

Abstract
The three-dimensional structures of leucine-rich repeat (LRR)-containing proteins from five different families were previously predicted based on the crystal structure of the ribonuclease inhibitor, using an approach that combined homology-based modeling, structure-based sequence alignment of LRRs, and several rational assumptions. The structural models have been produced based on very limited sequence similarity, which, in general, cannot yield trustworthy predictions. Recently, the protein structures from three of these five families have been determined. In this report we estimate the quality of the modeling approach by comparing the models with the experimentally determined structures. The comparison suggests that the general architecture, curvature, "interior/exterior" orientations of side chains, and backbone conformation of the LRR structures can be predicted correctly. On the other hand, the analysis revealed that, in some cases, it is difficult to predict correctly the twist of the overall super-helical structure. Taking into consideration the conclusions from these comparisons, we identified a new family of bacterial LRR proteins and present its structural model. The reliability of the LRR protein modeling suggests that it would be informative to apply similar modeling approaches to other classes of solenoid proteins.
Keywords: Crystal structure, leucine-rich repeat, molecular modeling, solenoid-like proteins, structural bioinformatics
Tandem arrays of characteristic leucine-rich repeat (LRR) motifs comprising ∼24 amino acids have been found in the primary structures of a large number of proteins. These proteins include many that participate in biologically important processes, such as hormone receptors, enzyme inhibitors, and proteins involved in cell adhesion (for review, see Kobe and Deisenhofer 1994, 1995; Buchanan and Gay 1996; Kobe 1996; Kajava 1998; Kobe and Kajava 2001). The first crystal structure of a LRR protein, ribonuclease inhibitor (RI) (Kobe and Deisenhofer 1993), showed that LRRs corresponded to structural units, consisting of a β-strand and an α-helix. The structural units are arranged so that all the β-strands and the helices are parallel to a common axis, resulting in a nonglobular, horseshoe-shaped molecule with curved parallel β-sheet lining the inner circumference of the horseshoe and the helices flanking its outer circumference. Therefore, the LRR proteins belong to a more general class of solenoid protein structures (Kobe and Kajava 2000).
The availability of the structure of RI and a large number of LRR sequences triggered studies focused on the analysis, classification (Claudianos and Campbell 1995; Buchanan and Gay 1996; Jones and Jones 1997) and molecular modeling of LRR proteins (Kajava et al. 1995; Moyle et al. 1995; Jiang et al. 1995; Bhowmick et al. 1996; Weber et al. 1996; Izzo 1997; Kajava 1998; Janosi et al. 1999; Montgomery et al. 2000; Hines et al. 2001). At least six families of LRR proteins, characterized by different lengths and consensus sequences of the repeats, have been identified (Kajava 1998). Eleven-residue segments of the LRRs (LxxLxLxxN/CxL), corresponding in RI to the β-strand and adjacent loop regions, are conserved in LRR proteins, whereas the remaining parts of the repeats (herein termed variable) may be very different (Kobe and Deisenhofer 1994; Kajava et al. 1995). Despite the differences, each of the variable parts contains two half-turns at both ends and a "linear" segment (as the chain follows a linear path overall), usually formed by a helix, in the middle. Therefore, although the invariant consensus sequence of the β-region is a characteristic feature of the entire LRR superfamily, the consensus sequences of the variable part suggest at least six specific families that may differ in the three-dimensional (3D) structure of the repeated unit. RI represents one of these families. The sequence–structure relationships of the remaining LRR families were examined by molecular modeling (Kajava 1998).
In general, prediction and modeling are not considered a source of reliable structural information. A structural model of any protein goes out of use as soon as its structure is determined by X-ray or NMR methods. However, the most valuable feature of the theoretical approaches resides in the fact that the prediction tests our understanding of protein structures. This is especially true when the structural model and principles underlying every modeling step are well documented. The comprehensively described modeling of LRR proteins (Kajava et al. 1995; Kajava 1998) provides an opportunity to test how adequate is our understanding of the principles of LRR structures as they are experimentally determined.
Recently, the 3D structures of eight new LRR proteins have been determined (Price et al. 1998; Hillig et al. 1999; Marino et al. 1999; Zhang et al. 2000; Liker et al. 2000; Schulman et al. 2000; Wu et al. 2000; Evdokimov et al. 2001) (Fig. 1 ▶). The yeast GTPase-activating protein rna1 (Hillig et al. 1999) is a member of RI-like family, and consequently resembles closely the structure of RI (Kobe and Deisenhofer 1993). The remaining LRR proteins belong to three LRR families (SDS22+, cysteine-containing (CC), and bacterial) for which no structural information had previously been available, except for the modeled structures (Kajava et al. 1995; Kajava 1998). In this paper we analyze the quality of the modeling by comparing the models with the experimentally determined structures. On the basis of this analysis and search of the latest sequence databases, we also present a new family of LRR proteins and its structural model.
Fig. 1.
