

Abstract
Histidine pKa values were measured in charge-reversal (K78E, K97E, K127E, and K97E/K127E) and charge-neutralization (E10A, E101A, and R35A) mutants of staphylococcal nuclease (SNase) by 1H-NMR spectroscopy. Energies of interaction between pairs of charges (ΔGij) were obtained from the shifts in pKa values relative to wild-type values. The data describe the distance dependence and salt sensitivity of pairwise coulombic interactions. Calculations with a continuum electrostatics method captured the experimental ΔGij when static structures were used and when the protein interior was treated empirically with a dielectric constant of 20. The ΔGij when rij ≤ 10 Å were exaggerated slightly in the calculations. Coulomb's law with a dielectric constant near 80 and a Debye-Hückel term to account for screening by the ionic strength reproduced the salt sensitivity and distance dependence of ΔGij as well as the structure-based method. In their interactions with each other, surface charges behave as if immersed in water; the Debye length describes realistically the distance where interactions become negligible at a given ionic strength. On average, charges separated by distances (rij) ≈5 Å interacted with ΔGij ≈ 0.6 kcal/mole in 0.01 M KCl, but ΔGij decayed to ≤0.10 kcal/mole when rij = 20 Å. In 0.10 M KCl, ΔGij ≈ 0.10 kcal/mole when rij = 10 Å. In 1.5 M KCl, only short-range interactions with rij ≤ 5 Å persisted. Although at physiological ionic strengths the interactions between charges separated by more than 10 Å are extremely weak, in situations where charge imbalance exists many weak interactions can cumulatively produce substantial effects.
Keywords: Electrostatics, Poisson-Boltzmann, determinants of pKa values, salt effects, ionic strength, coulombic interactions, staphylococcal nuclease
The structure, stability, and function of many proteins are governed by electrostatics. Therefore, knowledge of the pKa values of ionizable residues and of their molecular determinants is often required to understand the physical basis of stability and function. pKa values can be measured experimentally in small, soluble proteins, but in larger proteins and in membrane proteins they are experimentally inaccessible. In such cases, structure-based pKa calculations are useful for analysis of the relationship between protein structure and function. This approach is only productive if the reliability of the computational methods is established by comparison with experimental data. Here we present an experimental study of the magnitude, distance dependence, and salt sensitivity of the energy of coulombic interaction between pairs of surface charges in staphylococcal nuclease (SNase). The data are used to assess coulombic contributions to pKa values and to test the ability of a continuum method for structure-based pKa calculations to reproduce the experimental data.
The validity of physical models for calculation of electrostatic energies in proteins is usually established by comparison of measured and calculated pKa values. In calculations, the contributions from coulombic interactions between charged groups, and contributions related to the self-energies of the charged groups, are usually treated separately (Bashford and Karplus 1990; Sham et al. 1997). The self-energies account for interactions of charged groups with permanent dipoles and for differences between the hydration of charged groups in the protein and in water. Most charged groups in proteins have net favorable coulombic interactions and unfavorable self-energies; pKa values reflect the balance between these two terms.
To understand molecular determinants of pKa values, it is necessary to know how coulombic and self-energy contributions are parsed. This information is also necessary to establish that a computational method reproduces pKa values for the right physical reasons. Self-energies can be evaluated easily in the case of buried ionizable residues because the significant perturbation of their pKa values is dominated by the loss of hydration in the protein interior (Dwyer et al. 2000; Schutz and Warshel 2001). In the case of surface residues, the experimental dissection of the determinants of pKa values is considerably more difficult because these pKa values are similar to the values in model compounds in water, and the nature of the balance between coulombic energies and self-energies is not readily apparent. The relatively unperturbed pKa values of surface residues can be reproduced with very different physical models. Models in which the calculated coulombic and self-energy contributions are always small (Matthew et al. 1985) and models in which coulombic and self-energy contributions are large in magnitude and of opposite sign (Bashford and Karplus 1990) can both reproduce pKa values of surface residues. In the case of surface residues, agreement between calculated and measured pKa values is a required but not sufficient condition to establish that a model reproduces pKa values for the correct physical reasons.
Most previous studies of the magnitude of electrostatic interactions in proteins are based on comparisons of the stability of wild-type and mutant proteins where charged residues are substituted by neutral or oppositely charged ones (Serrano et al. 1990; Dao-pin et al. 1991a; Dao-pin et al. 1991b; Meeker et al. 1996; Blasie and Berg 1997; Grimsley et al. 1999; Ibarra-Molero et al. 1999; Loladze et al. 1999; Perl et al. 2000; Spector et al. 2000; Sanchez-Ruiz and Makhatadze 2001; Shaw et al. 2001). This approach can be unreliable because electrostatic interactions can be significant in denatured states of proteins (Oliveberg et al. 1994; Oliveberg et al. 1995; Swint-Kruse and Robertson 1995; Tan et al. 1995; Kuhlman et al. 1999; Pace et al. 2000; Whitten and García-Moreno E. 2000). This precludes interpretation of the effect of the mutations strictly in terms of the electrostatic properties of the native state. Furthermore, deconvolution of the effects of mutations on stability into electrostatic and non-electrostatic components is cumbersome.
These problems are avoided when the interaction energy between two charges (ΔGij) is obtained from the change in pKa values (ΔpKa) that results from removal or reversal of charge at other sites. pKa values can be measured by 1H-NMR spectroscopy, and ΔpKa can be interpreted in terms of ΔGij through the relation ΔGij = −1.36·ΔpKa (at 298 K). When hydrogen bonds or ion pairs are not involved, and when the mutations do not affect the conformation of the protein significantly, ΔGij is proportional to the contribution by the charge at the mutated site to the electrostatic potential at the ionizable site whose pKa is measured. This approach has been used previously (Russell et al. 1987; Akke and Forsen 1990; Loewenthal et al. 1993; Blasie and Berg 1997; Tishmack et al. 1997; Fisher et al. 1998; Forsyth et al. 1998; Fischer and Verma 1999; Forsyth and Robertson 2000). However, not enough values have been measured systematically in a single protein to be able to define the general trends in the distance dependence and salt sensitivity of ΔGij.
Our interest in understanding the range of coulombic interactions and their sensitivity to salt stems partly from a previous study that showed three of four histidines in SNase titrate with markedly depressed pKa values (Lee et al. 2002). The salt sensitivity of all histidines is substantial—three of them increase by almost a full unit between 0.01 and 1.5 M KCl. Structure-based pKa calculations with a continuum model captured the magnitude and the salt dependence of pKa values accurately when the protein interior was treated empirically as a medium of dielectric constant of 20. According to the calculations, the unusual electrostatic properties of these histidines reflect the cumulative effects of many weak, long-range interactions between charges separated by 10 Å or more. This would appear to contradict results from previous studies of charge–charge interactions in other proteins, which showed that long-range electrostatic interactions on the surface of proteins are extremely weak (Dao-pin et al. 1991b; Forsyth and Robertson 2000). Thus, it was of interest to measure ΔGij experimentally, both to understand the origins of the unusual salt sensitivity of pKa values in nuclease and to determine whether the calculations reproduced the pKa values for the right physical reasons. Toward this end, the pKa values of the four histidines in SNase were measured by 1H-NMR spectroscopy in charge-neutralization mutants and in single and double charge-reversal mutants. pKa values were also calculated with a continuum method based on the finite-difference solution of the Poisson-Boltzmann equation (FDPB) (Antosiewicz et al. 1994; Antosiewicz et al. 1996) to test the ability of this algorithm to reproduce the measured ΔGij quantitatively.
Results
1H-NMR spectroscopy was used to measure the pKa values of the four histidines in charge-reversal (K78E, K97E, K127E, and K97E/K127E) and in charge-neutralization (E10A, E101A, and R35A) mutants of SNase. The charge-reversal mutations allowed us to exaggerate the electrostatic consequences of altering the charge at a given site while minimizing the number of mutations. Experiments were performed under three ionic conditions for most mutants: no added salt (estimated to have an average ionic strength of 0.01 M), 0.10 M, and 1.5 M KCl. The location and conformation of the histidines (His-8, His-46, His-121, and His-124) and of the charged sites that were mutated are shown in Figure 1 ▶. The sites Lys-78, Lys-97, and Lys-127 were selected because these ionizable residues have high solvent-accessibility and do not appear to be involved in either ion pairing or hydrogen bonding interactions with any groups; thus, the mutations are unlikely to alter the structure significantly. Mutations at these sites allowed examination of charge–charge interactions over a wide range of distances. In contrast, E10A and E101A were selected to measure the magnitude of short-range interactions. R35A was a useful control because it is situated far from all four histidines. All proteins remained in the native state over the course of the titrations monitored by NMR spectroscopy. This was corroborated for all mutants studied by acid/base titrations monitored by intrinsic fluorescence and by the dispersion in the 1H-NMR spectra (data not shown).
