

Abstract
A previously proposed electron transfer (ET) pathway in the heme enzyme cytochrome c peroxidase has been excised from the structure, leaving an open ligand-binding channel in its place. Earlier studies on cavity mutants of this enzyme have revealed structural plasticity in this region of the molecule. Analysis of these structures has allowed the design of a variant in which the specific section of protein backbone representing a previously proposed ET pathway is accurately extracted from the protein. A crystal structure verified the creation of an open channel that overlays the removed segment, extending from the surface of the protein to the heme at the core of the protein. A number of heterocyclic cations were found to bind to the proximal-channel mutant with affinities that can be rationalized based on the structures. It is proposed that small ligands bind more weakly to the proximal-channel mutant than to the W191G cavity due to an increased off rate of the open channel, whereas larger ligands are able to bind to the channel mutant without inducing large conformational changes. The structure of benzimidazole bound to the proximal-channel mutant shows that the ligand accurately overlays the position of the tryptophan radical center that was removed from the wild-type enzyme and displaces four of the eight ordered solvent molecules seen in the empty cavity. Ligand binding also caused a small rearrangement of the redesigned protein loop, perhaps as a result of improved electrostatic interactions with the ligand. The engineered channel offers the potential for introducing synthetic replacements for the removed structure, such as sensitizer-linked substrates. These installed "molecular wires" could be used to rapidly initiate reactions, trap reactive intermediates, or answer unresolved questions about ET pathways.
Keywords: Protein engineering, ligand binding, protein cavities, electron transfer
A number of approaches have been used to obtain a detailed understanding of electron transfer (ET) in proteins. For example, photoactive centers introduced into a protein structure have been extensively used to study the kinetics of ET and its dependence on the length and conformation of the protein backbone (Beratan et al. 1990; Nocek et al. 1990; Millett et al. 1995). The results have shown that ET from donor to acceptor can be facilitated by the intervening protein medium (Farid et al. 1993) and can be mediated by electron tunneling through specific σ-bond pathways (Beratan et al. 1992; Gray and Winkler 1996; Winkler and Gray 1997). These features allow efficient ET to operate over long distances between cofactors that are buried in the core of proteins. Although many examples have been found that show specific protein pathways enhance electron transfer, recent analysis has suggested that it may not always be necessary for nature to evolve highly optimized ET pathways in order to achieve specificity of function (Page et al. 1999).
Further details about the importance of ET pathways might be addressed by combining photoinitiated ET with protein mutants in which the pathway itself or its surrounding environment is altered. However, because the most efficient ET pathways involve the protein backbone, direct and predictable alteration by mutagenesis has been limited. Unnatural peptide linkages, created using semisynthetic methods in place of the natural ET chain, hold promise (Dawson and Kent 2000) but remain technically challenging at present. Synthetic-model complexes have allowed studies of ET using a greater variability in the chemical makeup of the pathway (Mutz et al. 1999; Kornilova et al. 2000; Gibney and Dutton 2001), but only a few of these have attempted to mimic the natural context of the protein environment that intervenes between redox centers and sequesters them from solvent. Thus, limitations imposed by these approaches have prevented complete answers to a number of fundamental questions. For example, how much does a specific σ-bond tunneling pathway modulate the ET efficiency relative to the general chemical composition and protein environment? If a proposed ET pathway in a protein were to be replaced by a variety of synthetic structures within the same protein context, would they function normally?
Recent reports have demonstrated the use of sensitizer-linked substrates (SLS) to probe the buried active sites of redox proteins (Dmochowski et al. 1999; Wilker et al. 1999). A number of specifically designed SLS probes, consisting of ruthenium-based photosensitizers attached via hydrocarbon or aromatic linkers to substrate analogs, have been introduced into the substrate-binding site of a P450. The SLS probes were found to bind to P450cam such that the substrate analog of the SLS occupies the substrate-binding site of the protein, the ruthenium sensitizer remains on the surface of the protein, and the linker is threaded through the substrate access channel. These studies have shown that photoexcitation of bound SLS probes can be used to drive rapid oxidation or reduction of the buried heme center. This approach offers great potential for delivering electrons or holes directly into the buried active sites of enzymes to rapidly initiate reactions and study unstable intermediates. It is also possible that studies using SLS probes could be utilized to further our general understanding of ET in proteins through variation of the chemical composition of the linker. However, the SLS studies to date have been introduced into a preexisting protein channel that does not naturally serve to host an ET pathway. It thus may be of considerable interest to develop a protein system in which a natural ET pathway has been removed to allow installation of artificial SLS probes.