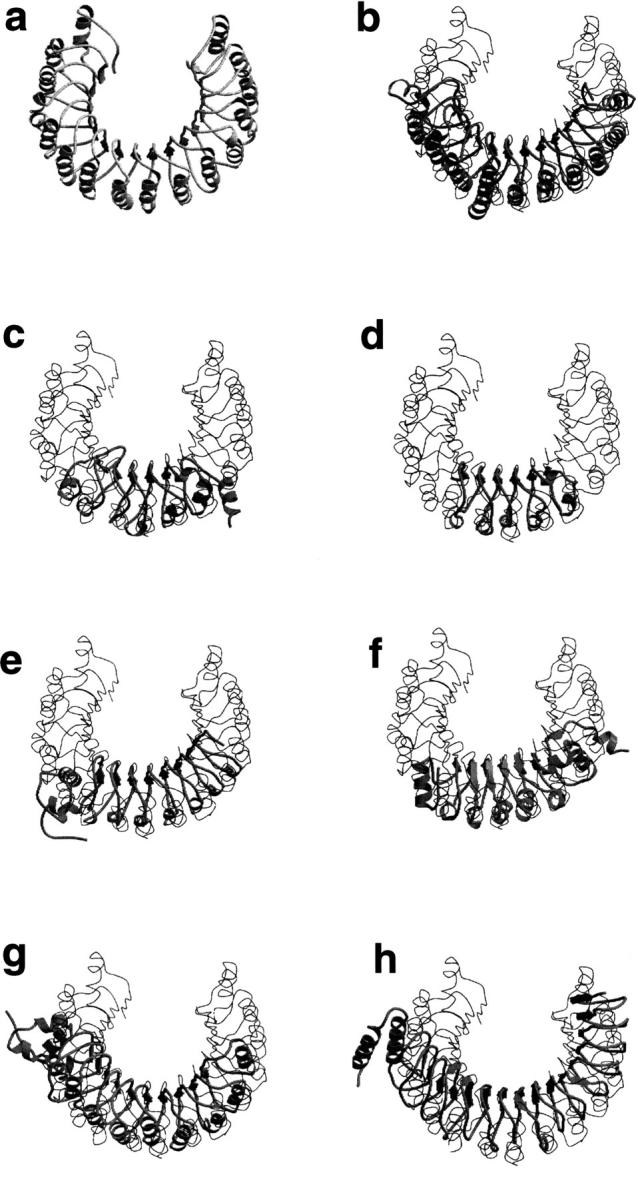
Superposition of the structures of (a) RI (PDB code 2BNH) and the recently determined LRR domains (ribbon): (b) rna1 (1YRG); (c) U2a (1A9N, residues 2–163), TAP protein is structurally very similar to U2a; (d) RabGGT(1DCE, residues 443–567); (e) internalin (1D0B); (f) dynein LC1(1DS9); (g) skp2 (1FQV, residues 107–400); (h) YopM. The structures were superimposed using Cα atoms of the β-strands.
Results and Discussion
Comparison of the models and the experimental structures from the SDS22+-like family
Our sequence analyses suggests that of the LRR proteins with available structures, U2a small nuclear protein (Price et al. 1998), TAP protein (Liker et al. 2000), Rab geranylgeranyltransferase (RabGGT) (Zhang et al. 2000), internalin B (InlB) (Marino et al. 1999), and light chain 1 (LC1) of dynein (Wu et al. 2000) belong to the LRR family, represented by the yeast SDS22+ protein (Malvar et al. 1992; Kajava et al. 1995) (Fig. 2 ▶). Inspection of these structures confirms that the less perfect the repetition of the sequence motif, the less regular the structure. Four of these proteins (U2a, TAP, RabGGT, and dynein LC1) have a small number (4 to 6) of imperfect repeats with the length ranging between 22 and 25 residues. In three dimensions, neither U2a, TAP, RabGGT, nor dynein LC1 have two identical LRR conformations within one domain. In contrast, internalin has seven well-conserved 22-residue repeats with almost the same conformation. The backbones of the 22-residue repeats from U2a, TAP, RabGGT, and internalin are very similar (Fig. 3a ▶). Surprisingly, however, some repeats that fit the consensus sequence of SDS22+-like LRRs show considerable differences in the backbone conformations of their "variable" regions, depending on the surrounding protein context (Table 1). There are several possible explanations, including the differences in the neighboring repeats and other flanking structures, and the presence of the conformationally restricted proline residues in certain positions. It may also be due to the difficulty to interpret the electron density in some flexible parts of the structures; for example, the B-factor of the backbone atoms of the U2a crystal structure is higher for residues 58–63 (∼50 Å2) compared to the adjacent regions (∼35 Å2). This is the region where the conformation of the 22-residue repeat of U2a protein differs most dramatically from the RabGGT and internalin repeats (Fig. 3a ▶). Furthermore, there are significant differences between the three structures determined crystallographically (Price et al. 1998; Marino et al. 1999; Zhang et al. 2000) on the one hand and the structure determined by NMR (dynein LC1; Wu et al. 2000) on the other (Fig. 3a ▶); smaller differences would usually be expected for structures with that degree of sequence similarity (Holm and Sander 1999).
Fig. 2.

Sequence alignments of the LRRs from proteins with the recently determined 3D structures; consensus sequences of the LRRs from SDS22+-like, typical, and RI-like protein families are also shown. Residues identical or conservatively substituted in >50% of the repeats of a given protein are printed in bold. The numbering refers to the position of the residue within the sequence. Boxes indicate residues directed into the interior of the known protein structures or models.
Fig. 3.