Fig. 1.

Stereoscopic ribbon diagram of staphylococcal nuclease (SNase), accession code 1stn (Hynes and Fox 1991). The highlighted side chains are the four histidines (His-8, His-46, His-121, and His-124), the three sites where Lys was replaced by Glu (Lys-78, Lys-97, and Lys-127), and the additional mutated sites (Arg-35, Glu-10, and Glu-101). Figure drawn with MOLSCRIPT and Raster3D.
Histidine p Ka values in the charge-reversal mutants K78E, K97E, and K127E
The pKa values of histidines in the K78E, K97E, and K127E mutants are listed in Table 1. The differences relative to the pKa values in the wild-type protein (pKaMUT-pKaWT) are presented in Figure 2 ▶. The pKa values in the mutants are higher than in the wild type, consistent with the replacement of unfavorable coulombic interactions between His and Lys with favorable interactions between His and Glu. With the exception of the dramatic effects of the mutations on the pKa of His-124, the effects of charge reversal are modest, even at low ionic strengths.
Table 1.
Histidine p Kavalues measured by 1H-NMR spectroscopya,b
| Salt concentration (M) | His-8 | His-46 | His-121 | His-124 | |
| Wild type | |||||
| 0.01c | 6.08 ± 0.02 | 5.66 ± 0.03 | 4.80 ± 0.04 | 5.31 ± 0.02 | |
| 0.10 | 6.52 ± 0.03 | 5.86 ± 0.04 | 5.30 ± 0.06 | 5.73 ± 0.02 | |
| 1.50 | 7.00 ± 0.03 | 6.14 ± 0.04 | 5.88 ± 0.03 | 6.24 ± 0.02 | |
| K78E | drij → | 12.6 Å | 30.1 Å | 8.9 Å | 12.6 Å |
| 0.01c | 6.42 ± 0.03 | 5.72 ± 0.02 | 4.98 ± 0.04 | 5.43 ± 0.03 | |
| 0.10 | 6.70 ± 0.03 | 5.83 ± 0.02 | 5.28 ± 0.02 | 5.71 ± 0.02 | |
| 1.50 | 7.05 ± 0.02 | 6.17 ± 0.03 | 5.95 ± 0.02 | 6.34 ± 0.04 | |
| K97E | drij → | 21.5 Å | 29.7 Å | 14.9 Å | 11.4 Å |
| 0.01c | 6.27 ± 0.04 | 5.73 ± 0.03 | 5.09 ± 0.04 | 5.76 ± 0.06 | |
| 0.10 | 6.54 ± 0.01 | 5.84 ± 0.03 | 5.37 ± 0.02 | 5.94 ± 0.04 | |
| 1.50 | 7.06 ± 0.01 | 6.18 ± 0.02 | 5.95 ± 0.03 | 6.33 ± 0.02 | |
| K127E | drij → | 23.7 Å | 29.0 Å | 10.4 Å | 5.0 Å |
| 0.01c | 6.25 ± 0.03 | 5.78 ± 0.03 | 4.99 ± 0.06 | 6.20 ± 0.05 | |
| 0.10 | 6.53 ± 0.02 | 5.85 ± 0.02 | 5.31 ± 0.02 | 6.39 ± 0.03 | |
| 1.50 | 7.04 ± 0.03 | 6.20 ± 0.04 | 5.95 ± 0.04 | 6.63 ± 0.04 | |
| K127A | drij → | 25.4 Å | 31.9 Å | 12.3 Å | 8.5 Å |
| 0.01c | 6.16 ± 0.03 | 5.73 ± 0.03 | 4.87 ± 0.05 | 5.70 ± 0.04 | |
| K97E/K127E | |||||
| 0.01c | 6.26 ± 0.01 | 5.74 ± 0.02 | 5.15 ± 0.04 | 6.60 ± 0.05 | |
| 0.10 | 6.56 ± 0.02 | 5.87 ± 0.02 | 5.43 ± 0.03 | 6.80 ± 0.04 | |
| 1.50 | 7.02 ± 0.02 | 6.22 ± 0.02 | 5.93 ± 0.04 | 6.70 ± 0.04 | |
| E10A | drij → | 3.8 Å | 31.6 Å | 14.2 Å | 20.6 Å |
| 0.01c | 5.72 ± 0.02 | 5.67 ± 0.02 | 4.87 ± 0.04 | 5.39 ± 0.02 | |
| 0.10 | 6.15 ± 0.02 | 5.85 ± 0.03 | 5.22 ± 0.03 | 5.76 ± 0.03 | |
| 1.50 | 6.73 ± 0.04 | 6.21 ± 0.05 | 5.93 ± 0.03 | 6.31 ± 0.03 | |
| E101A | drij → | 21.9 Å | 25.7 Å | 10.6 Å | 4.8 Å |
| 0.01c | 6.20 ± 0.02 | 5.80 ± 0.02 | 5.04 ± 0.03 | 4.84 ± 0.04 | |
| 0.10 | 6.45 ± 0.02 | 5.82 ± 0.03 | 5.20 ± 0.03 | 5.02 ± 0.03 | |
| R35A | drij → | 22.0 Å | 12.3 Å | 16.8 Å | 18.5 Å |
| 0.10 | 6.50 ± 0.01 | 5.91 ± 0.02 | 5.27 ± 0.04 | 5.71 ± 0.02 |
a All experiments were performed at 25°C. The wild-type data were obtained from Lee et al. (2002).
b Errors reflect goodness of fits to Equation 2. Standard deviation from multiple titrations of the wild-type protein in 0.10 M KCl is ± 0.05.
c Titrations performed in the absence of added salt, estimated to have 0.01 M ionic strength.
d rij calculated with the lstn structure (Hynes and Fox 1991) or in the case of mutants, in energy-minimized models. Distances were measured between histidine Cɛl and Nζ of Lys, Cζ of Arg, and Cδ of Glu.
Fig. 2.

Shift in histidine pKa values (ΔpKa = pKaMUT-pKaWT) of (A) His-8, (B) His-46, (C) His-121, and (D) His-124 measured by 1H-NMR spectroscopy in charge-reversal mutants in 0.01, 0.10, and 1.5 M KCl. For each salt condition the bars represent, from left to right, the effects of K78E, K97E, K127E, and K97E/K127E mutants. Error bars provided are standard errors of the fits of equation 2 to experimental data.
Histidine p Ka values in the charge-neutralization mutant K127A
The histidine pKa values of the K127A mutant are also listed in Table 1. In 0.01 M KCl, the pKa values for all four histidines in this mutant are approximately halfway between the pKa values in the wild type and in K127E. This is most clearly illustrated by the pKa of His-124, which responds to the change in charge at residue 127 in a linear fashion. This linear relation indicates that the primary consequence of the mutations at this position is to perturb coulombic contributions to the pKa values, as intended.
Histidine p Ka values in double charge-reversal mutant K97E/K127E
The pKa values of histidines in the K97E/K127E mutant are included in Table 1. The differences relative to the pKa values in the wild type are shown in Figure 2 ▶. The effects of the mutations K97E and K127E on the pKa values are largely additive, more so in 1.5 M and 0.10 M KCl than in 0.01 M KCl. For His-124, the effects are additive under all ionic conditions. Again, additivity indicates that the response to the mutations involved mostly coulombic effects. It is also consistent with the mutations having no significant structural consequences.
The salt sensitivity of the pKa value of His-124 in the double mutant is strikingly different than in the wild-type protein. In the double mutant, the pKa increases only by 0.07 units when salt concentration is increased from 0.01 M to 1.5 M, compared with an increase of 0.93 in the wild type. The salt sensitivity of His-124 in the double mutant is comparable to that of histidines in small, neutral peptides in water (Kao et al. 2000). The two Lys→Glu mutations in the neighborhood of His-124 are sufficient to neutralize the strongly unfavorable electrostatic environment that gave rise to the dramatic salt sensitivity of this histidine.
Histidine p Ka values in the charge-neutralization mutants E10A, E101A, and R35A
The pKa values of histidines in the E10A and E101A mutants are also included in Table 1. When the interaction between Glu-10 and His-8 was eliminated, the pKa of His-8 was depressed to values below the normal pKa of histidines in small peptides in water (Kao et al. 2000). The relatively normal pKa value of His-8 in the wild-type protein apparently reflects a balance between short-range attractive interactions (rij < 5 Å), and medium-range (rij < 10 Å) and long-range (rij > 10 Å) repulsive interactions. This is consistent with the indication by continuum calculations that the large salt dependence of this group, despite its normal pKa value, reflects substantial repulsive interactions with charges more than 10 Å away (Lee et al. 2002).
