

Abstract
Oligomerization of fibroblast growth factors (FGFs) induced on binding to heparin or heparan sulfate proteoglycan is considered to be crucial for receptor activation and initiation of biological responses. To gain insight into the mechanism of activation of the receptor by FGFs, in the present study we investigate the effect(s) of interaction of a heparin analog, sucrose octasulfate (SOS), on the structure, stability, and biological activities of a recombinant acidic FGF from Notophthalmus viridescens (nFGF-1). SOS is found to bind to nFGF-1 and significantly increase the thermodynamic stability of the protein. Using a variety of techniques such as size-exclusion chromatography, sedimentation velocity, and multidimensional nuclear magnetic resonance (NMR) spectroscopy, it is shown that binding of SOS to nFGF-1 retains the protein in its monomeric state. In its monomeric state (complexed to SOS), n-FGF-1 shows significant cell proliferation activity. 15N and 1H chemical shift perturbation and the intermolecular nuclear Overhauser effects (NOEs) between SOS and nFGF-1 reveal that the ligand binds to the dense, positively charged cluster located in the groove enclosed by β-strands 10 and 11. In addition, molecular modeling based on the NOEs observed for the SOS-nFGF-1 complex, indicates that SOS and heparin share a common binding site on the protein. In conclusion, the results of the present study clearly show that heparin-induced oligomerization of nFGF-1 is not mandatory for its cell proliferation activity.
Keywords: nFGF-1, β-barrel, stability, oligomerization, heparin binding, receptor
Fibroblast growth factors (FGFs) are a large family of monomeric growth factors that control proliferation, differentiation, and migration of a variety of cell types and can act as potent stimulators of angiogenesis and promote wound healing (Bikfalvi 1997; Conrad 1998; Friesel and Maciag 1999). At the cell surface, FGFs bind to two receptor classes with high and low affinity, respectively (Ornitz and Itoh 2001; Fernig and Gallagher 1994; Spivak-Kroizman et al. 1994). The high affinity receptors include a family of transmembrane signaling receptors with intrinsic protein kinase activity (Mohammadi et al. 1996; Plotnikov et al. 1999; Plotnikov et al. 2000; Stauber et al. 2000). Dimerization of the fibroblast growth factor receptor (FGFR) is proposed to be a key event in the transduction of the FGF-induced cellular signals (Ornitz 2000; Schlessinger et al. 2000). Dimerization of the extracellular domains of the receptor is believed to result in juxtaposition of the cytoplasmic domains and subsequent transautophosphorylation leading to the stimulation of the intrinsic catalytic activity of the receptor (Bellot et al. 1991; Schlessinger et al. 1995; McEwen et al. 1996; Ornitz et al. 1995). The low affinity receptor of FGFs on the cell surface is the glycosidic moiety of the heparan sulfate proteoglycan on the cell coat and basal membranes (Ornitz et al. 1992; Mach et al. 1993; Wong et al. 1995; Faham et al. 1996). It is generally believed that cell surface heparin-like molecules are required for FGFs binding to their high affinity receptor (Faham et al. 1996; Ornitz et al. 1995; Waksman and Herr 1998; Lyon and Gallagher 1998). Intact cells pretreated with heparanases fail to show FGF binding and consequently do not proliferate (Basilico et al. 1992; Moscatelli 1992). However, Roghani et al. (1994), expressing FGFR in mutant Chinese hamster ovary cells deficient in heparan sulfate synthesis, showed that basic FGF (FGF-2) per se does not require heparin/heparan sulfate to bind to FGFR. Similarly, recent studies of Venkataraman et al. (1999) and Davies et al. (1999) show that FGF-2 in the absence of heparin self-associates preferentially to a biologically active oligomerized state, and heparin is found to merely stabilize the oligomerized state of FGF.
Several mechanisms have been proposed to explain the role of heparin in the FGF-FGFR interactions that trigger cell proliferative responses. Proteoglycans such as heparin/heparan sulfate are believed to stabilize FGFs and protect them from heat inactivation and proteolytic degradation and consequently enhancing their (FGF's) functional efficacy (Sommer et al. 1989; Samuel et al. 2000). Saksela et al. (1988) indicated (on the basis of the plasminogen activator-mediated proteolytic activity on cultured endothelial cells) that FGF-2 bound to cell matrix proteoglycans serves as a reservoir of growth factors that can be released by enzymes that degrade the proteoglycans. In a related study, Ornitz et al. (1995) (investigating the optimal conditions for FGF-2 to elicit its desired mitogenic activity) proposed that heparin promotes oligomerization of FGFs by providing a template for binding of multiple growth factor molecules. Once oligomerized through heparin binding, two molecules of FGF have the potential to juxtapose two molecules of FGFR and consequently dimerize.
Sucrose octasulfate (SOS) is a polysulfated heparin analog and has been successfully used as an antiulcer drug (Szabo 1991;Volkin et al. 1993). SOS has been postulated to have effects on growth factor-mediated repair of gastric tissue (Volkin et al. 1993). Although the mechanism of action of this drug is poorly understood, it is proposed that the cell-repair activity of SOS stems from its in situ interaction with FGFs and protection of the growth factors against acid-induced inactivation (Szabo 1991).
In this study, we investigate the effect(s) of SOS and its derivatives on the structure, stability, and biological activity of acidic fibroblast growth factor (nFGF-1) derived from Newt (Notophthalamus viridescens). The data obtained herein clearly indicate that oligomerization of nFGF-1 is not a prerequisite for its cell proliferation activity. In addition, the results of this study indicate that polysulfonated ligands such as heparin and SOS might just confer protection against acid-induced inactivation and proteolytic agents and consequently provide increased opportunity to the FGF molecules to interact with their receptors.
Results and Discussion
Acidic fibroblast growth factors (FGFs-1) derived from the newt and the human sources bear more than 85% sequence homology (Patrie et al. 1997; Poulin et al. 1997). The mitogenic activities of FGFs from these two sources are mostly similar (Patrie et al. 1997). Homology modeling of the FGFs-1 analogue derived from newt (nFGF-1) and human (hFGF-1, Zhu et al. 1993) sources show that their three-dimensional structures bear very good resemblance (Suppl. Fig. S1). The secondary structural elements in the protein (nFGF-1) include 12 β-strands arranged into a β-trefoil motif. A dense cluster composed of positively charged residues located at the loop between β-strands 10 and 11 constitutes the heparin-binding site (Zhu et al. 1991 and 1993; Blaber et al. 1996).
The equilibrium thermal unfolding (monitored by far-ultraviolet circular dichroism [far-UV CD]) of nFGF-1 is examined in the presence of heparin and its polysulfated analogs such as SOS, sucrose heptasulfate (SHpS), and sucrose hexasulfate (SHxS). It could be discerned that all the ligands used increase the thermal stability of the protein. The order of stability of the protein in the presence of various ligands in terms of the Tm (temperature at which 50% of the molecules are unfolded, ΔGU = 0) is heparin (Tm = 63 ± 0.5°C), SOS (Tm = 62 ± 0.5°C), SHpS (Tm = 58 ± 0.4°C), and SHxS (Tm = 53 ± 0.3°C). The Tm for the thermal unfolding of the protein in the absence of any of the ligands is 42 ± 0.3°C. It appears that SOS and heparin stabilize the protein against thermal denaturation to a similar extent and among the sulfated derivatives of sucrose; the degree of stability offered to the protein appears to be a direct function of the extent of sulfation of the ligand.