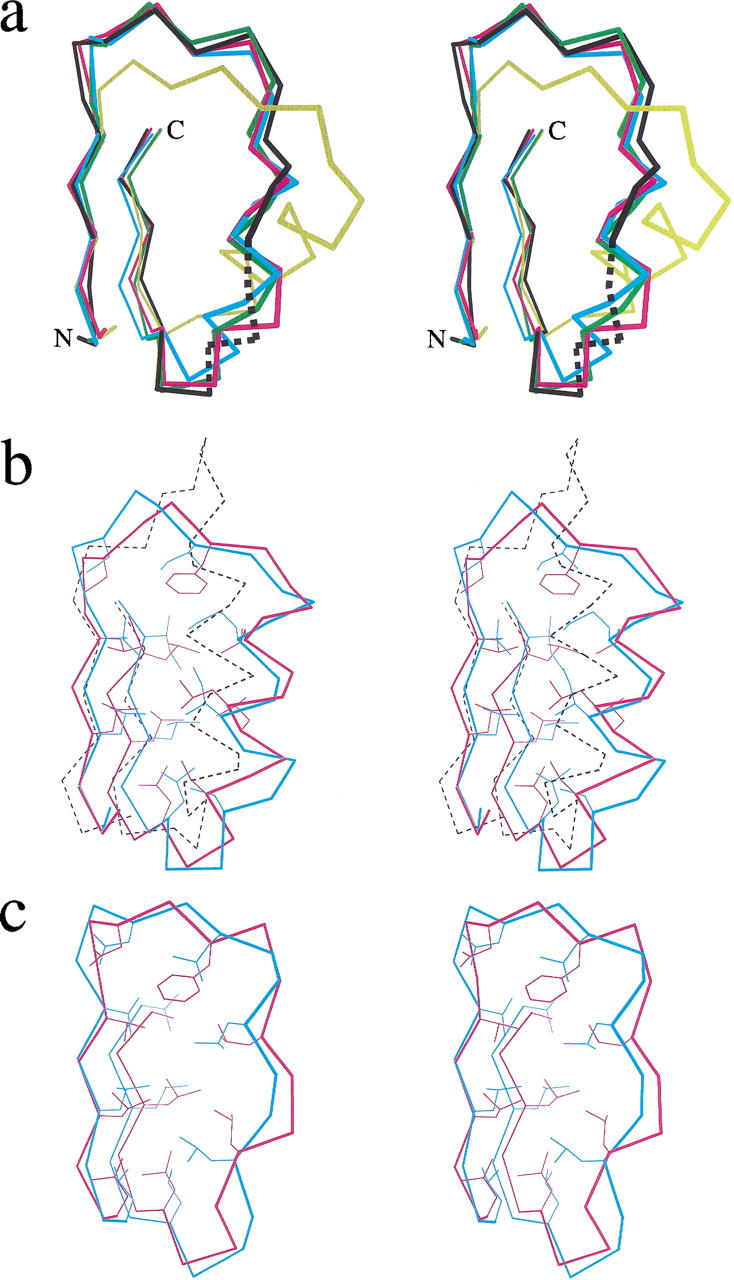
(a) Superposition of 28-residue long β×β units of the crystal structures of U2a (black), RabGGT (magenta), internalin (cyan), dynein LC1 (yellow), and the model of SDS22+ protein (green). Dotted line denotes the region of U2a structure with B-factors exceeding 50 Å2. All chosen repeats have the same 22-residue length and are located in the middle part (underlined on Fig. 2 ▶) of the LRR domains. The experimentally determined units were superimposed onto the modeled unit using all Cα-atoms, except for the dynein LC1 unit, which was superimposed using only the Cα-atoms of the β-strands. Only the Cα atoms are shown. (b) Stereodiagram showing the superposition of β×β units of the crystal structures of RI (dotted black), skp2 (magenta), and the model of ykk7 protein (cyan). The chosen repeat of skp2 has the same 26-residue length as the LRR from ykk7 protein and is located in the middle of the LRR domain (underlined on Fig. 4 ▶). The skp2 unit was superimposed on the modeled unit using all Cα-atoms, whereas the RI unit was superimposed on the CC LRRs using the Cα-atoms of the β-strands. All Cα atoms and the side chains of selected conserved residues are shown. (c) Superposition of β×β units of the crystal structure (magenta) and the model (cyan) of the YopM protein. The repeats have 20-residue length and are located in the middle of the LRR domains. The units were superimposed using all Cα-atoms. All Cα atoms and the side chains of selected conserved residues are shown.
Table 1.
Backbone conformations of modeled and observed LRRs

a The symbols α, αL, γ, β, and βp denote, respectively, residue backbone conformations closed to the right, left α-helical, and 310-helical conformations, the β-conformation and polyproline II helix conformations. Symbol ψ denotes unusual conformation with the dihedral angles φ = −140°; ξ = −160°.
b The arrows, lines, and rectangle denote β-strands, turn regions, and linear conformation of the variable region, respectively.
Before undertaking the experimental solution of four SDS22+-like LRR structures, a model of the 3D structure of LRR of the yeast SDS22+ protein was constructed (Kajava et al. 1995). The SDS22+ protein was chosen to represent this LRR family because it has regular and easily recognizable repeats and is one of the first identified LRR sequences from this family (Malvar et al. 1992). The β-structure portion of RI (Kobe and Deisenhofer 1993) was taken as a template for modeling the analogous part of SDS22+ LRRs. The modeling of the variable part of the LRRs was based on the assumption that the conserved residues of the LRR consensus patterns are important for structural integrity, rather than function. In the modeled structure, the conserved nonpolar residues are directed into the hydrophobic core, and the conserved polar residues are involved in specific hydrogen bonds in the interior of the structure; the residues in the nonconserved positions are exposed on the surface of the structure. The modeling showed that it was possible to build a horseshoe-shaped structure for the SDS22+ protein with the 22-residue LRRs (Kajava et al. 1995). The central part of the variable region was predicted to form a 5-residue helix; one amino-terminal residue of the helix was in α-helical conformation, whereas four others were in a 310-conformation (Table 1).
The comparison of the model with the crystal structures shows that the Cα-backbone of the model closely follows the backbone of the crystal structures (after superimposing β-strand–α-β-strand units from the model and central regions of the experimentally determined structures, the root mean square deviations (RMSD) of Cα atoms are 1.0, 1.1, and 1.5 Å for RabGGT, internalin, and U2a, respectively) (Fig. 3a ▶). The fact that the model fits the known structures as close as they match each other demonstrates the high accuracy of the prediction. Although the backbone conformations of the variable regions are similar, some differences exist in their linear segments (Table 1). The internalin structure is most similar to the SDS22+ model. Both molecules have several perfectly repeated 22-residue units (Fig. 3a ▶) and both structures have unusual helices that are closer to the 310-conformation than to the α-helical one. The modeling suggested that the second residue of the helix, which is frequently Asn, is necessary to form hydrogen bonds with NH-groups of the amino-terminal helical cap. None of the proteins with known structure has the conserved Asn in this position; however, internalin has conserved Asp in the position preceding the helix, and its side chain forms hydrogen bonds with the helical cap. This observation may be considered as a support of the prediction.