A shift of 0.07 ppm in the acid limit of the resonance of Cɛ1H of His-8 in E10A relative to the wild type indicates that the mutation alters the conformation of SNase slightly. We speculate that the short-range contact between Glu-10 and His-8 tethers His-8, and when Glu-10 is eliminated, the His-8 side chain assumes an altered conformation. The dramatic salt sensitivity of the pKa value of His-8 in E10A indicates that this histidine is still under the influence of the basic residues in the rest of the protein. The minor structural changes in E10A seem to propagate toward His-121 and His-124. Both of these histidines are far from Glu-10 (14.2 and 20.6 Å, respectively), yet they showed upward shifts in pKa values of nearly 0.1 units in 0.01 M ionic strength. The directions of these shifts are opposite of those expected from the loss of a negative charge. Figure 3 ▶ illustrates a network of interactions involving Lys-9, Glu-75, Tyr-93, and His-121 that might be disrupted when the interaction between His-8 and Glu-10 is broken in the E10A mutant. Perturbation to this network of hydrogen bonds and ion pairs may propagate instability to helix-3 where His-121 and His-124 reside. These conformational perturbations appear to be absent at high ionic strength, and they were not studied further.
Fig. 3.

Local electrostatic environment around His-121. Figure drawn using the 1stn structure by Hynes and Fox (1991) with MOLSCRIPT and Raster3D.
The E101A mutation is also very interesting. Glu-101 is the ionizable residue closest to His-124 (Hynes and Fox 1991), and it exerts a substantial influence on the pKa and on the salt sensitivity of this histidine. The pKa of His-124 in E101A in 0.01 M KCl is 0.47 units lower than in the wild type; in 0.10 M KCl it is lower by 0.71 units. Local restructuring about His-124 is also likely in the E101A mutant. This is indicated by the perturbation of the chemical shift of Cɛ1H of His-124 in the protonated state relative to the wild-type protein and to all other mutants examined in this study. This restructuring does not appear to influence the Cɛ1H chemical shift in the deprotonated state. The pKa values of His-8, His-46, and His-121 in E101A provide further evidence of minor structural changes because their pKa values are shifted upward by a small but statistically significant magnitude. This shift is in the direction opposite of that expected from the removal of a negatively charged residue. The pH titrations were repeated to show that the increase in pKa values did not reflect experimental error or incorrect setting of the ionic strength in the experiment. The titrations were perfectly superimposable; thus, we concluded that the observed shifts reflect a response of the E101A protein to the mutation. The nature of the effects of E10A and E101A indicates that the protein is more electrostatically relaxed in the mutants. The mechanism whereby these changes are transmitted in the protein is not obvious.
Arg-35 is far (17–22 Å) from all histidines except His-46; the Cζ atom of Arg-35 is 12 Å from His-46. The R35A mutation had no measurable consequences on any histidine pKa values in 0.10 M KCl and it was not studied further. His-46 served a similar function as a useful internal control. This histidine is situated in a flexible loop that extends into solution. Its pKa value has the lowest sensitivity to salt among the histidines. His-46 is located more than 28 Å from any of the charge-reversal sites. The minimal perturbations of the pKa values of His-46 in the mutants were expected.
Structure-based calculations of p Ka values and ΔGij
The pKa values of all histidines in all mutants, calculated by the method based on the numerical solution of the linearized Poisson-Boltzmann equation (FDPB), are listed in Table 2, and the contributions by individual pairwise coulombic interactions are listed in Table 3. The experimental and the calculated ΔpKa are compared in Figure 4 ▶. In these calculations a single protein conformation was used, and the protein interior was treated empirically with a dielectric constant of 20 to maximize agreement between calculated and measured values (Antosiewicz et al. 1994; Antosiewicz et al. 1996). Even with this relatively high dielectric constant, the calculations exaggerated the magnitude of some of the short-range interactions (His-8 in E10A and His-124 in E101), especially under conditions of low ionic strength. They also overestimated the magnitude of some medium- to long-range interactions (His-121 in K78E and His-121 in K127E). The pKa values calculated with the FDPB method are very sensitive to the details of the local microenvironment; some of these discrepancies between calculated and experimental pKa values might reflect subtle conformational changes in the mutants that are not captured correctly in the structural models of the mutants. They could probably be minimized by using protein dielectric constants higher than 20. Overall, the agreement between the calculated and the observed shifts in pKa values is very good.
Table 2.
pKavalues calculated with the FDPB method
| Salt concentration (M) | His-8 | His-46 | His-121 | His-124 |
| Wild type | ||||
| 0.01 | 5.96 | 4.98 | 4.51 | 5.43 |
| 0.10 | 6.35 | 5.43 | 5.18 | 5.96 |
| K78E | ||||
| 0.01 | 6.24 | 5.01 | 5.24 | 5.55 |
| 0.10 | 6.50 | 5.44 | 5.69 | 6.01 |
| K97E | ||||
| 0.01 | 6.09 | 5.01 | 4.74 | 5.98 |
| 0.10 | 6.40 | 5.44 | 5.31 | 6.30 |
| K127E | ||||
| 0.01 | 6.04 | 5.03 | 5.09 | 6.19 |
| 0.10 | 6.37 | 5.44 | 5.36 | 6.49 |
| K127A | ||||
| 0.01 | 6.00 | 5.01 | 4.81 | 5.78 |
| 0.10 | 6.36 | 5.44 | 5.38 | 6.20 |
| K97E/K127E | ||||
| 0.01 | 6.16 | 5.05 | 5.29 | 6.67 |
| 0.10 | 6.41 | 5.44 | 5.65 | 6.77 |
| E10A | ||||
| 0.01 | 5.04 | 4.97 | 4.45 | 5.41 |
| 0.10 | 5.58 | 5.44 | 5.16 | 5.96 |
| E101A | ||||
| 0.01 | 5.91 | 4.95 | 4.54 | 4.15 |
| 0.10 | 6.33 | 5.42 | 5.22 | 4.91 |
| R35A | ||||
| 0.01 | 6.02 | 5.15 | 4.64 | 5.53 |
| 0.10 | 6.38 | 5.39 | 5.28 | 6.03 |
Table 3.
Electrostatic energies of interaction between histidines and other ionizable groups (ΔGij) calculated at 0.10 M ionic strength with the FDPB methoda
| Contact | rij (Å)b | ΔGij (kcal/mole) | |
| His-8 | Glu-10 | 4.32 | 1.22 |
| Arg-81 | 8.13 | 0.43 | |
| Glu-75 | 8.50 | 0.40 | |
| Lys-28 | 8.20 | 0.36 | |
| Lys-9 | 9.07 | 0.29 | |
| Glu-73 | 9.86 | 0.28 | |
| His-46 | Glu-52 | 4.94 | 1.07 |
| Glu-43 | 5.33 | 0.93 | |
| Lys-49 | 5.75 | 0.64 | |
| Asp-40 | 9.73 | 0.41 | |
| Asp-21 | 10.27 | 0.37 | |
| Lys-48 | 7.78 | 0.35 | |
| Arg-35 | 11.09 | 0.34 | |
| Asp-19 | 10.54 | 0.28 | |
| Arg-87 | 11.86 | 0.27 | |
| His-121 | Glu-75 | 6.07 | 0.92 |
| His-124 | 6.30 | 0.76 | |
| Asp-77 | 7.27 | 0.73 | |
| Lys-9 | 8.14 | 0.49 | |
| Lys-78 | 8.77 | 0.36 | |
| Glu-101 | 9.92 | 0.34 | |
| Glu-122 | 10.49 | 0.27 | |
| His-124 | Glu-101 | 4.34 | 1.33 |
| His-121 | 6.30 | 0.76 | |
| Glu-75 | 10.35 | 0.35 | |
| Lys-9 | 9.49 | 0.32 | |
| Lys-127 | 9.49 | 0.28 | |
| Arg-105 | 10.81 | 0.26 |
a Interactions with |ΔGij| >0.25 kcal/mole are included.
b Distances measured between Nδ1 of His-8 and His-121, Nɛ2 of His-46 and His-124, and Nζ of Lyz, Cζ of Arg, Cɛ1 of His, Cγ of Asp, Cδ of Glu. The lstn structure was used.
Fig. 4.