SOS enhances the angiogenic and mitogenic activities of nFGF-1
It is well documented that FGFs promote cell proliferation. In the presence of SOS and heparin, the cell proliferation activity of nFGF-1 increases significantly (Fig. 1A ▶). The less sulfated analogs of heparin (SHpS and SHxS) are also found to enhance the mitogenic activity of nFGF-1 (data not shown). The degree of potentiation appears to be directly correlated to the extent of sulfation of the ligands. In the absence of the ligands, nFGF-1 shows weak mitogenic activity (Fig. 1A ▶). The low mitogenic activity of nFGF-1 (in the absence of the ligands) could be caused by partial heat inactivation at physiological temperature (free nFGF has a low Tm of 42 ± 0.3°C) or by proteolytic cleavage of free nFGF-1 at the cell surface (Saksela et al. 1988; Sommer and Rifkin 1989). This aspect is evident from the results of the limited proteolytic digestion experiments (using a cocktail of proteases) that show that nFGF-1 in its free form is highly susceptible to proteolytic cleavage (Suppl. Fig. S2). In contrast, the protein in the presence of heparin and the sulfated derivatives of sucrose is resistant to cleavage. The degree of protection against proteolytic cleavage is proportional to the extent of sulfation of the ligand. It should be mentioned that available reports on the effects of SOS on the mitogenic activity of FGFs are contradictory. Volkin et al. (1993), examining the antiulcer activity of SOS, reported that the cell proliferation activity of (hFGF-1) on Balb/c-3T3 mouse fibroblast cell lines is significantly enhanced in the presence of SOS. Interestingly, Spivak-Kroizman et al. (1994), investigating the role of proteoglycans and their synthetic analogs on the cell proliferation activity of hFGF-1, found that SOS completely inhibits the mitogenic activity of the protein in cultured Chinese hamster ovary and BaF3 cell lines. We are unable to currently offer a concrete explanation for the observed discrepancy, but it appears that the action(s) of SOS is/are dependent on the nature of the cultured cell lines used for the proliferation assay.
Fig. 1.

(A) Representation of the mitogenic activity (on NIH/3T3 cells) of nFGF-1 and nFGF-1 in the presence of various concentrations of heparin and sucrose octasulfate (SOS). The concentration of the protein used in the mitogenic assay was 1 ng/mL. About 3 × 105 cells/well were used in the assay of each of the samples. Appropriate positive and negative controls were used to evaluate the authenticity and sensitivity of the assay. (B) Stimulatory effects of SOS on nFGF-1-induced proliferating cellular nuclear antigen (PCNA) expression level. The PCNA protein levels of rat thoracic aorta smooth muscle cells pretreated with sodium chlorate for 48 h to deprive endogenous heparin were found to increase most significantly by nFGF-1 plus SOS (F + S) as compared with the negative control (0.5% serum). Although nFGF-1 plus heparin (F + H) showed an even higher expression level of PCNA as compared with the positive control (15% serum), such increment was much less than that of F + S. Treatment with nFGF-1 alone showed a slight enhancing effect on PCNA but not to a significant level.
The role of heparan sulfate in the FGF-mediated activation of the cell surface receptor is further evaluated using rat thoracic artery smooth muscle cells treated with sodium chlorate. Treatment with sodium chlorate inhibits sulfation of glycosamino glycans (Fannon et al. 2000). As the chlorate-treated cells are bereft of sulfated proteoglycans, it provides a conducive environment to specifically investigate the role of heparin in the cell proliferation activity of FGF-1. Mitogenic activity of nFGF-1 monitored using the proliferating cellular nuclear antigen (PCNA) assay reveals that the cell proliferation activity of the protein in the presence of SOS is slightly higher than that realized in the presence of heparin (Fig. 1B ▶). Presently we do not have a concrete explanation for the marginally higher mitogenic activity observed in the presence of SOS. The proliferation activity of nFGF-1 in the absence of the ligands (SOS/heparin) is weak (Fig. 1B ▶). As mentioned earlier, the low mitogenic activity of the free nFGF-1 could be caused by proteolytic digestion on the cell surface (Saksela et al. 1988). The results of the cell proliferation assay on the chlorate-treated cells clearly show that heparin per se is not a prerequisite for binding (of nFGF-1) and subsequent activation of its receptor. It should be mentioned that the present data is consistent with several earlier reports. Roghani et al. (1994) showed that FGFR expressed in Chinese hamster ovary deficient in heparan sulfate synthesis bind to human FGF-2. Similarly, Delehedde et al. (2000), using chlorate-treated rat mammary fibroblasts cells, showed that FGF-2 can bind to FGFR and trigger transient early phosphorylation of p42/42MAPK and p90RSK. Further, Fannon et al. (1996) used chlorate-treated (glycosaminoglycan-deficient) Balb/C3T3 fibroblasts to show that bFGF can stimulate DNA synthesis in the absence of heparan sulfate proteoglycans in a dose-dependent manner. More recently, Burgess and coworkers (Xue et al. 2000; Shireman et al. 2000) generated several mutants of FGF-1 that could show heparin-independent mitogenic activity.
SOS does not oligomerize nFGF-1
It is well known that dimerization of the FGFR is crucial for the transduction of FGF-induced signals (Plotnikov et al. 2000). It has been proposed that binding of heparin-like proteoglycans to FGFs induces a conformational change in the FGFs, which results in the formation dimers or oligomers (DiGabriele et al. 1998). FGFs in their dimeric/oligomeric state are considered to be biologically active and believed to bind to FGFR with high affinity and consequently trigger the transduction of the signal (Waksman et al. 1998; Pantoliano et al. 1994; Faham et al. 1998).
Size-exclusion chromatography is a useful technique to monitor the molecular association/dissociation and conformational changes in proteins on ligand binding. Under the experimental conditions used, free nFGF-1 elutes with a retention time of 94 ± 0.16 min (Fig. 2 ▶). The protein (nFGF-1) complexed to SOS has a retention time (90 ± 0.64 min) similar to the free form of nFGF-1. Thus, it appears that nFGF-1 on binding to the ligand (SOS) exists in a monomeric state. Interestingly, nFGF-1 bound to heparin elutes with a significantly lower retention time of 73 ± 0.17 min, indicating that the protein oligomerizes on binding to the proteoglycan (Fig. 2 ▶). Although the molecular state (monomer/oligomer) of nFGF-1 is different in the presence of heparin and SOS, the protein (nFGF-1) shows significant mitogenic activity when complexed to both the ligands (SOS and heparin). Thus, these results appear to indicate that oligomerization of the protein (nFGF-1) is not a prerequisite for its binding to the receptor (FGFR) and consequent cell proliferation activity.
Fig. 2.