Comparison of experimental and predicted structures of LRRs from the cysteine-containing family
The crystal structure of skp2 LRR domain (Schulman et al. 2000) is the first determined structure of a protein from the cysteine-containing (CC) LRR family. The CC LRRs are usually 26-residues long and have a distinctive consensus sequence (Fig. 4 ▶; Kajava 1998). LRRs of skp2 vary in length between 23 and 27 residues. Despite these length differences, the convex part of the horseshoe structure is formed by similar α-helices containing two to three turns. Figure 3b ▶ shows a superposition of the 26-residue LRR from skp2 (A244–A276) and one of the modeled 26-residue LRRs (residues 80–112; Kajava 1998) from the hypothetical 54.9-kD protein from Caenorhabditis elegans (P34284), revealing a good match between these structures. The 33-atom Cα backbone of the model closely follows the backbone of the crystal structure (RMSD) of model/skp2 = 1.25 Å using Cα atoms). Again, the backbone conformations are similar not only in the β region, but also in the linear segment of the variable part (Table 1). The model suggested that the α-helix of the CC LRR is shifted relative to the LRR of RI (Kajava 1998); the structure of skp2 confirmed this prediction (Fig. 3b ▶). The model is able to explain correctly almost all residue conservations within the family. The exception is Asp in the first position of the α-helix. In the model, this residue conservation was explained by formation of specific hydrogen bonds between the side chain and its own NH group and the NH of the adjacent LRR to avoid burial of peptide groups in the nonpolar environment. However, in the crystal structure, the backbone NH groups are hydrogen bonded by water molecules. The side chains of the aspartic acid residues are exposed to the solvent and their conservation may be important for functionally important intermolecular interactions.
Fig. 4.
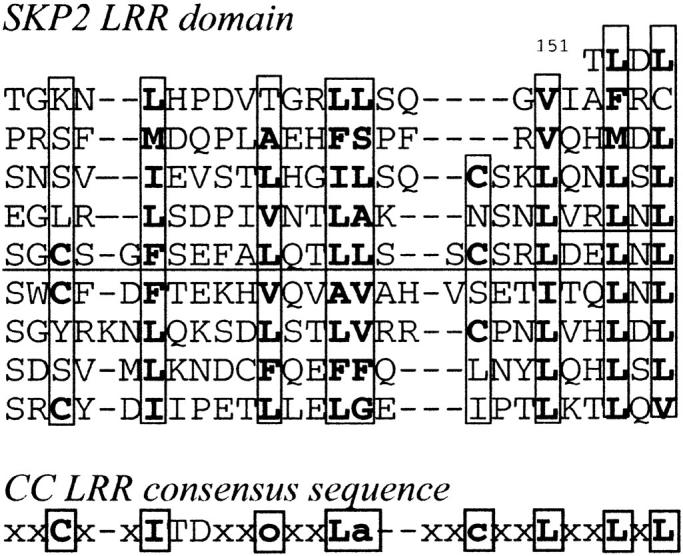
Sequence alignment of the LRRs from skp2 protein; the consensus sequence of LRRs from the CC LRR protein family is also shown. Residues identical or conservatively substituted in the repeats of skp2 and the CC LRR consensus sequence are in bold. The numbering refers to the position of the residue within the sequence. Boxes indicate residues directed into the interior of the known protein structures or models.
Comparison of experimental and predicted structures of LRRs from the bacterial family
The modeling suggested that the shortest known LRRs occurring in gram-negative bacteria have a horseshoe structure similar to other LRR proteins (Kajava 1998). The modeled 20-residue LRRs suggested a polyproline II helical conformation in the variable part, as opposed to α- or 310-helices found in other families (Table 1). The comparison of the model with the crystal structure of YopM from Yersinia pestis, a protein with 20-residue bacterial LRRs (Evdokimov et al. 2001) shows that the general architecture of the YopM structure was predicted correctly; both structures have a horseshoe shape with a linear part of the variable region adopting a polyproline conformation. The most striking difference is that, in contrast to the predicted flat horseshoe structure, it is twisted with its ends laying out of the plane. This twisted arrangement allows YopM molecules to wrap around each other and form tetramers in the crystal. However, it is not clear, whether the origin of the twist is intramolecular interactions of the YopM monomer or its crystal packing. The superposition of the modeled and observed 20-residue LRRs also reveals that small conformational differences for several residues lead to considerable differences in the orientation of the polyproline II helix segments (Fig. 3c ▶). The r.m.s.d. of the 26 Cα-atoms of the model and the structure is 1.57 Å. Further differences include the conserved Asp residue located on the polyproline II helix. In the model, the Asp side chain forms hydrogen bonds with two partially buried NH groups of the backbone. In contrast, the Asp side chain in the crystal structure faces the solvent and is probably important for the formation of intermolecular interactions.
Successes of leucine-rich repeat modeling
Prediction of the curvature
One basic assumption of the modeling was that all LRR structures contain a parallel β-sheet curved similarly to that observed in RI (Kobe and Deisenhofer 1993). LRRs have a highly conserved 11-residue stretch with the consensus sequence LxxLxLxxNxL, where the L positions can be occupied by bulky nonpolar residues, whereas the N position, in addition to asparagines, can also be occupied by cysteines. The polypeptide chain changes direction at a right angle preceding and after the four-residue β-region xLxL. Molecular modeling suggested that the curvature is caused by the first bulky nonpolar residue (usually leucine) and the conserved asparagine of the consensus pattern, which are directed into the interior of the structure. This was based on the comparative analysis of the sterically allowed distances between the neighboring β-strands and the half-turns of the LRRs. For this analysis, a fragment of the β-strand with the adjoining half-turns was taken from the RI crystal structure and duplicated by 4.8 Å translations (the optimal distance between β-strands) along the direction of the hydrogen bonds. Analysis of the generated model showed that the half-turns in the consecutive repeats made unfavorable short contacts with each other (Kajava et al. 1995). To alleviate these contacts, the β-sheet needed to be curved as the RI structure.