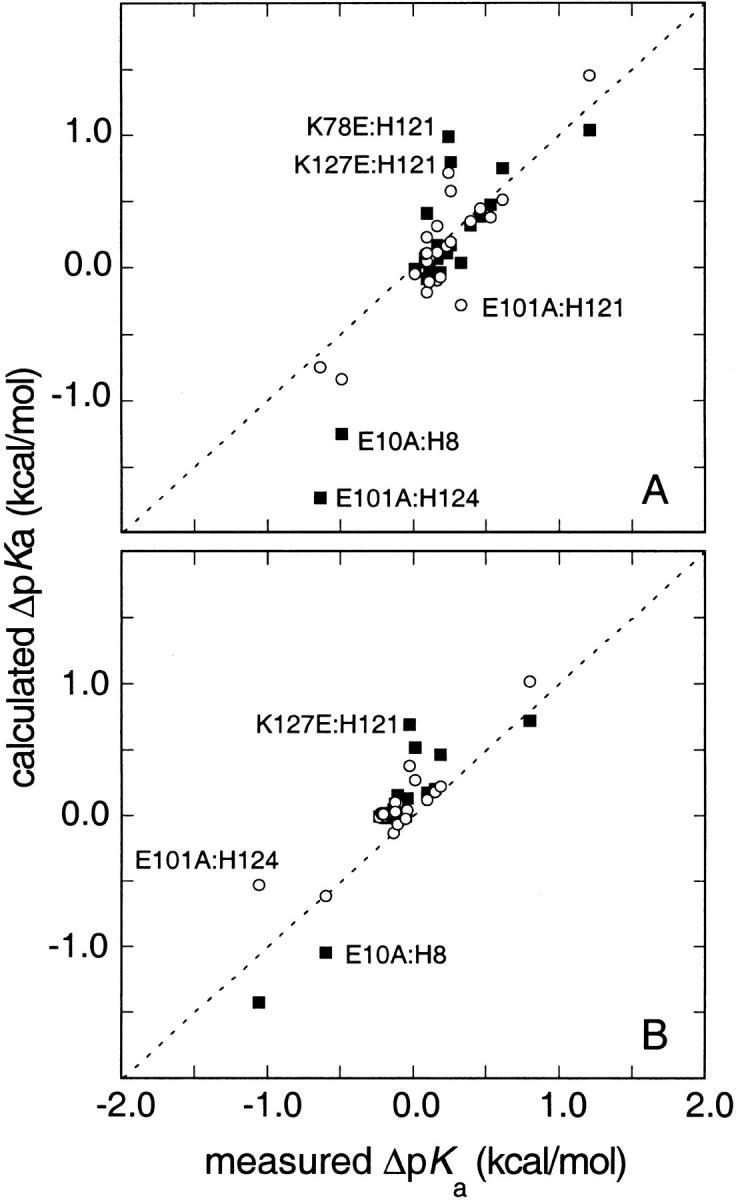
Comparison of measured and calculated ΔpKa = (pKaMUT-pKaWT) in charge-reversal and charge-neutralization mutants in (A) 0.01 M and (B) 0.10 M KCl, expressed in kcal/mole. Two sets of calculated ΔpKa are included: one obtained with the FDPB method (solid squares) and another obtained with Coulomb's law with a dielectric constant of 80 and a Debye-Hückel term to account for screening effects (open circles).
Discussion
The energy of interaction between two charges in water in salt concentration that is comparable to physiological conditions decays rapidly with increasing distance of separation and becomes less than 0.1 kcal/mole when the charges are separated by 8 to 10 Å. In principle, the strength and long-range character of electrostatic interactions on the surface of a protein can be magnified by the presence of the protein itself because the polarizability of proteins is much lower than the polarizability of water. In practice, this is not always borne out experimentally. Although cases of strong long-range interactions have been reported, weak interactions are more prevalent (Russell et al. 1987; Dao-pin et al. 1991a; Dao-pin et al. 1991b; Sancho et al. 1992; Loewenthal et al. 1993; Fisher et al. 1998; Grimsley et al. 1999; Shaw et al. 2001). The similarity between pKa values of ionizable groups in proteins and in water (Matthew et al. 1985), and the relatively minor consequences of substitution of ionizable residues with neutral ones on stability and on pKa values (references above), is consistent with weak coulombic interactions on the protein surface.
Computational studies have not provided clear insight about the strength and character of charge–charge interactions in proteins. The earliest model that successfully captured the ionization properties of surface residues of proteins was the semiempirical solvent accessibility-modified Tanford-Kirkwood (SATK) (Matthew et al. 1985). The interaction energies between individual pairs of charges (ΔGij) calculated with this model are always weak, even at short range, because the lowest effective dielectric constant sampled in the most common implementation of this method is 42 (Matthew et al. 1985). In the SATK model, the ΔGij are attenuated further by assuming they are linearly proportional to (1-SAij), where SAij is the normalized, averaged solvent accessibility of the interacting charges. Thus, the interaction energies tend toward 0 as solvent accessibility increases. In this model, ΔGij are extremely weak, particularly at long range, and pKa values are determined mostly by local coulombic interactions, consistent with some experimental observations (Dao-pin et al. 1991b).
In the structurally more realistic FDPB model, the effective dielectric constants that are sampled are much lower than in the SATK model (Warwicker and Watson 1982). The magnitude of ΔGij among surface charges calculated with the FDPB model when a single protein conformation is used and when the protein interior is treated with a dielectric constant of 4 can be substantial, even at long range. However, these calculations are not necessarily useful. They underestimate relaxation of the protein on ionization and they otherwise fail to capture the properties of water at the protein–water interface (Sham et al. 1998). It is known that the agreement between calculations and experiments can be improved by taking protein conformational relaxation into account explicitly, rather than subsuming these effects under a single-valued dielectric constant (You and Bashford 1995; van Vlijmen et al. 1998). Agreement can also be improved by treating the dielectric constant empirically with high values; the best agreement, when using a static structure, is obtained when values of 20 or higher are used (Antosiewicz et al. 1996). One goal of this study was to determine whether ΔGij calculated with this model using static structures and a protein dielectric constant of 20 are physically realistic over a wide range of distances and ionic strengths.
The unusual properties of the histidine pKa values of SNase allowed a stringent test of the structure-based calculations. Three of the four histidines titrate with depressed pKa values. On average, the pKa values are three times more sensitive to increases in ionic strength than histidines in model compounds and in other proteins; in three cases the pKa values increase by as much as 1 full unit between 0.01 M and 1.5 M KCl (Lee et al. 2002). This indicated that repulsive coulombic interactions contribute substantially to the histidine pKa values. However, no short-range (rij < 5 Å) contacts between histidines and basic residues are present in the crystallographic structure. The map of the distribution of charges and structure-based calculations with the FDPB model indicated that the dominant electrostatic influence on the histidines was exerted by charges more than 10 Å. This is in apparent conflict with the view, indicated by previous experimental work by others, that long-range electrostatic effects are too weak to influence the ionization properties of individual residues or the stability of proteins (Dao-pin et al. 1991b).
Distance dependence and salt sensitivity of charge–charge interactions
In charge-reversal mutants, the histidine pKa values were shifted upwards and their salt dependence was diminished relative to the wild type, as expected from the replacement of a repulsive coulombic interaction with an attractive one. The effects of each mutation were predominantly local, although the electrostatic effects of charge substitution could be sensed weakly at a distance of several angstroms. The functional dependence of ΔGij on distance of charge separation was sufficiently well defined to allow identification of the general trends.
Figure 5 ▶ describes the distance dependence of the ΔpKa, represented in terms of ΔGij. The standard deviations in pKa values listed in Table 1 translate to errors in ΔGij of approximately ±0.06 kcal/mole. The size of the error in rij is considerably more difficult to evaluate. Surface residues with long side chains are highly mobile and their average positions, as described by crystallographic structures, are only approximate. The error bar in the rij dimension could be large. Lys→Glu mutations were used, and Lys→Ala were avoided partly to minimize this error. Models of structures were used to determine rij, but it is not obvious that actual crystallographic structures of the mutants would have afforded a more precise measure of distances. The data for E10A and E101A were included in Figure 5 ▶, although the chemical shifts and the nature of the shifts in pKa values indicated that those mutations influenced the conformation of the protein slightly. Their omission would not change the analysis or the conclusions based on the data in Figure 5 ▶ in a significant way.
Fig. 5.

Distance dependence of the energy of interaction between pairs of charges obtained from normalized shifts in histidine pKa values expressed as interaction energies (ΔGij = −1.36 ΔpKa at 298K) in 0.01 M (solid circles), 0.10 M (solid triangles), and 1.5 M KCl (cross-hatched squares). Data for the mutants K78E, K97E, K127E, K127A, R35A, E10A, and E101A are included. ΔpKa measured in charge-reversal mutations were divided by two; those measured in E10A and E101A were multiplied by −1. In the Ala substitution mutants, the distances between Cɛ1 of the His and the Cδ of Glu, Nζ of Lys or Cζ of Arg were obtained from the wild-type structure (Hynes and Fox 1991). In the charge-reversal mutants, the Cδ of Glu in energy-minimized models of the mutants was used to calculate distances. Data for His-18 in barnase in seven mutants at 0.01 M ionic strength are plotted for comparison (open circles) (Loewenthal et al. 1993). The lines represent fits of equation 1 to the experimental data at ionic strengths of 0.01, 0.10, and 1.5 M.