One of the most well known of all proposed ET pathways is found in cytochrome c peroxidase (CCP), which belongs to a special class of enzymes that use a redox-active radical (Sivaraja et al. 1989). In a widely accepted model, the oxidized ferryl (Fe+4 = O) heme of CCP extracts an electron from Trp 191 to create a radical intermediate, which is then reduced by cytochrome c (cyt c) during enzyme turnover (Miller et al. 1995; Millett et al. 1995). In the crystal structure of the complex between CCP and yeast cyt c (Pelletier and Kraut 1992), the heme of cyt c is in contact with Ala 194 on the surface of CCP and would provide an efficient σ-bond tunneling pathway (W191-G192-A193-A194) between cyt c and the Trp 191 free-radical center (Fig. 1 ▶). Electron transfer from cyt c to the Trp 191 radical is very rapid in this complex, providing support for the operation of the proposed pathway (Liu et al. 1995). However, despite the compelling nature of these observations, much data indicate that this pathway may not be exclusively used (Nocek et al. 1996; Pappa et al. 1996) and direct evidence indicates that multiple cyt c-binding sites exist on CCP (Stemp and Hoffman 1993; Zhou and Hoffman 1994; Leesch et al. 2000; Nocek et al. 2000). Thus, even in this well-characterized case, many fundamental details about the specificity of the proposed ET pathway remain controversial.
Fig. 1.

The proposed electron transfer (ET) pathway for cytochrome c peroxidase (CCP) based upon the crystal structure of the complex of CCP with yeast cytochrome c (cyt c; left). The isolated hemes are shown (right), along with the proposed ET pathway of CCP extending from Ala 194 to Trp 191.
In this paper we describe the results of a protein redesign in which this proposed ET pathway in CCP has been excised from the structure, leaving in its place a binding channel that could be utilized for studies of SLS-initiated reactions. The structures and ligand-binding properties of this designed proximal-channel mutant are presented and discussed.
Results
Design of the proximal-channel mutant
Previous studies on mutants of CCP have suggested that the proposed ET pathway might be selectively designed out of the surrounding structure without deleterious structural consequences. This backbone segment (Fig. 1 ▶) consists of the sequence W191-G192-A193-A194 and extends from the surface where Ala 194 contacts the heme of cyt c down to the buried Trp 191 radical site that is in contact with the heme of CCP (Pelletier and Kraut 1992). Previous studies of the W191G mutant show that it contains an uncollapsed buried cavity that is capable of binding a number of heterocyclic cations (Fitzgerald et al. 1994; Musah and Goodin 1997; Musah et al. 1997, 2002). Cations bind in preference to neutral molecules due to a number of electrostatic interactions with the protein that stabilize the Trp 191 cation radical of the native protein (Fitzgerald et al. 1995; Jensen et al. 1998; Musah et al. 2002). We have also observed that larger ligands induce an alternate (open) conformation of the protein backbone near the W191G mutation that results in the displacement of the proposed ET chain from its normal (closed) conformation (Fig. 2 ▶; Fitzgerald et al. 1996; Cao et al. 1998). This displacement can be roughly thought of as a rotation around two hinge residues: a cis–trans isomerization of Pro 190 and the interchange of the main-chain and side-chain positions of Asn 195. From examination of these structural alternatives, the loop containing the proposed ET pathway was redesigned so that the protein backbone more efficiently spans the hinge points and cannot occupy its native position. To accomplish this, the P190G mutation was introduced to prevent it from locking the loop into discrete open or closed states. The other hinge point, Asn 195, was retained to entice the other end of the loop to adopt the alternate open conformation. Finally, the ET pathway sequence, WGAA, was shortened to GA to efficiently span the gap. Thus the original sequence, P190-W191-G192-A193-A194-N195, was replaced with G190-G191-A192-N193, resulting in a new sequence numbering from this point.
Fig. 2.

Stereo views of the structures illustrating the design of the proximal-channel mutant. The views in A and B are rotated by 90°. In blue is the structure of the W191G mutant in the closed conformation, in which the electron transfer pathway residues are in the same conformation as the wild-type enzyme. Shown in green is the structure of the same mutant in the open conformation with benzimidazole bound. The cis–trans isomerization of Pro 190 and the main-chain–side-chain interchange of Asn 195 serve as the hinge points for the conformational switch. Shown in white is the structure of the channel mutant in the absence of ligand.
Crystal structure of the proximal-channel mutant
The 1.8-Å crystal structure of the channel mutant was determined at 100 K (see Table 1), showing the accurate excision of the proposed ET chain and the creation of an open water-filled channel in its place. Shown in Figure 3 ▶ is the electron density of an Fo–Fc omit map for the proximal-channel mutant in the region of the redesigned loop superimposed upon the refined structure of this segment. The electron density was clear and continuous and allowed an unambiguous tracing of the chain. Changes in the structure were localized to the region of the redesigned loop and the newly formed channel. The conformation of the redesigned segment is compared with the open and closed forms of the loop of W191G in Figure 2 ▶. As anticipated, the greater flexibility of Gly 190 compared with proline allowed the protein backbone to approximately bisect the conformations observed in the open and closed conformation of W191G. Also consistent with the design, the protein backbone of Asn 193 was observed in a conformation similar to that of the open form of W191G (Fitzgerald et al. 1996). The ξ angle for Asn 193 in the proximal-channel mutant is 148°, compared with 16° and 175° for Asn 195 in the closed and open form of W191G, respectively. Finally, joining the ends with the shortened protein backbone leads to the effective removal of the ET pathway from the structure.