Remarkably, a number of investigators (Claudianos and Campbell 1995; Buchanan and Gay 1996; Heffron et al. 1998) proposed that the majority of LRR proteins have a β-helix structure similar to that first observed in pectate lyase (Yoder et al. 1993). The repeats of pectate lyase have similar lengths and contain a similar sequence motif (G/S)xxLxLxxNxL. However, the first bulky nonpolar residue of the 11-residue motif, which probably causes the curvature in LRR structures, is absent in pectate lyase. Instead, it typically has a small side chain in this position. Thus, modeling suggested that all LRR proteins, including the shortest bacterial LRR (Kajava 1998), have a curved superhelical structure and not a β-helical one. The crystal structures of U2 small nuclear protein, RabGGT, internalin, and YopM now provide strong support for this conclusion. The new data emphasize the utility of detailed analysis and modeling for structure prediction.
Interior/exterior orientation of side chains
In the modeled structures, the conserved nonpolar residues of the LRRs were directed into the hydrophobic core, based on the assumption that they are important for structural integrity, rather than functional reasons. At the same time, residues in the nonconserved positions were modeled to be exposed on the surface of the structure. The inspection of the LRR crystal structures supports the validity of this postulate. The interior/exterior orientations of the conserved apolar and nonconserved side chains were predicted correctly.
Backbone conformation
The backbone conformations of the modeled LRRs were generated based on the following considerations. First, the predictions of the interior/exterior orientations of the side chains were taken into account. Second, restraints on the general course of the polypeptide chain were imposed. Because each of the modeled variable fragments of the LRRs corresponds to a half-coil of the LRR superhelix, they must be bent to have two half-turns at both ends and a linear segment in the middle. A two-dimensional (2D) plot of the coil, which included information about possible side-chain orientations and the location of the half-turns, facilitated the conformational search. Third, the preference was given to the conformations that are frequently observed in protein structures and facilitate intrachain hydrogen bond formation (Efimov 1993). Possible conformations were also checked for their ability to accept proline, which is restricted to α or βP conformations, in positions where it was observed in at least one LRR.
The comparison of the modeled LRRs with analogous ones from the crystal structures reveals a good correspondence between the repeat backbones, especially those containing α or 310-helices (Fig. 3 ▶). In particular, the helix of the SDS22+ LRR model was predicted very similar to the observed 310-conformation (Fig. 3a ▶), and the model of CC LRR correctly predicted the α-helix and its shift relative to the helix of RI LRR (Fig. 3b ▶). The observed shift of the CC LRR helix confirms the prediction of the modeling that LRRs from the different families cannot occur concomitantly within one LRR domain (Kajava 1998). The orientations of the helices from different families, for example, from RI and CC, are different and cannot pack together well. In the case of the bacterial LRRs, the backbone conformation of the modeled and observed structures are also similar; however, the difference in the tilting of the polyproline helical segment is significant (Fig. 3c ▶).
Failures of leucine-rich repeat modeling
Predictions of side-chain rotamers and loop conformations
Side chains usually have a unique conformation and form a specific network of noncovalent contacts when occurring in the interior of a protein structure. The inability to predict the correct side-chain rotamer is a common problem for molecular modeling. Although the orientations of the conserved apolar side chains in the core of the LRR models were predicted correctly, the precise conformations and noncovalent contacts of some did not match the ones observed experimentally (Fig. 3b,c).
The model and the crystal structure of YopM show the highest deviation of the backbone conformations. In the crystal structure, the conserved Asn residue of YopM LRR has an unusual backbone conformation (φ = −140°; Ψ = −160°), because one of the basic approaches adopted for the modeling was to use the most frequently observed conformations, the true conformation was not anticipated. Many peptide groups of the loop regions do not form intrachain hydrogen bonds in the experimentally determined structures. Instead, they are connected by water molecules. This is another origin of difference between the modeled and observed backbone conformations.
Intercalation of the negatively charged side chains between the leucine-rich repeats
In the modeled structures of CC and bacterial LRRs, the conserved negatively charged Asp side chains were involved in specific hydrogen bonds with NH groups of the backbone. This prediction was based on the assumption that these conserved residues are important for structural integrity. However, this assumption was not correct because, in the determined structures, the Asp side chains are exposed to the solvent. Perhaps these residues may affect the structure in a different manner; for example, their charge repulsion may twist the overall LRR structure as in YopM (Evdokimov et al. 2001) or they may be important for function of the protein. The observation that the conserved negatively charged residues are exposed to the solvent and not involved in intramolecular hydrogen bonds can improve the results of the modeling of LRR proteins.
Prediction of the overall structures
The models containing two to three LRRs closely fit the experimentally determined structures. However, if we compare the overall structures consisting of 10 to 15 LRRs, the accumulation of small differences in LRR conformations and packing results in larger discrepancies. For example, the known horseshoe structures of internalin (Marino et al. 1999) and YopM (Evdokimov et al. 2001) have a right-handed twist (Kobe and Kajava 2000). The observed twist was not foreseen by the model. The crystal structures do not provide a clear explanation of the interactions responsible for the twisting. Both twisted LRR structures have a position in the variable region of LRR occupied by negatively charged residues (e.g., see Fig. 2 ▶ for LRRs of internalin). We propose that the repulsion of these side chains may lead to the twist; however, this explanation requires further analysis.
Identification and modeling of LRRs from a new bacterial family
The molecular modeling and analysis of newly determined structures of LRR proteins suggested that the presence of a bulky apolar residue in the first position of the conserved pattern LxxLxLxxN/CxL is sufficient to impart the characteristic horseshoe curvature (see Prediction of the curvature section). Taking this into consideration we modified the LRR sequence profiles (Kajava 1998) by increasing the importance of the first six residues LxxLxL. The profile search revealed a group of bacterial proteins with 23-residue repeats having a consensus pattern LxxLxLxxxLxxIgxxAFxxC/Nxx (Fig. 5a ▶). A protein from Treponema pallidum (TpLRR) was sequenced first among these proteins (Shevchenko et al. 1997); therefore, we name this group of proteins as the TpLRR family. The TpLRR family also includes BspA from Bacteroides forsythus (Sharma et al. 1998), PcpA from Streptococcus pneumoniae (Sanches-Beato et al. 1998), and a hypothetical protein GI:13622014 from Streptococcus pyogenes (Feretti et al. 2001).