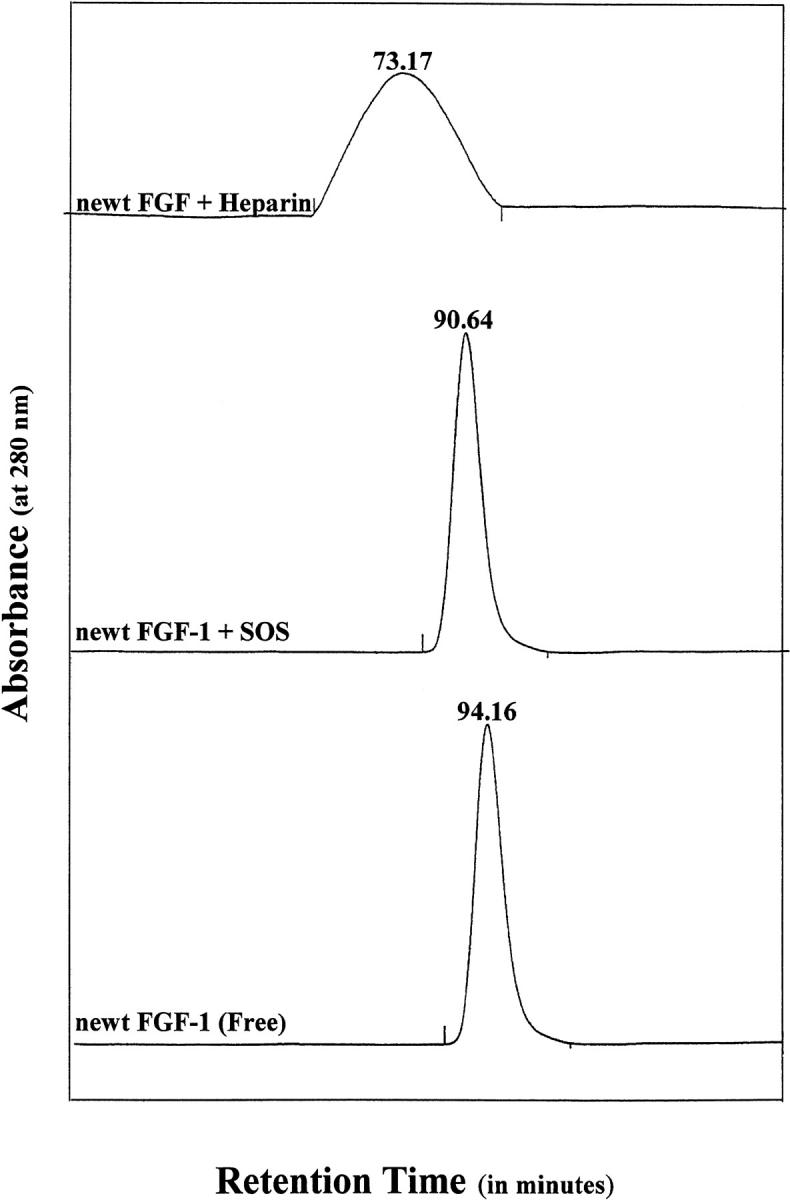
Size exclusion fast performance liquid chromatography (FPLC) profiles of free nFGF-1 (in 10 mM phosphate buffer [pH 7.2] containing 100 mM each of sodium chloride and ammonium sulfate) and when complexed to heparin and SOS. The corresponding retention times are indicated at the top of each peak. It could be observed that the elution volume of the nFGF-1 does not significantly change on binding to SOS, which indicates that the protein does not oligomerize in the presence of the ligand. However, the retention time of heparin-bound nFGF-1 is significantly lower than that observed for nFGF-1 alone or on complexation with SOS. These data are indicative of possible oligomerization of the protein in the presence of heparin. The elution of the protein(s) from the Superdex-100 column was monitored by the 280-nm absorbance of the eluate. The eluent used was 10 mM phosphate buffer containing 100 mM of NaCl. The flow rate of the eluent was 1 mL/min. The protein samples loaded were normalized by their absorbance at 280 nm.
The overall correlation time (τm), estimated using multidimensional nuclear magnetic resonance (NMR), is a useful relaxation parameter that would help in the assessment of the molecular status (monomeric/oligomeric) of the protein. τm values signify the rate of overall tumbling of the protein molecule in solution and it (τm) bears a near-linear relationship with the molecular weight of the protein (Lee et al. 1998). In this context, we measured the longitudinal (T1) and transverse relaxation (T2) rates of the backbone 15N nucleus using isotope- (15N) enriched nFGF-1 sample (Suppl. Fig. S3). The average τm value of nFGF-1 in its free form (in 10 mM phosphate buffer containing 100 mM NaCl and 100 mM ammonium sulfate) is estimated to be 9.35 ± 0.1 ns. Estimation of the average τm value of the protein (nFGF-1) in the presence of SOS (mixed in 1 : 1 ratio) did not significantly alter the overall correlation time (τm = 10.55 ± 0.1 ns). These results indicate that binding of SOS to nFGF-1 does not oligomerize the protein. In the event of oligomerization, the τm value of the protein in the ligand-bound state is expected to be approximately twice the value obtained for the protein in its monomeric form. Moy et al. (1997), investigating the backbone dynamics of hFGF-2 in the presence and absence of heparin, found that the τm of the protein in the heparin-bound state is almost double that of the value obtained for the native protein (hFGF-2) in its ligand-free state. Based on the τm value measurements and the data obtained using size-exclusion chromatography, it appears that SOS does not induce oligomerization of the protein (nFGF-1) but heparin does.
Sedimentation velocity studies provide useful information on the molecular mass of macromolecules and help to assess the presence of heterogeneous molecular states (of macromolecules) in equilibrium with each other (Prakash and Timasheff 1985 and 1986). In this context, we performed the sedimentation velocity experiments on nFGF-1 in its free and ligand-bound states. The protein boundary (in the absence and presence of SOS) moves with Svedberg constant (S20, w) values of 1.16 and 1.24 S, respectively (Fig. 3 ▶). Time-derivative analysis (Prakash and Timasheff 1986) of the sedimentation velocity data reveals that the protein in its free and SOS-bound states comprises single species corresponding to molecular weights of 15.2 and 17.0 kD, respectively (Fig. 3A,B ▶). These results clearly indicate that the protein exists as a monomer in the presence of SOS. Interestingly, the sedimentation velocity analysis on nFGF-1 bound to heparin (3 kD) showed that the protein exists in at least three high molecular mass forms corresponding to molecular weights, 70.1 kD, 83.0 kD, and 92.2 kD (Fig. 3C ▶). These results unambiguously show that nFGF-1 oligomerizes in the presence of heparin but not when bound to SOS. In summary, the results pre-sented thus far indicate that oligomerization of nFGF-1 apriori is not a mandatory requirement for its mitogenic activity.
Fig. 3.

Sedimentation boundary movement of nFGF-1, (A) in its free form, (B) in the presence of SOS, and (C) in the presence of heparin.
SOS-nFGF interaction sites
It is important to characterize the interaction site(s) of SOS on the protein (nFGF-1) to understand the physical basis for the differential action of SOS. Low-resolution crystal structure data exists on the binding of heparin and SOS to FGF (Zhu et al. 1991 and 1993; Blaber et al. 1996). Although these studies provide useful clues on the ligand-FGF interactions, no concrete conclusions could be drawn regarding the molecular state (monomer/oligomers) and the precise nature of the molecular interactions that operate between the protein and the ligand(s) in solution. In this context, we embarked on understanding the structural interactions stabilizing the nFGF-1-SOS complex using multidimensional NMR techniques.
Two-dimensional 1H-15N heteronuclear single quantum correlation (HSQC) spectrum of the free nFGF-1 was obtained in 10 mM phosphate buffer (pH 7.2) containing 100 mM each of sodium chloride and ammonium sulfate. The spectrum is well resolved and all the expected 126 cross-peaks could be observed in the fingerprint region of the spectrum (Suppl. Fig. S4). HSQC spectrum closely resembles the one reported for the hFGF-1 under similar conditions, which indicates that the structures of these two growth factor homologs are grossly similar (Ogura et al. 1999). Using a combination of various two- (total correlation spectroscopy [TOCSY], double quantum filtered-correlation spectroscopy [DQF-COSY], nuclear Overhauser effect spectroscopy [NOESY], and H1-N15 HSQC) and three- (TOCSY-HSQC and NOESY-HSQC) dimensional NMR spectra, we could successfully assign all the proton resonances in the protein. The secondary structure elements of the free nFGF-1 were delineated using the combined information obtained from the interstrand nuclear Overhauser effect (NOE) data and amide-proton exchange rates. The NOE connectivities indicate the existence of 12 antiparallel β-strands arranged into a β-trefoil motif (Suppl. Fig. S5). Thus, it appears that the three-dimensional structures of nFGF-1 and hFGF-1 are grossly similar. Residues constituting the putative high affinity receptor-binding site such as Tyr29, Arg49, Asn106, Tyr108, Leu147, and Leu149 are conserved in both hFGF-1 and nFGF-1 (Poulin et al. 1997; Springer et al. 1994). Comparison of the three dimensional structures of nFGF-1 and hFGF-1 show that orientation of the side chains of all the residues involved in the high affinity receptor-binding site are nearly similar. This aspect is consistent with the nearly identical mitogenic potency shown by nFGF-1 and hFGF-1 on the fibroblast (NIH/3T3) cells (Patrie et al. 1997). In addition to the high affinity receptor binding site, FGFs-1 possess a seven residue lower affinity receptor binding site spanning the loop located between beta strands 8 and 9 (Springer et al. 1994, residues, 114–120). With the exception of Lys115 in hFGF-1 (which is substituted by valine in that position in nFGF-1), the side chains of all the other residues in the lower affinity receptor binding site show similar spatial orientation(s). The side chain of Lys115 (in hFGF-1) is directed toward the hydrophobic lumen of the β-barrel (fig. not shown). In contrast, the nonpolar side chain of Val115 in nFGF-1 is solvent-exposed and is seen projecting toward the periphery of the growth factor molecule (fig. not shown). However, currently we are unable to comprehend the effects of the differential orientation of the side chains (of the residue at position 115 in hFGF-1 and nFGF-1) on the growth factor-receptor recognition process.