The data in Figure 5 ▶ are consistent with previous studies showing that the energy of interactions between surface-charged groups in proteins is small. Trends in the distance dependence of charge–charge interactions, not noted previously by others, are apparent because of the large number of data obtained. A coulombic potential, with a Debye-Hückel term derived from a model-dependent solution of the linearized Poisson-Boltzmann equation included to capture the effects of screening by counterions, was used to account for the general trends:
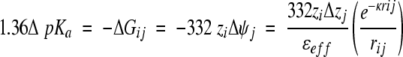 |
(1) |
The distance of separation between the two charges, rij, is the only structural information taken into account in this expression. Analysis of ΔGij with this function assumes that the full pKa shift is attributable to the pairwise interaction, which is a valid assumption for the predominantly medium- and long-range interactions described by the data included in Figure 5 ▶. In this expression, ψj refers to the electrostatic potential at site i caused by charge j; zi is the charge on the histidine, and zj is the charge at the charge-substituted site. When acidic (E10A, E101A) or basic (K127A, R35A) residues were replaced with Ala, Δzj was +1 or −1, respectively. For the Lys→Glu mutations, Δzj was −2. ΔGij is the free energy of interaction in units of kcal/mole and κ = 50.3middot;(I/ɛH20T)1/2 is the Debye parameter in units of Å−1, with I, ɛH20, and T, the ionic strength (M), dielectric constant of water, and temperature (K), respectively. ɛeff is the effective dielectric constant that modulates the magnitude of pairwise interactions among charges. In pure water ɛeff would be 78.5 (25°C). Values of ɛeff in proteins resolved by fitting subsume all dielectric relaxation processes involved in producing the observed ΔGij.
Equation 1 describes the experimental data very well despite the uncertainties in both ΔGij and rij. In ionic strengths of 0.1 M and 1.5 M, the shape of the curve is determined mostly by the exponential term, eκrij. At the lowest ionic strength studied, the rij−1 term in the coulombic potential is sufficient to capture the distance dependence of the measured data. When equation 1 was fitted to the data, the fitted values of ɛeff in 0.01 M and 0.10 M were 97 ± 12 and 82 ± 13, respectively. The fitted curves are shown in Figure 5 ▶. They are indistinguishable from the curves plotted with ɛeff fixed at 78.5, the dielectric constant of water. It is noteworthy that the fits to the data obtained in 1.5 M ionic strength are not as good as in 0.01 M or 0.10 M ionic strength. This indicates that the Debye-Hückel model underestimates ΔGij of short-range interactions under these conditions, which is not surprising. Nevertheless, the simple coulombic model with a Debye-Hückel term accurately reproduces the pairwise electrostatic potential, even under conditions of ionic strength of 0.10 M and higher, in which the Debye-Hückel theory is formally invalid. Others have observed previously that the magnitude of charge–charge interactions among surface residues can be reproduced with Coulomb's law (Akke and Forsen 1990; Kesvatera et al. 1994; Grimsley et al. 1999).
The utility of the fitted curves in Figure 5 ▶ is that they capture the general trends. For example, under conditions of no added salt, pairs of charges interact with ΔGij ≥ 0.1 kcal/mole up to rij = 20 Å. In 0.10 M ionic strength, only interactions within rij ≤ 12 Å have ΔGij ≥ 0.1 kcal/mole; however, short-range interactions can be as strong as in 0.01 M ionic strength. In 1.5 M ionic strength, only interactions with rij ≤ 5 Å persist. There is a rough correlation between distances beyond which ΔGij ≤ 0.1, and the Debye length, 1/κ. The Debye length is the distance from the central charge to the position of maximum fractional charge of the counterion cloud. In 0.01 M, 0.10 M, and 1.5 M ionic strength, the Debye length is approximately 30 Å, 10 Å, and 2.5 Å, respectively. The Debye length is a useful metric to describe how far the coulombic potential of a charge can be felt. The data in Figure 5 ▶ show that the distances beyond which charge–charge interactions on the surface of SNase become negligible at a given ionic strength are consistent with the Debye length at that ionic strength.
Comparison of measured and calculated energy of pairwise coulombic interactions
The comparison of the experimental ΔpKa with the values calculated with the FDPB method (Figure 4 ▶) shows that, overall, the calculations successfully reproduce the experimental pairwise interaction energies. The deviations between calculated and experimental values are smaller in 0.10 M ionic strength than in 0.01 M. They are especially noticeable for cases that involve short-range interactions, such as the interactions between Glu-10 and His-8, and Glu-101 and His-124. Note that these are also the two cases in which we suspect the mutations have structural consequences, so the problems in capturing the pKa values with the calculations might be related to the structures used in the calculations.
The slight deviations between experimental and calculated ΔpKa can be appreciated more easily in Figure 6 ▶, which illustrates the distance dependence of calculated ΔGij in ionic strengths of 0.01 M and 0.10 M and compares them with experimental values. The calculated ΔGij greater than 0.25 kcal/mole listed in Table 3 are included in this figure. The data in Figure 6 ▶ show very clearly that at 0.01 M and 0.10 M ionic strength, when rij < 12 Å, the calculations systematically exaggerate the magnitude of ΔGij by a factor of approximately two. When equation 1 was fitted through the calculated ΔGij, values of ɛeff of 53 and 36 were resolved for the data at ionic strengths 0.01 M and 0.10 M, respectively. This illustrates two things. First, although the shape of equation 1 is dominated by the presence of the exponential term in the denominator, this function is sensitive enough to detect the systematic exaggeration of the calculated ΔGij. Second, even when the protein interior was treated with a dielectric constant of 20, the ɛeff modulating the calculated ΔGij in the calculations are still considerably lower than the values of ɛeff ≈ 80 resolved from the experimental data.
Fig. 6.
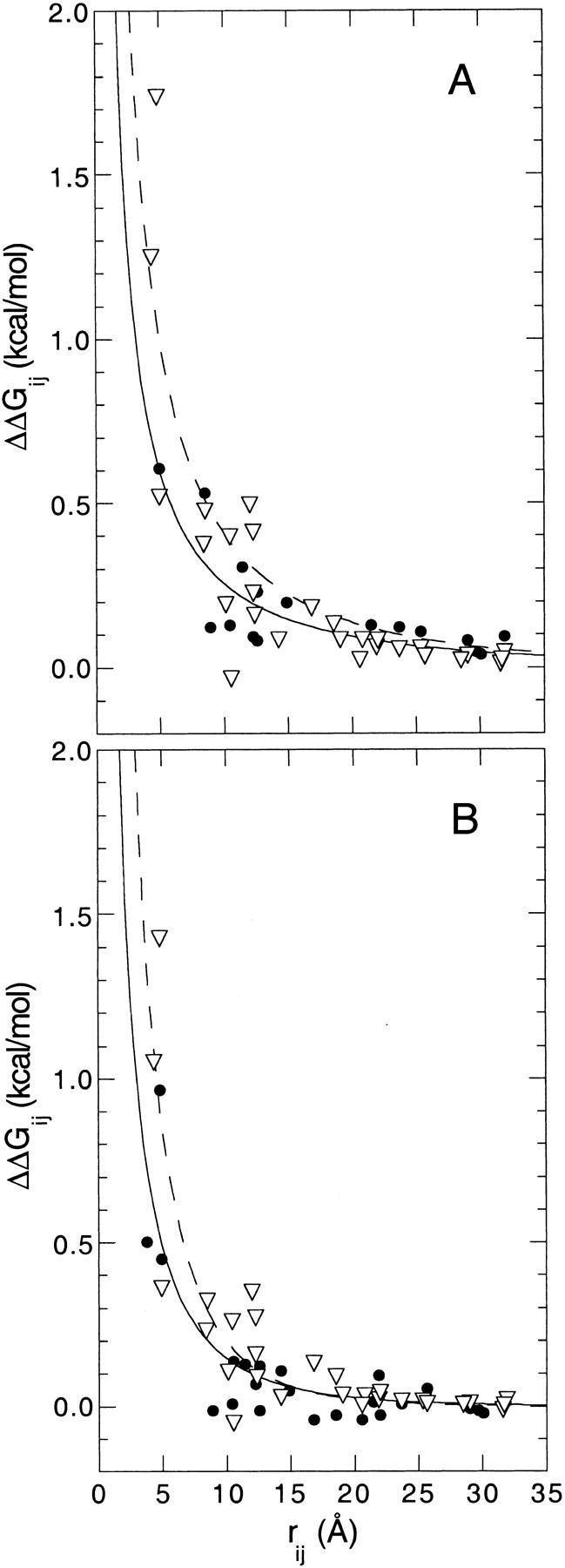
Comparison of the distance dependence of the energy of interaction between pairs of charges (ΔGij) measured experimentally (closed circles) and calculated with the FDPB method (open triangles) in (A) 0.01 M and (B) 0.10 M KCl. Also shown are the fits with equation 1 for the experimental (solid lines) and calculated (dashed lines) data, as described in the text.