Table 1.
Data collection and structure refinement statistics
| Structure | Channel mutant | + Benzimidazole |
| PDB code | 1KXN | 1KXM |
| Resolution (Å) | 1.8 (1.85–1.80) | 1.74 (1.80–1.74) |
| Unit cell (Å) | 106.8, 75.8, 51.1 | 106.3, 74.7, 50.9 |
| Unique reflections | 36741 (3208) | 33260 (2794) |
| Observations | 168657 (11517) | 105851 (4827) |
| % completeness | 91.2 (81.1) | 77.8 (40.1) |
| ≤I/σ> | 15.5 (3.1) | 28.5 (3.4) |
| Rsymm | 0.075 (0.55) | 0.046 |
| R (Fo > 4 σ) | 17.4% | 18.1% |
| R (all unique reflections) | 18.71% | 19.37% |
| Rfree | 19.4% | 21.4% |
| ≤overall B> (Å−2) | 21.2 | 16.9 |
| ≤main-chain B> (Å−2) | 19.8 | 15.2 |
| ≤side-chain B> (Å−2) | 23.2 | 18.9 |
| Rms bond (Å) | 0.009 | 0.020 |
| Rms ang (deg) | 0.029 | 0.024 |
| Residues | 290 | 290 |
| Cofactors and Ligands | Heme | Heme, benzimidazole |
| Waters | 347 | 353 |
Fig. 3.

Fo–Fc omit electron density, contoured at 3 and 5 σ, is shown in stereo for the proximal-channel mutant in the redesigned region. Superimposed is the refined structure of this segment.
The excision of the ET pathway leaves a deep open channel extending from the surface of the protein to the buried heme center. Shown in Figure 4A ▶ is the solvent-excluded surface for the ligand-free proximal-channel mutant, calculated with a 1.5-Å probe sphere radius and superimposed on the proposed ET pathway of wild-type CCP. This figure shows the extent of the deep invagination introduced into the protein structure and the remarkable registration of this channel with the proposed ET pathway. The channel appears to be fully hydrated, and Figure 5 ▶ shows the positions of eight water molecules that are observed to occupy the engineered channel. Finally, the average B value for atoms in the redesigned segment between Gly190 and Asn 193 (33.5 Å−2) is 58% higher than that of the overall structure (21.2 Å−2), indicating that this loop remains more mobile than the rest of the structure. However, with the exception of the Met 230 side chain, the B values for other atoms that line the interior of the created channel are not significantly larger than the overall average value, indicating that the interior channel walls are fairly rigid.
Fig. 4.

Solvent-excluded surface representation of the engineered proximal-channel mutant in its ligand-free (A) and benzimidazole-bound forms (B). For both A and B, the surface was calculated using a 1.5-Å radius probe sphere using the MSMS program. Clipping planes are used to generate a thin slice of the engineered channel in which the front surface of the invaginated channel is removed. The heme is shown in red and the ribbon representation of the protein is shown in brown. In A, the proposed electron transfer pathway and Trp 191 radical site from wild-type cytochrome c peroxidase is shown superimposed on this channel in blue. In B, the Fo–Fc omit electron density is shown at 7 σ (blue) for a crystal soaked in benzimidazole, in which no model for the ligand was included for several refinement cycles. The refined position of benzimidazole is also shown in yellow.
Fig. 5.
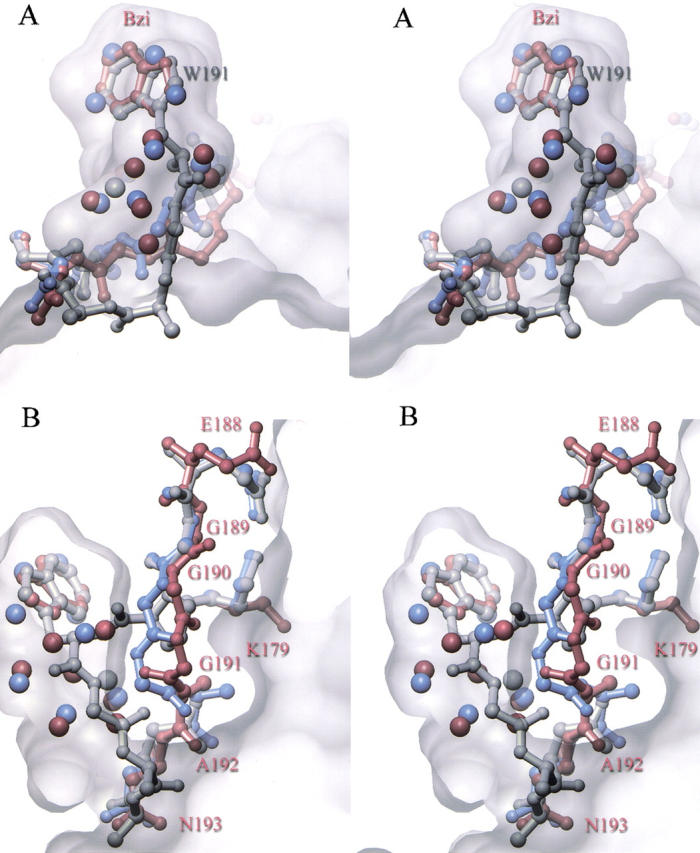
Stereo views of the binding of benzimidazole to the proximal-channel mutant. Shown in A (top) is the structure of wild-type cytochrome c peroxidase (gray) superimposed on the structure of the channel mutant in the absence (blue) and presence (red) of bound benzimidazole. The refined position of water molecules in the channel are shown as spheres, colored according to their respective structures as described above. Shown in B (bottom) is an alternate view to portray the shift in the protein backbone and loss of the Glu 188 hydrogen bond associated with benzimidazole binding. Superimposed on these structures is a semitransparent depiction of the solvent-excluded surface of the engineered channel.