Fig. 5.

A new TpLRR family. (a) A consensus sequence repeat pattern. The "C" position can also be occupied by asparagine, threonine, or serine; "x" is any residue, sites of insertions and deletions are denoted by + and −. (b) 2D plot of the predicted side-chain orientations within one coil of the LRR superhelix. Open circles denote any residues and magenta circles denote conserved residues. Location of the circles inside the coil contour indicates occurrence in the interior of the structure. (c) Structural model of TpLRR. The β-structural region is in blue; the α-helix is in green; and conserved residues are in magenta.
Applying the method used in the previous modeling work (Kajava et al. 1995) we build the 3D structure of one of these proteins—BspA from B. forsythus (Sharma et al. 1998). The modeling shows that proteins with such repeats can adopt the horseshoe structure (Fig. 5c ▶). It explains the observed residue conservation and insertion/deletions within the repeats. Thus, we identified a likely new family of the LRR proteins with 23-residue repeats and a "C/N inverted" C/NxxLxxLxL consensus pattern. The exact functions of these cell surface-associated proteins are currently unknown; however, it has been proposed that TpLRR protein may facilitate interactions between components of the T. pallidum cell envelope (Shevchenko et al. 1997). BspA of B. forsythus may play roles in its adherence to oral tissues and triggering of host immune responses (Sharma et al. 1998).
Conclusions
Comparison of the experimental structures of LRRs from three different families with the models (built before the experimental information was available; Kajava 1998) allows us to estimate the level of our understanding of LRR structures and the reliability of molecular modeling in this system. We compared the modeled and experimentally determined 3D structures from SDS22+-like, cysteine-containing, and bacterial LRR protein families. The comparison suggests that the general architecture, curvature, interior/exterior orientations of side chains, and even backbone conformation of the LRR structures can be predicted correctly. The inspection also validated several assumptions that underlay the modeling; these assumptions include the interior orientation of the conserved nonpolar residues of LRRs; the location of the nonconserved residues on the surface; and the assumption that the location of turns and linear segments within the repeat can be properly predicted.
There are at least two families of LRR proteins (typical and plant specific) for which no experimental structural information is currently available. Our comparison of models and structures suggests that when the LRR structures for these families are determined, the general structural characteristics of the models will be similar to the experimental ones.
The molecular modeling also suggested that the conserved pattern LxxLxL, which is shorter than the previously proposed LxxLxLxxN/CxL (Kobe and Deisenhofer 1994; Kajava et al. 1995), is sufficient to impart the characteristic horseshoe curvature to proteins with 20- to 30-residue repeats. Taking this into consideration, we identified a new family of bacterial LRR proteins and build its structural model.
On the other hand, the analysis revealed the limitations of molecular modeling and the limits of our current understanding of LRR structures. Building a model of an LRR protein, one should be aware that the correct side-chain rotamers may be difficult to predict, that it is difficult to discriminate when the conserved polar side chains are exposed to the solvent rather than involved in hydrogen bonding of the adjacent LRRs, and that the overall superhelical structure may in some instances have a right-handed twist.
Similar limitations exist for modeling of proteins in general. However, modeling of solenoid structures is both simpler and can be applied to proteins with more limited overall sequence similarity. The genome sequencing projects are revealing a considerable number of protein sequences with tandem repeats (Marcotte et al. 1999) and deciphering the sequence–structure–function relationship of such proteins promises to be a special subject of structural bioinformatics (Kajava 2001). We believe that it would be very informative to apply similar modeling approaches and analyses to other classes of solenoid proteins (Kobe and Kajava 2000).
Materials and methods
Sequence profile search
To identify additional LRR proteins we modified previously generated LRR sequence profiles (Kajava 1998), increasing the weight of the LxxLxL pattern and applied the sequence profile search (Bucher et al. 1996) against a recent release of the GENPEPT database (Benton 1990). In addition to the members of the already described families, we extracted a number of LRR proteins, which cannot be assigned to any known LRR families. These TpLRRs were aligned, a new profile was constructed, and the profile search was reapplied to enlarge collections of LRRs corresponding to this family. The profile contained more than two repeats being flanked by LxxLxL sequences of the β-regions. Construction of LRR profiles, spanning more than one repeat, increased the selectivity of the database search. The probability of error was P < 0.001, and was calculated by analyzing the score distribution obtained from a profile search against a regionally randomized version of the protein database, assuming an extreme value distribution (Hofmann and Bucher 1995).
Molecular modeling
The sequence analysis shows that the best template for constructing the TpLRR is a hybrid structure containing the α-helix–loop-β-strand region (positions 1–6 and 17–23 of the consensus sequence; Fig. 5 ▶) taken from the crystal structure of skp2 (Schulman et al. 2000), merged with a fragment from the typical LRR consisting of the loop region located before the α-helix (positions 10–16 of TpLRR). The modeling task was then limited to the construction of the three-residue connection between residues 6 and 10. The conformations of the analyzed connections were manually adjusted, by varying backbone torsion angles. The generated conformations were analyzed taking into account the requirements for side-chain orientations, sterical tension, and hydrogen bonding determined on the basis of the considerations described earlier (Kajava et al. 1995). The initial structure was constructed using the BIOPOLYMER and HOMOLOGY modules of Insight II program (Dayring et al. 1986). The resulting structure was subjected to the 300 steps of minimization based on the steepest descent algorithm with the backbone atoms of α-helical segments restrained to their starting positions with force constant K = 100. The next 500 steps of the refinement were performed without any restraints, using conjugate gradients algorithm. The CHARMM force field (Brooks et al. 1983) and the distance-dependent dielectric constant were used for the energy calculations. The program PROCHECK (Laskowski et al. 1993) was used to check the quality of the modeled structure. Figures 1 and 3 ▶ ▶ were generated by Molscript program (Kraulis 1991). The atomic coordinates of the modeled LRR structures are available over World Wide Web http://cmm.info.nih.gov/kajava.