In general, the pairwise energies of interaction calculated with equation 1 were comparable to those obtained using the FDPB structure-based method. Equation 1 underestimates the interaction energy at short range, for example, in the case of the response of His-124 to the E101A and K127E mutations in 0.10 M ionic strength. Because the Debye-Hückel calculations treat charges as point charges immersed in a high dielectric medium, they fail to capture the strengthening of coulombic interactions that would arise from the exclusion of water at the interface formed by two charges in direct contact.
The success of the simple coulombic model in reproducing the experimental ΔGij indicates that in the time scale of the equilibrium experiments used to measure pKa values, the environment sensed by surface residues is very water-like. This is not always reflected in the effective dielectric constants sampled in the FDPB calculations, which underestimate the dielectric response of water at this interface. This tendency of the FDPB method to exaggerate the magnitude of ΔGij in calculations with static structures, even when an artificially high dielectric constant of 20 is assigned to the protein, has been documented previously (Antosiewicz et al. 1994; Forsyth et al. 1998; Kao et al. 2000). To improve this situation it might be necessary to treat some aspects of the properties of water at the protein–water interface more explicitly, as proposed previously by others (Gibas and Subramaniam 1996), rather than to subsume the effects in empirical dielectric constants.
Molecular origins of the salt dependence of p Ka values of histidines
The dramatic increase in the pKa values of histidines in SNase with increasing salt concentration was interpreted previously in terms of screening of small contributions from medium- and long-range coulombic interactions (Lee et al. 2002). This interpretation was based partly on the distribution of positive and negative charges around each histidine, which showed that charge is balanced up to a distance 10 Å away from the histidines, but that beyond this distance this balance is broken. The experimental data in Figure 5 ▶ support this interpretation. They show conclusively that interactions between pairs of charges on the surface of SNase separated by rij ≥ 10 Å are very weak. However, these weak interactions are large enough that, in aggregate, they can have measurable effects on pKa values.
FDPB calculations reproduce the salt dependence of wild-type SNase histidine pKa values very well (Lee et al. 2002). The data in Figures 4, 5, and 6 ▶ ▶ ▶ showed that the calculations captured the experimental behavior for the right physical reasons. Surprisingly, although the calculations of salt dependence of pKa values in wild-type SNase were most successful for His-121, this is also the group for which the calculations most consistently appear to overestimate the magnitude of shifts caused by charge-reversal of neighboring ionizable residues (e.g., K78E and K127E) (Figure 4 ▶). The data indicate that the structure-based calculations may have fortuitously captured the balance of interactions involving His-121, although inaccurately computing the magnitude of individual pairwise interactions between this histidine and other charged groups. This discrepancy may reflect the complex electrostatic network in which His-121 is entangled (Fig. 3 ▶, Table 2) and the inherent difficulty of treating strongly coupled systems.
Comparison with other proteins
A unique aspect of this study is that a sufficiently large number of ΔGij values were measured, allowing description of the general trends in the distance dependence and salt sensitivity of pairwise coulombic interactions on the surface of nuclease. Comparison with data measured in other proteins indicates that the electrostatic properties of SNase are common to other proteins. The ΔGij values measured in barnase and subtilisin with an approach similar to ours are comparable to the values measured in SNase (Russell et al. 1987; Loewenthal et al. 1993). The values for barnase are included in Figure 5 ▶ for comparison with the values in SNase. Similarly, His-12, His-105, and His-119 in RNase A are 10, 15, and 4.7 Å from Asp-121, and the D121N mutation shifted their pKa values by 0.18, 0.16, and 0.02 units, respectively (Cederholm et al. 1991). In ovomucoid third domain, a Lys that is 5 to 11 Å from three acidic residues has little if any influence on their pKa values, even at 0.01 M ionic strength (Forsyth et al. 1998; Forsyth and Robertson 2000). The insensitivity of the acidic residue was surprising. According to FDPB calculations, the K34T and K34Q mutation shift the pKa values upward by 0.4 to 0.6 pKa units because of the loss of the favorable Coulombic interaction between Lys-34 and acidic residues, whereas the largest measured shifts were ∼0.15 pKa units. The agreement between ΔGij measured in SNase and in the ovomucoid third domain is excellent for rij > 10 Å, but at shorter range the interactions in the ovomucoid are even weaker than in nuclease for reasons that are not obvious. In general, the distance dependence of charge–charge interactions measured in SNase is characteristic of other proteins, and in all the cases studied, long-range interactions are always weak.
The situation is more complicated in active sites and in other contexts in which short-range interactions, or otherwise coupled effects, are involved. This is evident from measurements in the triple mutant K7A/R10A/K66A of RNaseA (Fisher et al. 1998). In the wild type, the pKa values of His-12, His-48, and His-119 are substantially depressed. His-48 is far from Lys-7, Arg-10, and Lys-66, and its pKa showed only minor shifts of ∼0.1 in response to the triple mutation. His-12 and His-119 are ∼6 to 14 Å from these basic residues and their pKa values are shifted between 0.4 and 0.8 units in 0.018 M ionic strength in the triple mutant. Much stronger short-range effects than those measured in SNase were also encountered in protein tyrosine phosphatase (Tishmack et al. 1997). ΔpKa of −2.36 and −1.69 pKa units were measured in ionic strengths of 0.13 M for His-72 in mutants in which acidic residues that are 3.3 Å and 4.3 Å were mutated to Ala (E23A and D42A). The pKa value of His-72 shifted downward by 2.99 pKa units in the E23A/D42A double mutant, a nonadditive response. His-66 did not respond to mutations at either Glu-23 (15.7 Å distant) or at Asp-42 (20.6 Å distant) individually or when combined in the double mutant, but when Glu-139 (3.6 Å from His-66) was mutated to Ala, the pKa value of His-66 dropped by 1.24 units. The shifts in histidine pKa values observed in charge-neutralization mutants of tyrosine phosphatase are much larger than the largest shifts observed in SNase because of charge-reversal under much lower counterion concentrations (namely, the increase of 0.91 in the pKa of His-124 in the K127E mutant at 0.01 M ionic strength). These groups in tyrosine phosphates are part of a complex network of electrostatic and hydrogen bonding interactions that are probably altered considerably in the mutants, thus accounting for the nonadditive effects of mutations E23A and D42A. The factors that influence the magnitude of interactions between charged groups at short range or in highly coupled networks are not well understood, which is unfortunate because they are of importance in catalysis, redox reactions, and proton transport.
Conclusions
The coulombic function that captured the experimental data is useful for summarizing the following general trends. Pairwise coulombic interactions among surface charges are weak at distances of interaction greater than 5 Å. Under ionic strengths near physiological values (0.10 M), the energy of interactions between charges separated by 10 Å is approximately 0.1 kcal/mole. This increases to 0.5 to 1.0 kcal/mole when the charges are separated by only 5 Å, which is similar to the energy of interaction in 0.01 M ionic strength at these short intercharge distances. In 0.01 M, interaction energies of 0.1 kcal/mole are still present when the charges are separated by 20 Å. At ionic strength 1.5 M, only charges separated by 5 Å or less have a measurable interaction, equivalent to 0.3 kcal/mole.
The pKa values of most surface residues in proteins are very similar to their values in small peptides in water (Matthew et al. 1985). Because pKa values reflect a balance between coulombic and self-energy effects, the observation that coulombic effects are weak implies that the contribution by the self-energy to the pKa values of surface residues must also be modest. The interface between proteins and water is apparently very water-like, and the state of hydration of surface residues seems to be unaffected by the presence of the protein.
The fact that long-range electrostatic interactions between pairs of charges are weak does not diminish the possibility that long-range electrostatic effects can influence the physical properties of proteins significantly. In proteins where the distribution of basic and acidic residues is not balanced, or under conditions of pH in which the number of positive or negative interactions is not balanced, weak long-range electrostatic interactions can, in aggregate, give rise to a considerable effect. This is the case with SNase. Each long-range, pairwise, repulsive interaction between a histidine and another basic residue is weak, but there are a large number of such interactions that are not balanced by similar interactions with acidic residues; thus, in aggregate, the weak, long-range, repulsive interactions produce a substantial effect. They contribute significantly to the depressed pKa values of histidines in SNase. They are also responsible for the dramatic salt sensitivity of these histidines because long-range coulombic interactions are screened very effectively by salt.