Two-dimensional NMR experiments provide an idea of the site(s) of interaction(s) of the ligand (SOS) to the protein. In general, protein-ligand interactions using one- and two-dimensional NMR spectroscopy could be discerned from the relative chemical shift changes between the free and bound forms of the protein. In this context, a series of 1H-15N HSQC spectra were recorded (at 25°C) at varying nFGF-1-SOS ratios (0–4.0). The chemical shift values of many proton resonances in the presence of SOS showed significant differences as compared with the free form of the protein (Fig. 4 ▶). The titration curves (based on the 1H chemical shift changes) are hyperbolic and tend to saturate when the molar ratio of SOS to nFGF-1 is 1 : 1 (Suppl. Fig. S6). This aspect apparently indicates a 1 : 1 binding of the polysulfated ligand to the protein. The average binding constant of SOS as estimated from the titration curves is 4 ± 0.14 μM. It appears that binding of SOS to nFGF-1 is much weaker than that of heparin. The binding constant of heparin to FGF is reported to be in the nanomolar range (Pantoliano et al. 1994).
Fig. 4.

1H-15N heteronuclear single quantum correlation (HSQC) spectrum of nFGF-1 in 10 mM phosphate buffer (pH 7.2) containing 100 mM ammonium sulfate and 100 mM NaCl. The cross-peaks representing the free form of the protein are indicated in black. The cross-peaks in the HSQC spectrum obtained in the presence of SOS are indicated in red. The labeled residues are those that undergo significant changes in their chemical shift values when the protein binds to SOS in a 1 : 1 ratio. It could be deduced that most of the cross-peaks that show significant change(s) in their chemical shift values are those of the positively charged residues located in the putative heparin-binding site in the protein.
A critical comparison of the HSQC spectra of the protein obtained in the presence (at 1 : 1 protein-ligand ratio) or absence of SOS indicates that most of the cross-peaks show no significant change in their chemical shift values on addition of SOS. The amide protons of only a few residues such as Asn32, Gly33, Lys130, Thr131, K132, Gly134, Arg136, Thr137, His138, Phe139, Lys142, and Ala143 show appreciable changes in their chemical shift values indicating that these residues are proximal to the SOS-binding site (Fig. 4 ▶). Interestingly, increasing the SOS to protein ratio even to about 10 : 1 caused no further change(s) in the HSQC spectra. This aspect not only implies the saturation of the ligand-binding site(s) in the protein but also indicates that the ligand (SOS) only binds to specific site(s) on the protein. The residues involved in the binding of SOS to nFGF-1 were mapped based on 15N and 1H chemical shift perturbation. A plot of the weighted average of the 15N and 1H chemical shift changes of residues in FGF on binding to SOS is depicted in Figure 5 ▶. It could be observed that residues in nFGF-1 show both upfield and downfield chemical shifts (15N and 1H) on binding to SOS. The majority of the residues that show maximal change in chemical shifts are located at the C-terminal end (residues, 120–145) of the nFGF-1 molecule (Fig. 5 ▶). However, in addition to the C-terminal residues, Asn32 and Gly33 that are located at the N-terminal end of the truncated form of nFGF-1 (as used in the present study) show significant chemical shift perturbation. This observation appears reasonable because nFGF-1 is a β-barrel protein and the residues at the N-terminal end (such as Asn32 and Gly33) are expected to be spatially close to the putative heparin-binding site located at the C-terminal end of the growth-factor molecule. The crystal and NMR structures of the truncated form of hFGF also support this contention (Zhu et al. 1991 and 1993; Ogura et al. 1999). Available crystal structure data of the human FGF-2 complexed to heparin di- and trisaccharides indicate that the ligands interact with the residues lying in the pseudohairpin developed by β-strands 10 and 11 (Ornitz et al. 1995). These residues include Lys130, Arg136, His138, and Lys142. Interestingly, all these residues show significant chemical shift changes when nFGF-1 binds to SOS. These results strongly indicate that heparin and SOS share a common binding site.
Fig. 5.

Weighted average (of 15N and 1H) chemical shift differences of residues in the free and SOS-complexed forms of nFGF-1. Profound changes in the chemical shift values could be observed for residues located in the C-terminal region of the nFGF-1 molecule. Most of the residues showing significant changes in the chemical shift values possess positively charged side chains. The difference in the chemical shift values was estimated from the HSQC spectra of nFGF-1 in its free and SOS-bound states. The single letter code represents residues, which show significant weighted average chemical shift differences.
Clinching evidence for protein-ligand interaction(s) based on NMR experiments comes from the observation of intermolecular NOEs between the protons of the ligand and the protein. Analysis of the NOEs obtained in the NOESY spectrum of the nFGF-1-SOS complex shows that there are at least four unambiguous intermolecular NOEs characterizing the complex between the ligand and the protein. These include Lys126 CɛH-H`, Lys130 CΔH-H3`, Lys130CɛH-H2, and Lys142CγH-H3` (Fig. 6 ▶). As the intermolecular NOEs observed are between the terminal side-chain protons of the positively charged residues in nFGF-1 and SOS, it appears that the protein-ligand interaction is mostly governed by the electrostatic contacts. This aspect probably accounts for the weak and fewer number of intermolecular NOEs observed between nFGF-1 and SOS. In addition, no intermolecular NOEs characterizing the interactions between the backbone of the protein and SOS could be observed.
Fig. 6.

Intermolecular nuclear Overhauser effect (NOE) cross-peaks that are observed between nFGF-1 and the SOS molecule in the nuclear Overhauser effect spectroscopy (NOESY) spectra recorded with a mixing time of 200 ms.
Structure of the nFGF-1-SOS complex
Molecular modeling of the nFGF-1-SOS interaction(s) was attempted to obtain a visual concept of the topology of the SOS-binding site and the structural interactions in operation at the ligand-nFGF-1 interface. The proposed model is in good agreement with the experimental NMR data (Fig. 7 ▶). Interestingly, the total CHARMm energy of the free form of nFGF-1 (−6266 k.cal.mole−1) was lower than that of the SOS-bound form of the protein (−6793 k.cal.mole−1). This further indicates that extra stabilization of the protein occurs on its binding to SOS. In addition, this result is in conformity with the conclusions drawn from the results of the equilibrium unfolding experiments. The structure of the nFGF-1-SOS complex depicts the SOS molecule to be lodged in the groove formed between β-strands 10 and 11. This groove is densely populated with positively charged residues and appears to provide a conducive environment for the polysulfated (negatively charged) ligands such as SOS to interact strongly with the protein (Fig. 7 ▶). In the absence of the ligand, the close proximity of positively charged residues appear to experience a force of repulsion that consequently destabilizes the protein. Binding of polyanionic ligands (such as SOS) appears to screen the repulsion(s) among the positively charged residues in the `cationic cluster'. Critical analysis of the modeled structure of the nFGF-1-SOS complex reveals that the SOS exactly fits into the groove comprising the `cationic cluster', and the eight sulfate groups of the ligand (SOS) appear to be engaged in electrostatic interactions with the positively charged side chains of residues in the groove (Fig. 7 ▶). The nonavailability of unbalanced negative charges in SOS probably disfavors the dimerization/oligomerization of nFGF-1. On the other hand, owing to the size and availability of negative charges, heparin probably promotes oligomerization of FGF by interacting with more than one protein molecule in a "beads on a string" manner.