Ligand binding to the proximal-channel mutant
To examine the feasibility of utilizing the engineered proximal-channel mutant to bind SLS probes, we have performed a preliminary characterization of the ligand-binding properties of the engineered channel. The demonstrated ability of the buried cavity of W191G to bind small heterocyclic cations (Fitzgerald et al. 1994; Musah and Goodin 1997; Musah et al. 1997, 2002) suggests that similar compounds might bind to the much larger open channel of the redesigned mutant. Ligand-binding titrations for W191G and the proximal-channel mutant are compared in Table 2. The titrations were performed in the absence of potentially competing counter ions, and at pH 4.5 to favor the protonated cationic forms of the ligands (Fitzgerald et al. 1995). Ligand binding was detected by the small perturbation of the Soret absorption band that accompanies occupation of the cavity (Fitzgerald et al. 1994; Musah et al. 2002). Several interesting observations are evident from the data. First, the open channel of the proximal-channel mutant appears to retain an affinity for ligands that bind W191G, but most ligands bind to the proximal-channel mutant with a 10- to 50-fold reduced affinity compared with W191G. One exception to this trend is benzimidazole, which is the highest affinity ligand for the proximal-channel mutant and binds with a roughly equal affinity to W191G. The other exception is 2-n-propylimidazole, which does not appear to bind to W191G but does bind weakly to the proximal-channel mutant. Our data suggest that benzimidazole, and presumably the other ligands, bind to the channel mutant as the protonated cations and not the deprotonated neutral forms. Isothermal titration calorimetry (Fig. 6 ▶) was used to show that the affinity of benzimidazole for the proximal-channel mutant (Kd = 0.14 mM) was reduced in the presence of 150-mM potassium ion (Kd = 0.36 mM). This behavior is similar to that observed for the W191G cavity mutant, where it was shown that potassium ions competitively inhibit heterocyclic cation binding to the cavity (Fitzgerald et al. 1995). In addition, binding of benzimidazole to the channel mutant appears to be lost as the pH is raised from 4.5 to 6, with which the titration of the ligand induces only small changes in the heme spectra that cannot be fit well to a binding isotherm. This behavior is also similar to that reported for the progenitor W191G cavity for a number of ligands with accessible pKas (Fitzgerald et al. 1994; Musah et al. 2002).
Table 2.
Ligand dissociation constants for W191G and the proximal channel mutant determined from the perturbation of the Soret absorption band upon titration of the ligand into protein
| Kd (mM) | ||
| Ligand | W191Ga | Channel mutant |
| Benzimidazole | 0.15 | 0.13 |
| Imidazo[1,2-a]pyridine | 0.09 | 0.4 |
| 4-aminopyridine | 0.05 | 0.41 |
| 2-aminopyridine | 0.07 | 0.46 |
| 3,4,5-trimethylthiazole | 0.13 | 0.51 |
| 3-methylthiazole | 0.11 | 1.5 |
| 3-aminopyridine | 0.07 | 2.1 |
| 1,2-dimethylimidazole | 0.03 | 2.2 |
| 2,3,4-trimethylthiazole | 0.56 | 2.4 |
| 1-vinylimidazole | 0.14 | 6.4 |
| 2-n-propylimidazole | – | 15.8 |
a Taken from Musah et al. (2002).
Fig. 6.

Binding of benzimidazole to the proximal-channel mutant by isothermal titration calorimetry is shown in the presence and absence of potassium ion. Potassium, which serves as a competitive inhibitor of cationic ligand binding to W191G, was removed from the protein samples by gel filtration in 100 mM bis-Tris propane/citric acid (pH 4.5) and the titrations carried out in the same buffer. Experimental conditions are given in Materials and Methods. Fits to the binding titrations gave Kd = 0.14 mM, ΔG = −5.2 kcal/mole, ΔH = −13 kcal/mole, and ΔS = 26 cal mole−1 °K−1 in the absence of potassium, and Kd = 0.36 mM, ΔG = −4.7 kcal/mole, ΔH = −5 kcal/mole, and ΔS = 1 cal mole−1 °K−1 in the presence of 150 mM potassium ion.