Acknowledgments
We thank D.S. Waugh and A.G. Evdokimov for providing us with the coordinates of the crystal structure of YopM protein before the publication of this structure. B.K. is a Wellcome Senior Research Fellow in Medical Science in Australia.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
3D, three-dimensional
CC, cysteine-containing
LRR(s), leucine-rich repeat(s)
PS, plant-specific
RI, ribonuclease inhibitor
TpLRR, Treponema pallidum LRR
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.4010102.
References
- Benton, D. 1990. Recent changes in the GenBank On-line Service. Nucleic Acids Res. 18 1517–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhowmick, N., Huang, J., Puett, D., Isaacs, N.W., and Lapthorn, A.J. 1996. Determination of residues important in hormone binding to the extracellular domain of the luteinizing hormone/chorionic gonadotropin receptor by site-directed mutagenesis and modeling. Mol. Endocrinol. 10 1147–1159. [DOI] [PubMed] [Google Scholar]
- Brooks, B.R., Bruccoleri, R.E., Olafson, B.D., States, D.J., Swaminathan, S., and Karplus, M. 1983. CHARMM: A program for macromolecular energy minimization, and dynamics calculations. J. Computat. Chem. 4 187–217. [Google Scholar]
- Buchanan, S.G. and Gay, N.J. 1996. Structural and functional diversity in the leucine-rich repeat family of proteins. Prog. Biophys. Mol. Biol. 65 1–44. [DOI] [PubMed] [Google Scholar]
- Bucher, P., Karplus, K., Moeri, N., and Hofmann, K. 1996. A flexible motif search technique based on generalized profiles. Comput. Chem. 20 3–23. [DOI] [PubMed] [Google Scholar]
- Claudianos, C. and Campbell, H.D. 1995. The novel flightless-I gene brings together two gene families, actin-binding proteins related to gelsolin and leucine-rich-repeat proteins involved in Ras signal transduction. Mol. Biol. Evol. 12 405–414. [DOI] [PubMed] [Google Scholar]
- Dayring, H.E., Tramonato, A., Sprang, S.R., and Fletterick, R.J. 1986. Interactive program for visualization and modeling of proteins, nucleic acids and small molecules. J. Mol. Graph. 4 82–87. [Google Scholar]
- Efimov, A.V. 1993. Standard structures in proteins. Prog. Biophys. Mol. Biol. 60 201–239. [DOI] [PubMed] [Google Scholar]
- Evdokimov, A.G., Anderson, D.E., Routzahn, K.M., and Waugh, D.S. 2001. Crystal structure of Yopm-leucine rich effector protein from Yersinia pestis. J. Mol. Biol. 312 807–821. [DOI] [PubMed] [Google Scholar]
- Ferretti, J.J., McShan, W.M., Adjic, D., Savic, D., Savic, G., Lyon, K., Primeaux, C., Surorov, A.N., Kenton, S., Lai, H., Lin, S., Jia, H.G., Najar, F.Z., Ren, Q., Zhu, H., Song, L., White, J., Yuan, X., Clifton, S.W., Roe, B.A., and McLaughlin, R.E. 2001. Proc. Natl. Acad. Sci U.S.A. 98 4658–4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heffron, S., Moe, G.R., Sieber, V., Mengaud, J., Cossart, P., Vitali, J., and Jurnak, F. 1998. Sequence profile of the parallel beta helix in the pectate lyase superfamily. J. Struct. Biol. 122 223–235. [DOI] [PubMed] [Google Scholar]
- Hillig, R.C., Renault, L., Vetter, I.R., Drell, T. 4th, Wittinghofer, A., and Becker, J. 1999. The crystal structure of rna1p: A new fold for a GTPase-activating protein. Mol. Cell 3 781–791. [DOI] [PubMed] [Google Scholar]
- Hines, J., Skrzypek, E., Kajava, A.V., and Straley, S.C. 2001. Structure–function analysis of Yersinia pestis YopM's interaction with alpha-thrombin to rule on its significance in systemic plague and to model YopM's mechanism of binding host proteins. Microb. Pathog. 30 193–209. [DOI] [PubMed] [Google Scholar]
- Hofmann, K. and Bucher, P. 1995. The FHA domain: A putative nuclear signalling domain found in protein kinases and transcription factors. Trends Biochem. Sci. 20 347–349. [DOI] [PubMed] [Google Scholar]
- Holm, L. and Sander, C. 1999. Protein folds and families: Sequence and structure alignments. Nucleic Acids Res. 27 244–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iozzo, R.V. 1997. The family of the small leucine-rich proteoglycans: Key regulators of matrix assembly and cellular growth. Crit. Rev. Biochem. Mol. Biol. 32 141–174. [DOI] [PubMed] [Google Scholar]
- Janosi, J.B., Ramsland, P.A., Mott, M.R., Firth, S.M., Baxter, R.C., and Delhanty, P.J. 1999. The acid-labile subunit of the serum insulin-like growth factor-binding protein complexes. Structural determination by molecular modeling and electron microscopy. J. Biol. Chem. 274 23328–23332. [DOI] [PubMed] [Google Scholar]
- Jiang, X., Dreano, M., Buckler, D.R., Cheng, S., Ythier, A., Wu, H., Hendrickson, W.A., and El Tayar, N. 1995. Structural predictions for the ligand-binding region of glycoprotein hormone receptors and the nature of hormone-receptor interactions. Structure 3 1341–1353. [DOI] [PubMed] [Google Scholar]
- Jones, D.A. and Jones, J.D.G. 1997. The role of leucine-rich repeat proteins in plant defences. Adv. Botanical Res. 24 90–167. [Google Scholar]