Fig. 7.

Graphical Representation and Analysis of Structural Properties (GRASP) representation of the nFGF-1-SOS complex. SOS (indicated in yellow) could be observed to be lodged in the putative heparin-binding site (shown in blue). The negatively charged ligand (SOS) appears to stabilize the (nFGF-1) molecule (indicated in green) through electrostatic interactions with the closely spaced positively charged residues in the heparin-binding site. Binding of SOS appears to decrease the repulsion force(s) operating in the "cationic cluster."
Is oligomerization mandatory for FGF action(s)?
The crystal structures of FGF/receptor complex (in the absence of heparin) show that FGF interacts extensively with D2 and D3 domains and the flexible loop linking these structural domains. Interestingly, the FGF molecules bound on the opposite surfaces of the dimeric structure of the receptor domains have no direct structural contacts. In this context, it appears that heparin-induced oligomerization of FGF is not crucial for its binding to the receptor. In addition, several mutants of FGF-1 have recently been reported to show heparin-independent cell proliferation activity, indicating that proteoglycan(s)-mediated oligomerization of FGF may not be a prerequisite for the cell proliferation activity shown by FGF. In the context of the results obtained in the present study and the available literature, it may not be far-fetched to believe that FGF binds to its receptor in both its monomeric (in the presence of SOS) and dimeric/oligomeric (induced by heparin) states. In the monomeric state, FGF possibly triggers cell proliferation activity by binding to the dimeric state of the receptor, which could be formed transiently because of the dynamics of the membrane.
It should be mentioned that the results of the present study in no way dispel the role of heparin in the FGF-FGFR interaction. The proteoglycan(s)-induced oligomerization of FGF is probably a natural mechanism adopted by the cell to overcome heat inactivation and proteolytic action and consequently provide greater opportunity for the FGF molecules to interact with their high affinity receptors.
Materials and methods
Plasmid DNA was prepared using a miniprep kit obtained from Qiagen. EcoR I and Xba I were purchased from New England Biolabs. Ampicillin and chloramphenicol were procured from AMERSCO. Ingredients for the LB medium were obtained from DIFCO Laboratories. Low-molecular-weight heparin (Mr ∼3000 Da), phenylmethylsulfonyl fluoride (PMSF), aprotinin, pepstatin, leupeptin, triton X-100, β-mercaptoethanol, and α-chymotrypsin were obtained from Sigma Chemical Company. SOS, SHpS, and SHxS were purchased from Toronto Research Chemicals. Heparin-Sepharose was procured from Amersham-Pharmacia. All other chemicals used were of high quality analytical grade. All solutions were made in Milli Q water.
Protein purification
Recombinant newt acidic fibroblast growth factor (nFGF-1) was prepared from transformed Escherichia coli BL21(DE3)pLysS. The nFGF-1 DNA construct consisting of 486 base pairs was inserted between the NdeI and Bam H1 restriction sites. The expressed protein was purified on a heparin-Sepharose affinity column over an NaCl gradient (0–1.5 M). Desalting of the purified protein was achieved by ultrafiltration using an Amicon setup. The purity of the protein was assessed using SDS-PAGE. The first 22 residues of the full form of nFGF-1 were digested by subjecting the expressed full form of nFGF-1 to the action of chymotrypsin. Chymotrypsin digestion was performed by incubating the column material (heparin-Sepharose containing the bound protein) with the enzyme (at an enzyme to protein ratio of 20 : 1) in 10 mM phosphate buffer (pH 7.2) containing 0.85 M NaCl. The incubated mixture was stirred mildly at room temperature for 3 h. The incubated material was repacked into a column and was washed with 10 mM phosphate containing 0.85 M NaCl until absorbance of the eluate plateaued to a steady baseline. Truncated nFGF-1 was later eluted with 10 mM phosphate buffer (pH 7.2) containing 1.5 M NaCl. The homogeneity of the truncated nFGF-1 sample was checked by SDS-PAGE. The authenticity of the truncated sample was verified by ES-Mass analysis. The protein concentration was estimated based on the extinction coefficient value of the protein at 280 nm. It should be stated that the truncated newt FGF-1, which we label as nFGF-1, is used in all the experiments described ahead in the present work.
Preparation of isotope-enriched nFGF-1
15N-isotope labeling was achieved using M9 minimal medium containing 15NH4Cl. To realize maximal expression yields, the composition of the M9 medium was modified by the addition of a cocktail mixture of vitamins. The expression host strain E. coli BL21(DE3)pLysS is a vitamin B1-deficient host and hence the medium was supplemented with thiamine (vitamin B1). Protein-expression yields were in the range of 25–30 mg/L of the isotope-enriched medium. Purification and chymotrypsin digestion methods to obtain truncated nFGF-1 were the same as described in the previous section. The extent of 15N labeling was verified by ES-Mass analysis.
Circular dichroism
The thermal unfolding of nFGF-1 is monitored by far UV CD. The far UV CD spectra were measured using an Aviv 62DS spectropolarimeter. Samples of nFGF-1 at 100 μg/mL in 10 mM phosphate buffer (containing 100 mM NaCl) mixed with appropriate amounts of the ligands in a 1 : 1 ratio were placed into 1-mm pathlength cells with the cell temperature controlled by a Peltier device.
Mitogenic activity
The mitogenic assay was performed on NIH/3T3 cells using the method reported by Patrie et al. (1997). NIH/3T3 cells maintained in Dulbecco's-modified Eagle's medium (DMEM) supplemented with 10% calf serum and penicillin/streptomycin. Cells were seeded in 24-well plates at a density of 20 × 105 cells/well. At ∼80% confluency, the cells were washed once with phosphate-buffered saline (PBS) and placed in low serum media (DMEM, 0.5% calf serum, penicillin/streptomycin) for 24 h. The cells were stimulated with recombinant nFGF-1 in the presence of appropriate concentrations of the ligand. The mitogenic activity was estimated using a cell cytometer based on emission of the propidium bromide dye bound to the DNA within the cell.
Rat thoracic artery smooth muscle cells (A10, CCRC) were cultured in DMEM to evaluate the stimulation effects of SOS and heparin on FGF mitogenic activity. The cells were first treated with sodium chlorate to deprive endogenous heparin as described previously (Fannon and Nugent 1996). Briefly, 35,000 cells/cm2 for sodium-chlorate treatment were plated on culture dishes for 4 h before addition of 50 mM of sodium chlorate. Following 48 h of sodium-chlorate treatment, cells were treated with 1 ng/mL of FGF plus 20 μg/mL of heparin, 1 ng/mL of FGF plus 8.6μg/mL of SOS, and 1 ng/mL of FGF alone for additional 24 h. Cells cultured in 0.5% and 15% serum dialyzed served as negative and positive controls, respectively. Cells were thereafter resuspended in the lysis buffer containing 75 mM NaCl, 2 mM D, L-dithiothreitol (DTT), 2 mM EDTA, 0.2 μM benzamidine, 0.5 mM PMSF, 1 μM pepstatin A, and 50 mM triethanolamine (TEA)/HCl, pH7.4. The lysate was then subjected to 7.5% (w/v) SDS/PAGE gel for protein separation, followed by immunoblotting using an antibody against 36 kD PCNA (Upstate Biotechnology), and followed by reacting with a biotinylated antirabbit IgG at a 1 : 5000 dilution. Detection of protein levels was performed using an ECL kit (Amersham Pharmacia Biotech) according to the protocols provided by the manufacturer.