Crystal structure of benzimidazole bound to the proximal-channel mutant
A structure was determined at 100-K and at 1.7-Å resolution for a crystal of the channel mutant after soaking in 30 mM benzimidazole. Difference Fourier maps showed the clear occupation of benzimidazole in the channel (Fig. 4B ▶). It is remarkable that the benzimidazole ring occupies essentially the same position as the indole ring of the Trp 191 side chain of the wild-type enzyme, differing by only an ∼5° rotation. In so doing, it replaces four of the ordered solvent molecules that were observed to occupy this region of the channel in the absence of ligand. The other four water molecules that were observed in the empty channel remain after binding benzimidazole. Three additional water molecules were seen in the benzimidazole-bound structure that were not seen in the empty channel. Benzimidazole interacts with the channel mutant in much the same way that it interacts with W191G (Fitzgerald et al. 1996), forming a hydrogen bond with Asp 235 (d = 2.5 Å) and with one of the water molecules introduced with the ligand.
Ligand binding appears to induce a small but significant change in the conformation of the protein backbone between Gly 189 and Gly 191, with the largest movement of 2.5 Å at the Cα of Gly 191 (Fig. 5B ▶). As a result of this movement, the peptide carbonyl oxygen of Gly 191 rotates ∼180° to face in toward the bound benzimidazole. In addition, a hydrogen-bonding interaction that is observed between Glu 188 and Lys 179 in both wild-type CCP and in the empty-channel mutant has now been broken. As a result of this small conformational change, the mouth of the channel opening at the protein surface is somewhat wider, but its overall dimensions remain relatively unchanged. The average B value for the redesigned segment between Gly 190 and Asn 193 for the benzimidazole bound form (25.2 Å−2) is 49% higher than that of the overall structure (16.9 Å−2), indicating that it is subject to a similar increase in static or thermal disorder in the bound and unbound conformations.
Discussion
In this study, we have characterized the removal of the most often proposed ET pathway from CCP, in which a template channel is created that is capable of binding complementary ligands. Important for the success of this redesign were earlier studies, in which ligand binding to the W191G-cavity-forming mutant caused movements in a protein loop (Fitzgerald et al. 1996; Cao et al. 1998). This unmasked the intrinsic structural plasticity of a region between two hinge residues, Pro 190 and Asn 195, and allowed a redesign in which a shorter and more flexible sequence was introduced that could not occupy the position of the native structure. It is remarkable that such an extended channel can be introduced into the structure of a protein without significant collapse. The empty channel is occupied by eight ordered water molecules, so it is possible that most of the enthalpic hydrogen-bonding interactions that were lost upon the removal of the structure are recovered by interactions with solvent. Significant rigidity of the protein structure surrounding the removed segment may also contribute to the lack of collapse. As has been observed in a number of smaller cavity-forming mutants (Eriksson et al. 1992a,b; McRee et al. 1994; Morton and Matthews 1995; Hirst and Goodin 2000; Hirst et al. 2001a,b; Musah et al. 2002), it is apparent that the protein is unable to recover the energy cost of cavity creation by repacking of the surrounding structure.
The general affinity of the proximal-channel mutant for binding cationic ligands was similar to W191G, and observed differences in specificity were easily rationalized from the structures. For smaller ligands, the overall lower affinities of the channel mutant compared with the buried W191G cavity most likely result from increased rates of ligand dissociation for the open channel (Fitzgerald et al. 1996). Larger ligands behave differently due to their poor binding to W191G. For example, 2-n-propylimidazole is too large to bind to the cavity of W191G but is able to bind weakly to the engineered channel. In addition, benzimidazole binds with a similar affinity to both proteins because it is the compound that induced the movement of the loop that inspired the redesign and, therefore, presumably has similar off rates. The crystal structure of benzimidazole bound in the channel at the position of the original Trp 191 side chain shows that the enlarged channel retains the template-directed specificity seen for W191G (Musah et al. 2002). As observed for benzimidazole binding to W191G, ordered solvent at the binding site is displaced by the ligand and an additional water is introduced to enable the ligand to fully satisfy its hydrogen-bonding capability. Finally, we propose that the small backbone rearrangement associated with benzimidazole binding, in which the backbone carbonyl of Gly 191 turns to direct its C = O vector toward the ligand, may result in a more favorable electrostatic interaction with the cationic ligand (Fitzgerald et al. 1995; Jensen et al. 1998).
The fairly precise removal of the proposed ET pathway from CCP and the ability of the resulting engineered channel to bind ligands shows the potential for replacing this important section of the enzyme with artificial structures. We propose that an SLS with an attached heterocyclic cation might act as bait to drive specifically designed probes into the channel with high specificity. These novel structures could lead to new methods for rapidly generating and studying intermediates by both photochemical and electrochemical means and they may provide a novel way to further the study of electron transfer in these enzymes.