- Kajava, A.V. 1998. Structural diversity of leucine-rich repeat proteins. J. Mol. Biol. 277 519–527. [DOI] [PubMed] [Google Scholar]
- ———. 2001. Proteins with repeated sequence: Structural prediction and modeling. J. Struct. Biol. 134 132–144. [DOI] [PubMed] [Google Scholar]
- Kajava, A.V., Vassart, G., and Wodak, S.J. 1995. Modeling of the three-dimensional structure of proteins with the typical leucine-rich repeats. Structure 3 867–877. [DOI] [PubMed] [Google Scholar]
- Kobe, B. 1996. Leucines on a roll. Nat. Struct. Biol. 3 977–980. [DOI] [PubMed] [Google Scholar]
- Kobe, B. and Deisenhofer, J. 1993. Crystal structure of porcine ribonuclease inhibitor, a protein with leucine-rich repeats. Nature 366 751–756. [DOI] [PubMed] [Google Scholar]
- ———. 1994. The leucine-rich repeat: A versatile binding motif. Trends Biochem. Sci. 19 415–421. [DOI] [PubMed] [Google Scholar]
- ———. 1995. Proteins with leucine-rich repeats. Curr. Opin. Struct. Biol. 5 409–416. [DOI] [PubMed] [Google Scholar]
- Kobe, B. and Kajava, A.V. 2000. When protein folding is simplified to protein coiling: The continuum of solenoid protein structures. Trends Biochem. Sci. 25 509–515. [DOI] [PubMed] [Google Scholar]
- ———. 2001. The leucine-rich repeat as a protein recognition motif. Curr. Opin. Struct. Biol. 11 725–732. [DOI] [PubMed] [Google Scholar]
- Kraulis, P.J. 1991. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallog. 24 946–950. [Google Scholar]
- Laskowski, R.A., McArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Cryst. 26 282–291. [Google Scholar]
- Liker, E., Fernandez, E., Izaurralde, E., and Conti, E. 2000. The structure of the mRNA export factor TAP reveals a cis arrangement of a non-canonical RNP domain and an LRR domain. EMBO J. 19 5587–5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malvar, T., Biron, R.W., Kaback, D.B., and Denis, C.L. 1992. The CCR4 protein from Saccharomyces cerevisiae contains a leucine-rich repeat region which is required for its control of ADH2 gene expression. Genetics 132 951–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotte, E.M., Pellegrini, M., Yeates, T.O., and Eisenberg, D. 1999. A census of protein repeats. J. Mol. Biol. 293 151–160. [DOI] [PubMed] [Google Scholar]
- Marino, M., Braun, L., Cossart, P., and Ghosh, P. 1999. Structure of the lnlB leucine-rich repeats, a domain that triggers host cell invasion by the bacterial pathogen L. monocytogenes. Mol. Cell. 4 1063–1072. [DOI] [PubMed] [Google Scholar]
- Montgomery, J., Ilg, T., Thompson, J.K., Kobe, B., and Handman, E. 2000. Identification and predicted structure of a leucine-rich repeat motif shared by Leishmania major proteophosphoglycan and Parasite Surface Antigen 2. Mol. Biochem. Parasitol. 107 289–295. [DOI] [PubMed] [Google Scholar]
- Moyle, W.R., Campbell, R.K., Rao, S.N., Ayad, N.G., Bernard, M.P., Han, Y., and Wang, Y. 1995. Model of human chorionic gonadotropin and lutropin receptor interaction that explains signal transduction of the glycoprotein hormones. J. Biol. Chem. 270 20020–20031. [DOI] [PubMed] [Google Scholar]
- Price, S.R., Evans, P.R., and Nagai, K. 1998. Crystal structure of the spliceosomal U2B"–U2A` protein complex bound to a fragment of U2 small nuclear RNA. Nature 394 645–650. [DOI] [PubMed] [Google Scholar]
- Sanchez-Beato, A.R., Lopez, R., and Garcia, J.L. 1998. Molecular characterization of PcpA: A novel choline-binding protein of Streptococcus pneumoniae. FEMS Microbiol. Lett. 164 207–214. [DOI] [PubMed] [Google Scholar]
- Schulman, B.A., Carrano, A.C., Jeffrey, P.D., Bowen, Z., Kinnucan, E.R., Finnin, M.S., Elledge, S.J., Harper, J.W., Pagano, M., and Pavletich, N.P. 2000. Insights into SCF ubiquitin ligases from the structure of the Skp1–Skp2 complex. Nature 408 381–386. [DOI] [PubMed] [Google Scholar]
- Shevchenko, D.V., Akins, D.R., Robinson, E., Li, M., Popova, T.G., Cox, D.L., and Radolf, J.D. 1997. Molecular characterization and cellular localization of TpLRR, a processed leucine-rich repeat protein of Treponema pallidum, the syphilis spirochete. J. Bacteriol. 179 3188–3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber, I.T., Harrison, R.W., and Iozzo, R.V. 1996. Model structure of decorin and implications for collagen fibrillogenesis. J. Biol. Chem. 271 31767–31770. [DOI] [PubMed] [Google Scholar]
- Wu, H., Maciejewski, M.W., Marintchev, A., Benashski, S.E., Mullen, G.P., and King, S.M. 2000. Solution structure of a dynein motor domain associated light chain. Nature Struct. Biol. 7 575–579. [DOI] [PubMed] [Google Scholar]
- Yoder, M.D., Keen, N.T., and Jurnak, F. 1993. New domain motif: The structure of pectate lyase C, a secreted plant virulence factor. Science 260 1503–1507. [DOI] [PubMed] [Google Scholar]
- Zhang, H., Seabra, M.C., and Deisenhofer, J. 2000. Crystal structure of Rab geranylgeranyltransferase at 2.0 A resolution. Structure Fold Des. 8 241–251. [DOI] [PubMed] [Google Scholar]