y., Zhou, N.E., Kay, C.M., and Hodges, R.S. 1993. Packing and hydrophobicity effects on protein folding and stability: Effects of β-branched amino acids, valine and isoleucine, on the formation and stability of two-stranded α-helical coiled coils/leucine zippers. Protein Sci. 2 383–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolkiewski, M., Redowicz, M.J., Korn, E.D., Hammer III, J.A., and A. Ginsburg. 1997. Two-state thermal unfolding of a long dimeric coiled-coil: The Acanthamoeba myosin II rod. Biochemistry 36 7876–7883. [DOI] [PubMed] [Google Scholar]