Materials and methods
Protein expression and purification
The proximal-channel mutant was constructed in the Escherichia coli expression plasmid pT7CCP by oligonucleotide site-directed mutagenesis as previously described (Fitzgerald et al. 1994). The mutation was verified by automated DNA sequencing of the entire coding region. Protein was overexpressed in E. coli BL21(DE3), purified, reconstituted with heme, and crystallized as previously described (Fitzgerald et al. 1994). UV-Vis absorption spectra were used to calculate protein concentration based on its molar absorptivity at 412 nm (ɛ = 103.6 mM−1 cm−1).
Binding measurements
Samples of protein were prepared for binding measurements by removal of all cations from the structure, which have been shown to interfere with ligand binding to the W191G cavity (Fitzgerald et al. 1995). Redissolved protein crystals in phosphate buffer were loaded onto an equilibrated PD-10 column that was equilibrated and eluted with 100 mM bis-Tris propane/citric acid (pH 4.5). Ligand solutions were prepared in the same buffer. Ligand binding to the protein was measured either by measuring the small perturbation of the heme Soret band or by isothermal titration calorimetry as previously described (Musah et al. 2002).
X-ray crystallography
Single crystals of the proximal-channel mutant were grown in sitting drops by vapor diffusion against 2-methyl-2,4-pentanediol (MPD; Wang et al. 1990). Drops containing 0.06 mM protein, 80–120 mM potassium phosphate (pH 6.0), and 20% MPD were equilibrated against 25% MPD at 15°C until crystal growth was complete (1–2 d). Crystals were mounted in nylon loops and frozen directly in a liquid nitrogen cryo-stream held at 100 K. For the benzimidazole-bound structure, crystals were soaked in artificial mother liquor containing 30 mM benzimidazole (pH 4.5) for 5 min before mounting and freezing. X-ray diffraction data was collected at 100 K on a Siemens area detector using Cu Kα radiation from the rotating anode of a Rigaku X-ray generator. Data were processed using the Xengen (Howard et al. 1985) program suite. Fourier difference maps were created for each data set using an initial model constructed from the W191G structure (Fitzgerald et al. 1994; PDB entry 1CMQ) with the loop residues (189–196) omitted. The models were refined with iterative cycles of least squares refinement using SHELXL (Sheldrick and Schneider 1997) and manual fitting using XFIT (McRee 1999).
Acknowledgments
We thank Prof. H.B. Gray, Dr. Duncan McRee, Prof. Judy Hirst, Drs. Alycen Nigro and Stephan Vetter, and Alex Dunn for advice and helpful discussions. This work was supported in part by grants GM41049 and GM48495 from the NIH to D.B.G, by NIH National Research Service Awards postdoctoral fellowships GM20703 to A.-M.A.H. and GM17844 to R.A.M, and by predoctoral fellowships from the DOE and the La Jolla Interfaces in Science program to R.J.R.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.4870102.
Note added in proof
The crystallographic coordinates for the structures presented in this work have been deposited with the Protein Data Bank, Research Collaboratory for Structural Bioinformatics (RCSB), (http://www.rcsb.org) as entries 1KXN and 1KXM.
References
- Beratan, D.N., Onuchic, J.N., Betts, J.N., Bowler, B.E., and Gray, H.B. 1990. Electron-tunneling pathways in ruthenated proteins. J. Am. Chem. Soc. 112 7915–7921. [Google Scholar]
- Beratan, D.N., Onuchic, J.N., Winkler, J.R., and Gray, H.B. 1992. Electron-tunneling pathways in proteins. Science 258 1740–1741. [DOI] [PubMed] [Google Scholar]
- Cao, Y., Musah, R.A., Wilcox, S.K., Goodin, D.B., and McRee, D.E. 1998. Protein conformer selection by ligand binding observed with crystallography. Protein Sci. 7 72–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson, P.E. and Kent, S.B.H. 2000. Synthesis of native proteins by chemical ligation. Ann. Rev. Biochem. 69 923–960. [DOI] [PubMed] [Google Scholar]
- Dmochowski, I.J., Crane, B.R., Wilker, J.J., Winkler, J.R., and Gray, H.B. 1999. Optical detection of cytochrome P450 by sensitizer-linked substrates. Proc. Natl. Acad. Sci. 96 12987–12990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson, A.E., Baase, W.A., Wozniak, J.A., and Matthews, B.W. 1992a. A cavity-containing mutant of T4 lysozyme is stabilized by buried benzene. Nature 355 371–373. [DOI] [PubMed] [Google Scholar]