The magnitude of pairwise energies of interaction calculated with the structure-based continuum method using a static structure and treating the protein interior with a dielectric constant of 20 are in overall good agreement with the experimental values. The calculations correctly identified the extreme salt sensitivity of the histidines as originating from the salt sensitivity of many weak, long-range electrostatic effects. It is noteworthy that the salt and distance dependence of ΔGij were also described very well with a coulombic potential with a Debye-Hückel term to account for screening by the ionic strength. These simple functions captured the measured energetics as accurately as (and in a few notable cases, more accurately than) the structure-based FDPB calculations. This indicates that the FDPB calculations still underestimate the relaxation of solvent and protein on ionization, even when the protein interior is treated artificially with a dielectric constant of 20. The simple coulombic expressions might be useful in computationally demanding calculations in very large systems, or within molecular dynamics simulation, in which it is often desirable to capture hydration and ionic double-layer effects implicitly. These simple functions will also be useful to guide rational attempts to modulate the stability of proteins by optimizing charge–charge interactions (Loladze et al. 1999; Spector et al. 2000; Sanchez-Ruiz and Makhatadze 2001; Shaw et al. 2001).
Materials and methods
Proteins
Clones of K78E were a gift from Professor Wesley Stites. Those of E10A, E101A, K127A, and R35A were constructed by Professor David Shortle (Meeker et al., 1996). All other mutants, K97E, K127E, K97E/K127E, and K78E/K97E/K127E were made using the Stratagene Quikchange site-directed mutagenesis kit. Mutated plasmids were transformed into the AR120 Escherichia coli cell line for expression. Protein purification and sample preparation were performed by procedures described previously (Whitten and García-Moreno E. 2000).
NMR experiments
Assignments of resonances corresponding to each of four histidines in wild-type SNase have been determined previously by Markley and coworkers by means of site-directed mutagenesis (Alexandrescu et al. 1988). We have found that relative to one another, the histidine resonance chemical shift positions corresponding to the fully deprotonated states at basic pH values are largely invariant with respect to changes in salt condition. By comparing spectra of wild-type and mutant proteins, resonances for each of the four histidines were identified unambiguously for all mutants studied.
Sample preparation and NMR experiments to obtain pKa values of histidine residues were performed as previously reported (Kao et al. 2000; Fitch et al. 2002). Protein concentrations in the NMR experiments were 0.6 to 1.0 mM (10 to 17 mg/mL) in D2O solutions.
The pKa values were obtained from nonlinear least squares fitting of a modified Henderson-Hasselbach equation to the titration curve described by the pH dependence of the chemical shifts for resonances assigned to the histidine Cɛ1H (Markley 1975):
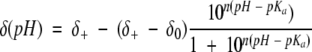 |
(2) |
δ+ and δo refer to the chemical shifts of the acidic and basic asymptotes of the titration, respectively. n is a Hill coefficient that improves the goodness of fit by allowing the transition steepness to vary. pKa values are based on the uncorrected pH readings measured in D2O solutions (Glasoe and Long 1960).
Previous systematic studies of SNase histidine pKa values at 0.10 M KCl revealed no significant concentration-dependent effects over the range of protein concentrations typically used in our experiments. In the present study, experiments under conditions of no added salt, 0.10 M, and 1.5 M KCl were performed. Repetitions of titrations in the absence of added salt and at high salt concentrations over the 0.4 mM to 1.0 mM SNase concentration range yielded precisely superimposable protonation curves for all four histidines. Typical reproducibility trials under all three salt conditions yield experimental errors that are comparable to the standard error of fits of equation 2 to experimental data. Under conditions of no added salt, the ionic strength resulting from the introduction of small amounts of DCl and KOD titrants over the course of the pH titration was estimated to be 0.01 M.
Structure-based calculations of p Ka values and pairwise energies of interaction, ΔGij
pKa calculations were performed with the finite difference Poisson-Boltzmann method (FDPB) described previously (Antosiewicz et al. 1994; Antosiewicz et al. 1996). Electrostatic potentials were calculated with the University of Houston Brownian Dynamics package (Davis et al. 1991). Charge states were determined with the statistical thermodynamic algorithm by Gilson (Gilson 1993). The implementation used here follows the procedure described previously for SNase (Fitch et al. 2002). The adjustable parameters that were used are: ɛin = 20, ɛout = 78.5, T = 298 K, Stern layer = 2.0 Å, and probe radius = 1.4 Å.
Structural models of the mutants were built by amino acid substitutions in the 1stn structure (Hynes and Fox 1991) using the Molecule Builder function within the Insight II software package (Accelrys Inc.). The mutated sidechain position was energy minimized using the CHARMm program (Accelrys Inc.) against a fixed background with protonatable sites in their neutral states, as discussed previously for SNase (Fitch et al. 2002).
Acknowledgments
The authors gratefully acknowledge gifts of overproducing strains from Professors Wesley Stites (University of Arkansas) and David Shortle (Johns Hopkins University). The authors thank Ms. Mei-Lin Zimmerman for technical assistance. This work was supported by NSF grant MCB-9600991 to B.G.-M.E.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.4700102.
References
- Accelrys Inc. CHARMm, www.accelrys.com.
- Accelrys Inc. InsightII, www.accelrys.com.
- Akke, M. and Forsen, S. 1990. Protein stability and electrostatic interactions between solvent exposed charged side chains. Proteins 8 23–29. [DOI] [PubMed] [Google Scholar]
- Alexandrescu, A.T., Mills, D.A., Ulrich, E.L., Chinami, M., and Markley, J.L. 1988. NMR assignments of the 4 histidines of staphylococcal nuclease in native and denatured states. Biochemistry 27 2158–2165. [DOI] [PubMed] [Google Scholar]
- Antosiewicz, J., McCammon, J.A., and Gilson, M.K. 1994. Prediction of pH-dependent properties of proteins. J. Mol. Biol. 238 415–436. [DOI] [PubMed] [Google Scholar]
- ———. 1996. The determinants of pKas in proteins. Biochemistry 35 7819–7833. [DOI] [PubMed] [Google Scholar]
- Bashford, D. and Karplus, M. 1990. pKa's of ionizable groups in proteins: Atomic detail from a continuum electrostatic model. Biochemistry 29 10219–10225. [DOI] [PubMed] [Google Scholar]
- Blasie, C.A. and Berg, J.M. 1997. Electrostatic interactions across a beta-sheet. Biochemistry 36 6218–6222. [DOI] [PubMed] [Google Scholar]
- Cederholm, M.T., Stuckey, J.A., Doscher, M.S., and Lee, L. 1991. Histidine pKa shifts accompanying the inactivating Asp121Asn substitution in a semisynthetic bovine pancreatic ribonuclease. Proc. Natl. Acad. Sci. 88 8116–8120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dao-pin, S., Sauer, U., Nicholson, H., and Matthews, B.W. 1991a. Contributions of engineered surface salt bridges to the stability of T4 lysozyme determined by directed mutagenesis. Biochemistry 30 7142–7153. [DOI] [PubMed] [Google Scholar]
- Dao-pin, S., Sóderlind, E., Baase, W.A., Wozniak, J.A., Sauer, U., and Matthews, B.W. 1991b. Cumulative site-directed charge–change replacements in bacteriophage T4 lysozyme suggest that long-range electrostatic interactions contribute little to protein stability. J. Mol. Biol. 221 873–887. [DOI] [PubMed] [Google Scholar]
- Davis, M.E., Madura, J.D., Luty, B.A., and McCammon, J.A. 1991. Electrostatics and diffusion of molecules in solution—simulations with the University of Houston Brownian Dynamics program. Comput. Phys. Commun. 62 187–197. [Google Scholar]
- Dwyer, J., Gittis, A., Karp, D., Lattman, E., Spencer, D., Stites, W., and García-Moreno E., B. 2000. High apparent dielectric constants in the interior of a protein reflect water penetration. Biophys. J. 79 1610–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer, S. and Verma, C.S. 1999. Binding of buried structural water increases the flexibility of proteins. Proc. Natl. Acad. Sci. 96 9613–9615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher, B., Schultz, L., and Raines, R. 1998. Coulombic effects of remote subsites on the active site of ribonuclease A. Biochemistry 37 17386–17401. [DOI] [PubMed] [Google Scholar]