Proteolytic activity
Protection of nFGF-1 by heparin and analogs against the action of proteases was evaluated by incubating nFGF-1 at a concentration of 0.5 mg/mL (in the presence and absence of heparin and its analogs) with 0.25 mg/mL of a cocktail mixture of proteases (chymotrypsin, pepsin, and protease V) in 10 mM phosphate buffer containing 100 mM NaCl. The protease action was stopped for heating the mixture at 90°C for 10 min. The products of the protease action(s) were analyzed by SDS-PAGE. The degree of protection offered by heparin or its analogs was estimated by measuring the intensity of the band (on SDS-PAGE) corresponding to nFGF-1 (remaining after protease digestion) using a scanning densitometer. The intensity of the band corresponding to nFGF-1 not subjected to protease treatment was considered as a control for 100% protection.
Size-exclusion chromatography
Gel filtration experiments were performed at room temperature on a Superdex-100 column (using an AKTA FPLC device purchased from Amersham Pharmacia). The protein sample was incubated with the ligand (SOS/heparin) in 10 mM phosphate buffer containing 100 mM each of sodium chloride and ammonium acetate for 3 h at 4°C before loading onto the Superdex-100 column. All the protein samples were normalized to 0.5 absorbance units before loading onto the column. Ten mM phosphate buffer containing 100 mM NaCl was used as the eluent. The flow rate of the eluent was set at 1 mL/min using a peristaltic pump. Protein peaks were detected by their 280 nm absorbance. Under the experimental conditions used, no shrinkage of the resin was observed.
Sedimentation velocity experiments
Sedimentation velocity experiments were performed on a Beckman-Coulter Optima XL-A analytical ultracentrifuge equipped with An-60 Ti analytical rotor and standard double sector cells. Experiments were performed at 4°C at 50,000 rpm for 20 h. The Svedberg constant (S20, w) values were estimated using standard methods. The sedimentation boundary movement was traced by the absorbance of the protein using an inbuilt UV detector. The molecular weights of the protein in the presence and absence of the ligands were estimated using the time-derivative analysis method (Prakash and Timasheff 1985 and 1986). The concentration of the protein used was 1.5 mg/mL.
NMR experiments
NMR experiments were performed on a Bruker DMX 600 MHz spectrometer. In all two-dimensional 1H NMR experiments (TOCSY [Bax and Davies 1985], DQF-COSY [Rance et al. 1983], and NOESY [Kumar et al. 1981]), the concentration of the protein (nFGF-1) used was about 3 mM. For the two- (HSQC [Marion et al. 1989a]) and three- (HSQC-TOCSY [Marion et al. 1989b] and HSQC-NOESY [Marion et al. 1989c]) dimensional heteronuclear NMR experiments, the concentration of the protein used was 1.0 mM (Table 1). The protein samples were prepared in 10 mM phosphate buffer containing 0.1 mM mercaptoethanol and 0.1 mM EDTA in the presence of SOS by repeated exchange using Centricon ultrafiltration cartridges. All NMR data were acquired at a temperature of 25°C. Unless otherwise stated, solvent suppression was achieved by presaturation of the water signal during the relaxation delay, and quadrature detection in the indirectly detected dimensions was obtained with States-TPPI phase cycling.
Table 1.
List of two- and three-dimensional NMR experiments recorded on nFGF-1
| A. Through bond correlation experiments on uniformly labeled 15N sample | |
| 2D 1H-COSY | 3D HNCO |
| 2D 1H-15N HSQC | 3D HNHA |
| 2D 1H-13C HSQC | 3D NH(CA)CO |
| 2D 1H-TOCSY (75 ms mixing time) | 3D HNCA |
| 2D 1H-15N TOCSY-HSQC (150 ms mixing time) | 3D HCCH-TOCSY |
| 3D NH(CO)CA | |
| 3D CBCANH | |
| 3D CBCA(C)NH | |
| 3D C(CO)NH | |
| B. Through space correlation experiments | |
| (NMR) Nuclear magnetic resonance; (TOCSY) total correlation spectroscopy; (NOESY) nuclear Overhauser effect spectroscopy; (HSQC) heteronuclear single quantum correlation; (COSY) correlated spectroscopy. | |
| 2D 1H-NOESY (200 ms mixing time) | |
| 3D 15N-edited NOESY (150 ms mixing time) | |
| 3D 13C-edited NOESY (120 ms mixing time) | |
1H-15N NOESY-HSQC and (1H) NOESY spectra were recorded using a mixing time of 200 ms. All two-dimensional spectra were obtained with 2048 complex data points in t2 (detection period) and 512 points in t1 (evolution period). 3D NMR spectra (3D NOESY-HSQC and 3D TOCSY-HSQC) were obtained with 128, 64, and 2048 complex data points in the F1, F2, and F3 dimensions, respectively. Spectra were processed on Silicon Graphics workstations using the UXNMR and Aurelia software. All spectra were referenced indirectly to 3-(trimethylsilyl) propionate, sodium salt, using the ratio of 0.101329118 (for 15N/1H), respectively, for the zero-point frequency (Wishart et al. 1995).
T1 and T2 relaxation measurements
All 15N T1 and T2 relaxation measurements for nFGF-1 were performed in duplicate. Water suppression in the T1 and T2 experiments was performed with the WATERGATE sequence and water flipback pulses (Grziesiek and Bax 1993). All 2D spectra with 15N indirectly detected dimensions were collected with appropriate refocusing delays to allow for spectra without any phase correction. A Carr-Purcell-Meiboom-Gill (CPMG) spin-echo sequence (Meibloom et al. 1958) was applied during the transverse relaxation period of the T2 experiment. The Τ1 variable delay periods were 60, 140, 240, 360, 520, 720, and 1200 ms and the Τ2 CPMG periods were 16.2, 32.4, 48.6, 64.8, 81, 113.4, 145.8, and 194.4 ms.
T1 and T2 values were determined by fitting the measured peak heights to the two-parameter equation, I(t) = Io exp(-t/T1, 2). I(t) is a function of the variable delay period `t' (Fig. 3 ▶). The Levenberg-Marquardt algorithm (Press et al. 1986) was used to determine the optimum values of T1/T2 by minimizing the goodness of fit parameter (χ2). The overall correlation time (τm) was determined by using residues that had 15N T1/T2 ratios within one standard deviation.
Molecular modeling
The modeling of nFGF-1/SOS complex was performed in different steps. Minimizations were performed using the CHARMm energy function. The crystal structure coordinates of SOS were obtained from the Cambridge Data Bank, U.K. SOS molecule was built using the CHEMNOTE model building facility that is available within QUANTA (Molecular Simulations Inc.). SOS molecule was then subjected to Powell energy minimization (500 steps). The energy-minimized SOS molecule was then placed on the heparin-binding domain of nFGF-1 (average structure). Both the SOS and nFGF-1 molecules were subjected to Adopted Newton-Raphson restraint energy minimization with the inclusion of the available intermolecular NOE constraints (obtained from the NOESY spectrum). The restraint energy minimization process was repeated for few cycles to obtain the lowest energy structure and avoid unnecessary bad contacts in the molecule. During this process the atomic restraints that had been imposed on the nFGF-1 molecule were relaxed.
Acknowledgments
This work was supported by the National Science Council, Taiwan, and the Dr. C.S. Tsong Memorial Medical Research Foundation.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.2270102.
References
- Basilico, C. and Moscatelli, G. 1992. The FGF family of growth factors and oncogenes. Adv. Cancer. Res. 59 115–165. [DOI] [PubMed] [Google Scholar]
- Bax, A. and Davies, D.G. 1985. MLEV-17-based two-dimensional homonuclear magnetization transfer spectroscopy. J. Magn. Reson. 65 355–360. [Google Scholar]
- Bellot, F., Crumley, G., Kaplow, J.M., Schliessenger, J., Jaye, M., and Dionne, C.A. 1991. Ligand-induced transphosphorylation between different FGF receptors. EMBO. J. 10 2849–2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bikfalvi, A., Klein, S., Pintucci, G, and Rifkin, D.B. 1997. Biological roles of fibroblast growth factor-2. Endocr. Res. 18 26–45. [DOI] [PubMed] [Google Scholar]
- Blaber, M., Disalor, J., and Thomas K.A. 1996. X-ray crystal structure of human acidic fibroblast growth factor. Biochemistry 35 2086–2094. [DOI] [PubMed] [Google Scholar]
- Conrad, E.H. 1998 Heparin-binding proteins, Academic Press, San Diego, CA.