- Eriksson, A.E., Baase, W.A., Zhang, X.J., Heinz, D.W., Blaber, M., Baldwin, E.P., and Matthews, B.W. 1992b. Response of a protein structure to cavity-creating mutations and its relation to the hydrophobic effect. Science 255 178–183. [DOI] [PubMed] [Google Scholar]
- Farid, R.S., Moser, C.C., and Dutton, P.L. 1993. Electron transfer in proteins. Curr. Opin. Struct. Biol. 3 225–233. [Google Scholar]
- Fitzgerald, M.M., Churchill, M.J., McRee, D.E., and Goodin, D.B. 1994. Small molecule binding to an artificially created cavity at the active site of cytochrome c peroxidase. Biochemistry 33 3807–3818. [PubMed] [Google Scholar]
- Fitzgerald, M.M., Trester, M.L., Jensen, G.M., McRee, D.E., and Goodin, D.B. 1995. The role of aspartate-235 in the binding of cations to an artificial cavity at the radical site of cytochrome c peroxidase. Protein Sci. 4 1844–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald, M.M., Musah, R.A., McRee, D.E., and Goodin, D.B. 1996. A ligand-gated, hinged loop rearrangement opens a channel to a buried artificial protein cavity. Nat. Struct. Biol. 3 626–631. [DOI] [PubMed] [Google Scholar]
- Gibney, B.R. and Dutton, P.L. 2001. De novo design and synthesis of heme proteins. Adv. Inorg. Chem. 51 409–455. [Google Scholar]
- Gray, H.B. and Winkler, J.R. 1996. Electron transfer in proteins. Annu. Rev. Biochem. 65 537–561. [DOI] [PubMed] [Google Scholar]
- Hirst, J. and Goodin, D.B. 2000. Unusual oxidative chemistry of Nω-hydroxyarginine and N-hydroxyguanidine catalyzed at an engineered cavity in a heme peroxidase. J. Biol. Chem. 275 8582–8591. [DOI] [PubMed] [Google Scholar]
- Hirst, J., Wilcox, S.K., Ai, J., Moënne-Loccoz, P., Loehr, T.M., and Goodin, D.B. 2001a. Replacement of the axial histidine ligand with imidazole in cytochrome c peroxidase. 2. Effects on heme coordination and function. Biochemistry 40 1274–1283. [DOI] [PubMed] [Google Scholar]
- Hirst, J., Wilcox, S.K., Williams, P.A., Blankenship, J., McRee, D.E., and Goodin, D.B. 2001b. Replacement of the axial histidine ligand with imidazole in cytochrome c peroxidase. 1. Effects on structure. Biochemistry 40 1265–1273. [DOI] [PubMed] [Google Scholar]
- Howard, A.J., Nielson, C., and Xuong, N.H. 1985. Software for a diffractometer with multiwire area detector. In Methods in Enzymology (eds. H.W. Wyckoff et al.), pp. 452–472. Academic Press, Orlando, FL. [DOI] [PubMed]
- Jensen, G.M., Bunte, S.W., Warshel, A., and Goodin, D.B. 1998. Energetics of cation radical formation at the proximal active site tryptophan of cytochrome c peroxidase and ascorbate peroxidase. J. Phys. Chem. B 102 8221–8228. [Google Scholar]
- Kornilova, A.Y., Wishart, J.F., Xiao, W., Lasey, R.C., Fedorova, A., Shin, Y.K., and Ogawa, M.Y. 2000. Design and characterization of a synthetic electron-transfer protein. J. Am. Chem. Soc. 122 7999–8006. [Google Scholar]
- Leesch, V.W., Bujons, J., Mauk, A.G., and Hoffman, B.M. 2000. Cytochrome c peroxidase–cytochrome c complex: Locating the second binding domain on cytochrome c peroxidase with site-directed mutagenesis. Biochemistry 39 10132–10139. [DOI] [PubMed] [Google Scholar]
- Liu, R.Q., Hahm, S., Miller, M., Durham, B., and Millett, F. 1995. Photooxidation of Trp-191 in cytochrome c peroxidase by ruthenium cytochrome c derivatives. Biochemistry 34 973–983. [DOI] [PubMed] [Google Scholar]
- McRee, D.E. 1999. XtalView/Xfit—A versatile program for manipulating atomic coordinates and electron density. J. Struct. Biol. 125 156–165. [DOI] [PubMed] [Google Scholar]
- McRee, D.E., Jensen, G.M., Fitzgerald, M.M., Siegel, H.A., and Goodin, D.B. 1994. Construction of a bisaquo heme enzyme and binding by exogenous ligands. Proc. Natl. Acad. Sci. 91 12847–12851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, M.A., Vitello, L., and Erman, J.E. 1995. Regulation of interprotein electron transfer by Trp 191 of cytochrome c peroxidase. Biochemistry 34 12048–12058. [DOI] [PubMed] [Google Scholar]
- Millett, F., Miller, M.A., Geren, L., and Durham, B. 1995. Electron transfer between cytochrome c and cytochrome c peroxidase. J. Bioenerg. Biomembr. 27 341–351. [DOI] [PubMed] [Google Scholar]