- Fitch, C.A., Karp, D.A., Lee, K.K., Stites, W.E., Lattman, E.E., and García-Moreno E., B. 2002. Experimental pKa values of buried residues: Analysis with continuum methods and role of water penetration. Biophys. J., in press. [DOI] [PMC free article] [PubMed]
- Forsyth, W.R., Gilson, M.K., Antosiewicz, J., Jaren, O.R., and Robertson, A.D. 1998. Theoretical and experimental analysis of ionization equilibria in ovomucoid third domain. Biochemistry 37 8643–8652. [DOI] [PubMed] [Google Scholar]
- Forsyth, W.R. and Robertson, A.D. 2000. Insensitivity of perturbed carboxyl pKa values in the ovomucoid third domain to charge replacement at a neighboring residue. Biochemistry 39 8067–8072. [DOI] [PubMed] [Google Scholar]
- Gibas, C.J. and Subramaniam, S. 1996. Explicit solvent models in protein pKa calculations. Biophys. J. 71 138–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilson, M.K. 1993. Multiple-site titration and molecular modeling: Two rapid methods for computing energies and forces for ionizable groups in proteins. Proteins 15 266–282. [DOI] [PubMed] [Google Scholar]
- Glasoe, P.K. and Long, F.A. 1960. Use of glass electrodes to measure acidities in deuterium oxide. J. Phys. Chem. 64 188–190. [Google Scholar]
- Grimsley, G., Shaw, K., Fee, L., Alston, R., Huyghues-Despointes, B., Thurlkill, R., Scholtz, J., and Pace, C. 1999. Increasing protein stability by altering long-range coulombic interactions. Protein Sci. 8 1843–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes, T.T. and Fox, R.O. 1991. The crystal structure of staphylococcal nuclease refined at 1.7 Å resolution. Proteins 10 92–105. [DOI] [PubMed] [Google Scholar]
- Ibarra-Molero, B., Loladze, V.V., Makhatadze, G.I., and Sanchez-Ruiz, J.M. 1999. Thermal versus guanidine-induced unfolding of ubiquitin. An analysis in terms of the contributions from charge–charge interactions to protein stability. Biochemistry 38 8138–8149. [DOI] [PubMed] [Google Scholar]
- Kao, Y.-H., Fitch, C.A., Bhattacharya, S., Sarkisian, C.J., Lecomte, J.T.J., and García-Moreno E., B. 2000. Salt effects on ionization equilibria of histidines in myoglobin. Biophys. J. 79 1637–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kesvatera, T., Jönsson, B., Thulin, E., and Linse, S. 1994. Binding of Ca2+ to Calbinding D9K: Structural stability and function at high salt concentration. Biochemistry 33 14170–14176. [DOI] [PubMed] [Google Scholar]
- Kuhlman, B., Luisi, D., Young, P., and Raleigh, D. 1999. pKa values and the pH dependent stability of the N-terminal domain of L9 as probes of electrostatic interactions in the denatured state. Differentiation between local and nonlocal interactions. Biochemistry 38 4896–4903. [DOI] [PubMed] [Google Scholar]
- Lee, K.K., Fitch, C.A., Lecomte, J.T.J., and García-Moreno E., B. 2002. Electrostatic effects in highly changed proteins: Salt sensitivity of pKa values of histidines in staphylococcal nuclease. Biochemistry, in press. [DOI] [PubMed]
- Loewenthal, R., Sancho, J., Reinikainen, T., and Fersht, A.R. 1993. Long-range surface charge–charge interactions in proteins. Comparison of experimental results with calculations from a theoretical method. J. Mol. Biol. 232 574–583. [DOI] [PubMed] [Google Scholar]
- Loladze, V., Ibarra-Molero, B., Sanchez-Ruiz, J., and Makhatadze, G. 1999. Engineering a thermostable protein via optimization of charge–charge interactions on the protein surface. Biochemistry 38 16419–16423. [DOI] [PubMed] [Google Scholar]
- Markley, J. 1975. Observation of histidine residues in proteins by means of nuclear magnetic resonance spectroscopy. Acc. Chem. Res. 8 70–80. [Google Scholar]
- Matthew, J.B., Gurd, F.R.N., García-Moreno E., B., Flanagan, M.A., March, K.L., and Shire, S.J. 1985. pH-dependent properties in proteins. CRC Crit. Rev. Biochem. 18 91–197. [DOI] [PubMed] [Google Scholar]
- Meeker, A.K., García-Moreno E., B., and Shortle, D. 1996. Contributions of the ionizable amino acids to the stability of staphylococcal nuclease. Biochemistry 35 6443–6449. [DOI] [PubMed] [Google Scholar]
- Oliveberg, M., Vuilleumier, S., and Fersht, A. 1994. Thermodynamic study of the acid denaturation of barnase and its dependence on ionic strength: Evidence for residual electrostatic interactions in the acid/thermally denatured state. Biochemistry 33 8826–8832. [DOI] [PubMed] [Google Scholar]
- Oliveberg, M., Arcus, V.L., and Fersht, A.R. 1995. pKa values of carboxyl groups in the native and denatured states of barnase: The pKa values of the denatured state are on average 0.4 units lower than those of model compounds. Biochemistry 34 9424–9433. [DOI] [PubMed] [Google Scholar]
- Pace, C., Alston, R., and Shaw, K. 2000. Charge–charge interactions influence the denatured state ensemble and contribute to protein stability. Protein Sci. 9 1395–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perl, D., Mueller, U., Heinemann, U., and Schmid, F.X. 2000. Two exposed amino acid residues confer thermostability on a cold shock protein. Nat. Struct. Biol. 7 380–383. [DOI] [PubMed] [Google Scholar]
- Russell, A.J., Thomas, P.G., and Fersht, A.R. 1987. Electrostatic effects on modification of charged groups in the active site cleft of subtilisin by protein engineering. J. Mol. Biol. 193 803–813. [DOI] [PubMed] [Google Scholar]
- Sanchez-Ruiz, J.M. and Makhatadze, G.I. 2001. To charge or not to charge? Trends Biotechnol. 19 132–135. [DOI] [PubMed] [Google Scholar]
- Sancho, J., Serrano, L., and Fersht, A.R. 1992. Histidine residues at the N- and C-termini of alpha-helices: Perturbed pKas and protein stability. Biochemistry 31 2253–2258. [DOI] [PubMed] [Google Scholar]
- Schutz, C.N. and Warshel, A. 2001. What are the dielectric "constants" of proteins and how to validate electrostatic models? Proteins 44 400–417. [DOI] [PubMed] [Google Scholar]
- Serrano, L., Horovitz, A., Avron, B., Bycroft, M., and Fersht, A.R. 1990. Estimating the contribution of engineered surface electrostatic interactions to protein stability by using double-mutant cycles. Biochemistry 29 9343–9352. [DOI] [PubMed] [Google Scholar]
- Sham, Y.Y., Chu, Z.T., and Warshel, A. 1997. Consistent calculations of pKa's of ionizable residues in proteins: Semi-microscopic and microscopic approaches. J. Phys. Chem. B. 101 4458–4472. [Google Scholar]
- Sham, Y.Y., Muegge, I., and Warshel, A. 1998. The effect of protein relaxation on charge–charge interactions and dielectric constants of proteins. Biophys. J. 74 1744–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw, K.L., Grimsley, G.R., Yakovlev, G.I., Makarov, A.A., and Pace, C.N. 2001. The effect of net charge on the solubility, activity, and stability of ribonuclease Sa. Protein Sci. 10 1206–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector, S., Wang, M., Carp, S., Robblee, J., Hendsch, Z., Fairman, R., Tidor, B., and Raleigh, D. 2000. Rational modification of protein stability by the mutation of charged surface residues. Biochemistry 39 872–879. [DOI] [PubMed] [Google Scholar]
- Swint-Kruse, L. and Robertson, A.D. 1995. Hydrogen-bonds and the pH-dependence of ovomucoid 3rd domain stability. Biochemistry 34 4724–4732. [DOI] [PubMed] [Google Scholar]
- Tan, Y.J., Oliveberg, M., Davis, B., and Fersht, A.R. 1995. Perturbed pKa-values in the denatured states of proteins. J. Mol. Biol. 254 980–992. [DOI] [PubMed] [Google Scholar]
- Tishmack, P.A., Bashford, D., Harms, E., and Van Etten, R.L. 1997. Use of 1H-NMR spectroscopy and computer simulations to analyze histidine pKa changes in a protein tyrosine phosphatase: Experimental and theoretical determination of electrostatic properties in a small protein. Biochemistry 36 11984–11994. [DOI] [PubMed] [Google Scholar]
- van Vlijmen, H.W.T., Schaefer, M., and Karplus, M. 1998. Improving the accuracy of protein pKa calculations: Conformational averaging versus the average structure. Proteins 33 145–158. [DOI] [PubMed] [Google Scholar]
- Warwicker, J. and Watson, H.C. 1982. Calculation of the electric potential in the active site cleft due to a-helix dipoles. J. Mol. Biol. 157 671–679. [DOI] [PubMed] [Google Scholar]
- Whitten, S.T. and García-Moreno E., B. 2000. pH dependence of stability of staphylococcal nuclease: Evidence of substantial electrostatic interactions in the denatured state. Biochemistry 39 14292–14304. [DOI] [PubMed] [Google Scholar]
- You, T. and Bashford, D. 1995. Conformation and hydrogen ion titration of proteins: A continuum electrostatic model with conformational flexibility. Biophys. J. 69 1721–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]