- Davies, J., Venkataraman, G., Shriver, Z., Raj, P.A, and Sasisekharan, R. 1999. Oligomeric self-association of basic fibroblast growth factor in the absence of heparin-like glycosaminoglycans. Biochem. J. 341 613–629. [PMC free article] [PubMed] [Google Scholar]
- Delehedde, M., Sere, M., Sergeant, N., Wartelle, I., Lyon, M., Rudland, P.S., and Fernig, D.G. 2000. Fibroblast growth factor-2 stimulation of p42/44 MAPK phosphorylation and 1KB degradation is regulated by heparan sulfate/heparin in rat mammary fibroblasts, J. Biol. Chem. 275 33905–33910. [DOI] [PubMed] [Google Scholar]
- DiGabriele, A.D., Lax, I., Chen, D.I., Svahn, C.M., Jaye, M., Schlessinger, J., and Hendrickson, W.A. 1998. Structure of a heparin-linked biologically active dimer of fibroblast growth factor. Nature 393 812–817. [DOI] [PubMed] [Google Scholar]
- Faham, S., Hileman, R.G., Fromm, J.R., Linhardt, R.J., and Rees, D.C. 1996. Heparin structure and interactions with basic fibroblast growth factor. Science 271 1116–1120. [DOI] [PubMed] [Google Scholar]
- Faham, S., Linhardt, J., and Rees, D.C. 1998. Diversity does make a difference: Fibroblast growth factor-heparin interactions. Curr. Opin. Struct. Biol. 8 578–586. [DOI] [PubMed] [Google Scholar]
- Fannon, M., Forsten, K.E., and Nugent, M.A. 2000. Potentiation and inhibition of bFGF binding by heparin: A model for regulation of cellular response. Biochemistry 39 1434–1445. [DOI] [PubMed] [Google Scholar]
- Fannon, M. and Nugent., M.A. 1996. Basic fibroblast factor binds its receptors is internalized and stimulates DNA synthesis in Balb/C3T3 cells in the absence of heparan sulfate. J. Biol. Chem. 271 17949–17956. [DOI] [PubMed] [Google Scholar]
- Fernig, D.G and Gallagher, J.T. 1994. Fibroblast growth factors and their receptors: An information network controlling tissue growth, morphogenesis and repair. Prog. Growth Factor. Res. 5 353–377. [DOI] [PubMed] [Google Scholar]
- Friesel, R and Maciag, T. 1999. Fibroblast growth factor prototype release and fibroblast growth factor receptor signaling. Thromb. Haemost. 82 748–754. [PubMed] [Google Scholar]
- Grziesiek, S. and Bax, A. 1993. The importance of not saturating H2O in protein NMR. Application to sensitivity enhancement and NOE measurements. J. Am. Chem. Soc. 115 12593–12597. [Google Scholar]
- Kumar, A., Wagner, G., Ernst, R.R, and Wuthrich, K. 1981. Build up rates of the Nuclear Overhauser Effect measured by two-dimensional proton magnetic resonance spectroscopy: Implications for studies of protein conformation. J. Am. Chem. Soc. 103 3654–3658. [Google Scholar]
- Lee, C.S., Kumar, T.K.S., Lian, L.Y., Cheng, J.Y., and Yu, C. 1998. Main-chain dynamics of cardiotoxin II from Taiwan cobra (Naja naja atra) as studied by carbon-13 NMR at natural abundance: Delineation of the role of functionally important residues. Biochemistry 37 155–164. [DOI] [PubMed] [Google Scholar]
- Lyon, M. and Gallagher, J.T. 1998. Bio-specific sequences and domains in heparan sulphate and the regulation of cell growth and adhesion. Matrix Biol. 17 485–493. [DOI] [PubMed] [Google Scholar]
- Mach, H., Volkin, D.B., Burke, C.J., Middaugh, C.R., Linhardt, R.J., Fromm., J.R., Loganathan, D., and Mattson, L. 1993. Nature of the interaction of heparin with acidic fibroblast growth factor. Biochemistry 32 5480–5489. [DOI] [PubMed] [Google Scholar]
- Marion, D., Ikura, M, and Bax, A.. 1989. Improved solvent suppression in one- and two-dimensional NMR spectra by Convolution Time-Domain data. J. Magn. Reson. 84 425–430. [Google Scholar]
- Marion, D., Ikura, M., Tuchudin, R., and Bax, A. 1989. Rapid recording of 2D NMR spectra without phase cycling. Application to the study of hydrogen exchange in proteins. J. Magn. Reson. 85 393–399. [Google Scholar]
- Marion, D., Kay, L.E., Sparks, S.W., Torchia, D.A, and Bax, A. 1989. Three-dimensional heteronuclear NMR of 15N-labeled proteins. J. Am. Chem. Soc. 111 1515–158. [Google Scholar]
- McEwen, D.G., MacArthar, C.A., Coulier, F., Gao, G.X., and Goldfarb, M. 1996. Receptor specificity of the fibroblast growth factor family. J. Biol. Chem. 271 15292–15297. [DOI] [PubMed] [Google Scholar]
- Meibloom, S. and Gill, D. 1958. Modified spin-echo method for measuring nuclear spin relaxation times. Res. Sci. Instrum. 29 688. [Google Scholar]
- Mohammadi, M., Schlessinger, J., and Hubbard, S.R. 1996. Structure of the FGF receptor tyrosine kinase domain reveals a novel autoinhibitory mechanism. Cell 86 577–587. [DOI] [PubMed] [Google Scholar]
- Moscatelli. D. 1992. Basic fibroblast growth factor (bFGF) dissociates rapidly from heparan sulfates but slowly from receptors. Implementations for mechanisms of bFGF release from pericellular matrix. J. Biol. Chem. 267 25803–25809. [PubMed] [Google Scholar]
- Moy, F.J., Safran, M., Seddon, A.P., Kitchen, D., Bohlen, P., Aviezer, D., Yayon, A., and Powers, R. 1997. Properly oriented heparin-decasaccharide-induced dimers are the biologically active form of basic fibroblast growth factor. Biochemistry 36 4782–4791. [DOI] [PubMed] [Google Scholar]
- Ogura, K., Nagata, H., Habuchi, H., Kimata, K., Tate, S., Ravera, M.W., Jaye, M., Schlessinger, J., Inagaki, F. 1999. Solution structure of human acidic fibroblast growth factor and interaction with heparin-derived hexasaccharide., J. Biomol. NMR 13 11–24. [DOI] [PubMed] [Google Scholar]
- Ornitz, D.M. 2000. FGFs, heparan sulfate and FGFRs: Complex interactions essential for development. Bioessays 22 108–112. [DOI] [PubMed] [Google Scholar]
- Ornitz, D.M. and Itoh, N. 2001. Fibroblast growth factors. Genome Biol. 2 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ornitz, D.M., Herr, A.B., Nilsson, M., Westman, J., Svahn, C.M., and Waksman, G. 1995. FGF binding and FGF receptor activation by synthetic heparan-derived di- and trisaccharides. Science 268 432–436. [DOI] [PubMed] [Google Scholar]
- Ornitz, D.M., Yahon, A., Flanagan, J.G., Savhn, C.M., Levi, E, and Leder, P. 1992. Heparin is required for cell-free binding of basic fibroblast growth factor to a soluble receptor and for mitogenesis in whole cells. Mol. Cell. Biol. 12 240–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantoliano, M.W., Horlick, R.A., Springer, B.A., Van Dyk, D.E., Tobery.T., Wetmore, D.P., Lear, J.D., Nahapetian, A.T., Bradley, J.D, and Sisk, W.P. 1994. Multivalent ligand-receptor binding interactions in the fibroblast growth factor system produce a cooperative growth factor and heparin mechanism for receptor dimerization. Biochemistry 33 10229–10248. [DOI] [PubMed] [Google Scholar]
- Patrie, K.M., Botelho, M.J., Ray, S.K., Mehta, V.B., and Chiu, I.M. 1997. Amphibian FGF-1 is structurally and functionally similar to but antigenically distinguishable from its mammalian counterpart. Growth Factors 14 39–57. [DOI] [PubMed] [Google Scholar]
- Plotnikov, A.N., Hubbard, S.R., Schlessinger, J., and Mohammad, M. 2000. Crystal structures of two FGF-FGFR complexes reveal the determinants of ligand-receptor specificity. Cell 101 413–424. [DOI] [PubMed] [Google Scholar]
- Plotnikov, A.N., Schlessinger, J., Hubbard, S.R., and Mohammadi, M. 1999. Structural basis for FGF receptor dimerization and activation. Cell 90 641–650. [DOI] [PubMed] [Google Scholar]
- Poulin, M.I., Botelho, M.J., and Chiu, I.M. 1997. Cloning and interspecies comparisons of three newt (Notophthalmus viridescens) fibroblast growth factor receptor sequences. Mol. Cell. Biochem. 1745 11–19. [DOI] [PubMed] [Google Scholar]
- Prakash, V. and Timasheff, S.N. 1985. Vincristine-induced self-association of calf brain tubulin. Biochemistry 24 5004–5010. [DOI] [PubMed] [Google Scholar]
- ———. 1986. Criteria for distinguishing self-associations in velocity sedimentation. Methods Enzymol. 130 3–6. [DOI] [PubMed] [Google Scholar]
- Press, W.M., Flatnnery, B.P., Teskolsky, S.A., and Vetterling, W.T. 1986. Numerical recipes. Cambridge University Press, Cambridge, MA.