- Morton, A. and Matthews, B.W. 1995. Specificity of ligand binding in a buried nonpolar cavity of T4 lysozyme: Linkage of dynamics and structural plasticity. Biochemistry 34 8576–8588. [DOI] [PubMed] [Google Scholar]
- Musah, R.A. and Goodin, D.B. 1997. Introduction of novel substrate oxidation into a heme peroxidase by cavity complementation: Oxidation of 2-aminothiazole and covalent modification of the enzyme. Biochemistry 36 11665–11674. [DOI] [PubMed] [Google Scholar]
- Musah, R.A., Jensen, G.M., Rosenfeld, R.J., Bunte, S.W., McRee, D.E., and Goodin, D.B. 1997. Variation in strength of a CH to O hydrogen bond in an artificial cavity. J. Am. Chem. Soc. 119 9083–9084. [Google Scholar]
- Musah, R.A., Jensen, G.M., Bunte, S.W., Rosenfeld, R.J., and Goodin, D.B. 2002. Artificial protein cavities as specific ligand-binding templates: Characterization of an engineered heterocyclic cation binding site that preserves the evolved specificity of the parent protein. J. Mol. Biol. 315 845–857. [DOI] [PubMed] [Google Scholar]
- Mutz, M.W., Case, M.A., Wishart, J.F., Ghadiri, M.R., and McLendon, G.L. 1999. De novo design of protein function: Predictable structure–function relationships in synthetic redox proteins. J. Am. Chem. Soc. 121 858–859. [Google Scholar]
- Nocek, J.M., Liang, N., Wallin, S.A., Mauk, A.G., and Hoffman, B.M. 1990. Low-temperature conformational transitional within the <Zn-cytochrome-C peroxidase, cytochrome-C>electron-transfer complex. J. Am. Chem. Soc. 112 1623–1625. [Google Scholar]
- Nocek, J.M., Zhou, J.S., Forest, S.D., Priyadarshy, S., Beratan, D.N., Onuchic, J.N., and Hoffman, B.M. 1996. Theory and practice of electron transfer within protein–protein complexes: Application to the multidomain binding of cytochrome c by cytochrome c peroxidase. Chem. Rev. 96 2459–2489. [DOI] [PubMed] [Google Scholar]
- Nocek, J.M., Leesch, V.W., Zhou, J.S., Jiang, M., and Hoffman, B.M. 2000. Multidomain binding of cytochrome c peroxidase by cytochrome c: Thermodynamic vs. microscopic binding constants. Isr. J. Chem. 40 35–46. [Google Scholar]
- Page, C.C., Moser, C.C., Chen, X., and Dutton, P.L. 1999. Natural engineering principles of electron tunnelling in biological oxidation-reduction. Nature 402 47–52. [DOI] [PubMed] [Google Scholar]
- Pappa, H.S., Tajbaksh, S., Saunders, A.J., Pielak, G.J., and Poulos, T.L. 1996. Probing the cytochrome c peroxidase–cytochrome c electron transfer reaction using site specific crosslinking. Biochemistry 35 4837–4845. [DOI] [PubMed] [Google Scholar]
- Pelletier, H. and Kraut, J. 1992. Crystal structure of a complex between electron transfer partners, cytochrome-c peroxidase and cytochrome-c. Science 258 1748–1755. [DOI] [PubMed] [Google Scholar]
- Sheldrick, G.M. and Schneider, T.R. 1997. SHELXL: High resolution refinement. Methods Enzymol. 277 319–343. [PubMed] [Google Scholar]
- Sivaraja, M., Goodin, D.B., Smith, M., and Hoffman, B.M. 1989. Identification by ENDOR of Trp191 as the free-radical site in cytochrome c peroxidase compound ES. Science 245 738–740. [DOI] [PubMed] [Google Scholar]
- Stemp, E.D. and Hoffman, B.M. 1993. Cytochrome c peroxidase binds two molecules of cytochrome c: Evidence for a low-affinity, electron-transfer-active site on cytochrome c peroxidase. Biochemistry 32 10848–10865. [DOI] [PubMed] [Google Scholar]
- Wang, J.M., Mauro, J.M., Edwards, S.L., Oatley, S.J., Fishel, L.A., Ashford, V.A., Xuong, N.H., and Kraut, J. 1990. X-ray structures of recombinant yeast cytochrome-c peroxidase and 3 heme–cleft mutants prepared by site-directed mutagenesis. Biochemistry 29 7160–7173. [DOI] [PubMed] [Google Scholar]
- Wilker, J.J., Dmochowski, I.J., Dawson, J.H., Winkler, J.R., and Gray, H.B. 1999. Substrates for rapid delivery of electrons and holes to buried active sites in proteins. Angew. Chem. Int. Ed. Engl. 38 90–92. [Google Scholar]
- Winkler, J.R. and Gray, H.B. 1997. Electron tunneling in proteins: Role of the intervening medium. J. Biol. Inorg. Chem. 2 399–404. [Google Scholar]
- Zhou, J.S. and Hoffman, B.M. 1994. Stern-Volmer in reverse: 2:1 Stoichiometry of the cytochrome c cytochrome c peroxidase electron-transfer complex. Science 265 1693–1696. [DOI] [PubMed] [Google Scholar]