- Rance, M., Sorensen, O.W., Bodenhaussen, G., Wagner, G., Ernst, R.R., and Wuthrich, K. 1983. Improved spectral resolution in COSY 1H NMR spectra of proteins via double quantum filtering. Biochem. Biophys. Res. Commun. 113 967–974. [DOI] [PubMed] [Google Scholar]
- Roghani, M., Mansukhani, A., Deil'Erat, P., Beilosta, P., Basilico, C., RifKin, D.B., and Moscatelli, D. 1994. Heparin increases the affinity of basic fibroblast growth factor for its receptor but is not required for binding. J. Biol. Chem. 269 3976–398. [PubMed] [Google Scholar]
- Saksela, O., Moscatelli, D., Summer, A., and RifKin, D.B. 1988. Endothelial cell-derived heparan sulfate binds basic fibroblast growth factor and protects it from proteolytic degradation. J. Cell. Biol. 107 743–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel, D., Kumar, T.K.S., Srimathi, T., Hsieh, H.C., and Yu, C. 2000. Identification and characterization of an equilibrium intermediate in the unfolding pathway of an all beta-barrel protein. J. Biol. Chem. 275 34968–34975. [DOI] [PubMed] [Google Scholar]
- Schlessinger, J., Lax, I., and Lemmon, M. 1995. Regulation of growth factor activation by proteoglycans: What is the role of the low affinity receptors? Cell 83 357–360. [DOI] [PubMed] [Google Scholar]
- Schlessinger, J., Plotnikov, A.N., Ibrahimi, O.A., Eliseenkova, A.V., Yeh, B.K., Yayon, A., Lin hardt, R.J., and Mohammadi, M. 2000. Crystal structure of a ternary FGF-FGFR heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Mol. Cell 6 743–750. [DOI] [PubMed] [Google Scholar]
- Shireman, P.K., Xue, L., Madox, E., Burgers, W.H., and Greisler, H.P. 2000. The S130K fibroblast growth factor-1 mutant induces heparin independent proliferation and is resistant to thrombin degradation in fibrin glue. J. Vasc. Surg. 31 382–390. [DOI] [PubMed] [Google Scholar]
- Sommer, A. and Rifkin, D.B. 1989. Interaction of heparin with human basic fibroblast growth factor: Protection of the angiogenic protein from proteolytic degradation by a glycosaminoglycan. J. Cell. Physiol. 138 215–220. [DOI] [PubMed] [Google Scholar]
- Springer, B.A., Dantoiliano, M.W., Barbera, F.A., Gunyuzlu, D.L., Thompson, L.D., Herbilin, W., Rosenfeld, S.A., and Book, G.W. 1994. Identification and concerted function of two receptor binding surfaces on basic fibroblast growth factor required for mitogenesis. J. Biol. Chem. 259 26873–26884. [PubMed] [Google Scholar]
- Spivak-Kroizman, T., Lemmon, M.S., Dikic, I., Ladbury, J.E., Pinchasi, D., Huang, J., Jaye, M., Crumley, G., Schlessinger, J., and Lax, I. 1994. Heparin-induced oligomerization of FGF molecules is responsible for FGF receptor dimerization, activation, and cell proliferation. Cell 79 1015–1024. [DOI] [PubMed] [Google Scholar]
- Stauber, D.J., Di Gabriele, A.D., and Hendrickson, W.A. 2000. Structural interactions of fibroblast growth factor receptor with its ligands. Proc. Natl. Acad. Sci. 97 49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo, S. 1991. The mode of action of sucralfate: The 1 × 1 × 1 mechanism of action. Scand. J. Gastroentrol. 185(Suppl. 26): 7–12. [DOI] [PubMed] [Google Scholar]
- Venkataraman, G., Shriver, Z., Davis, J.C., and Sasisekharan, R. 1999. Fibroblast growth factors 1 and 2 are distinct in oligomerization in the presence of heparin-like glycosaminoglycans. Proc. Natl. Acad. Sci. 96 1892–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkin, D.B., Verticelli, A.M., Marfia, K.E., Burkie, C.J., Mach, H., and Middaugh, C.R. 1993. Sucralfate and soluble sucrose octasulfate bind and stabilize acidic fibroblast growth factor. Biochim. Biophys. Acta. 1203 18–26. [DOI] [PubMed] [Google Scholar]
- Waksman, G. and Herr, A.B. 1998. New insights into heparin-induced FGF oligomerization. Nat. Struct. Biol. 5 527–530. [DOI] [PubMed] [Google Scholar]
- Wishart, D.S., Bigan, C.G., Yao, J., Abildgard, F., Dyson, H.J., Oldfield, E., Markley, J.L., and Sykes, B.D. 1995. 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J. Biomolec. NMR 6 135–140. [DOI] [PubMed] [Google Scholar]
- Wong, P., Hampton, B., Szylobrgt, E., Gallager, A.M., Jaye, M., and Burgers, W.H. 1995. Analysis of putative heparin-binding domains of fibroblast growth factor-2 using site-directed mutagenesis and peptide analogues. J. Biol. Chem. 270 25805–25811. [DOI] [PubMed] [Google Scholar]
- Xue, L., Shireman, P.K., Hampton, B., Burgers, W.H., and Greisler, H.P. 2000. The cysteine-free fibroblast growth factor 9 mutant induces heparin-independent proliferation of endothelial cells and smooth muscle cells. J. Surg. Res. 92 255–260. [DOI] [PubMed] [Google Scholar]
- Zhu, X., Hsu, B.T., and Rees, D.C. 1993. Structural studies of the binding of the anti-ulcer drug sucrose octasulfate to acidic fibroblast growth factor. Structure 1 27–34. [DOI] [PubMed] [Google Scholar]
- Zhu, X., Komiya, H., Chirno, A., Fahamo. S., Fox, G.M., Arakawa, T., Hsu, B.T., and Rees, D.C. 1991. Three-dimensional structures of acidic and basic fibroblast growth factors. Science 253 90–93. [DOI] [PubMed] [Google Scholar]