

Abstract
2-Hydroxy-6-oxo-7-methylocta-2,4-dienoate hydrolase (CumD) from Pseudomonas fluorescens IP01 hydrolyzes a meta-cleavage product generated in the cumene (isopropylbenzene) degradation pathway. The crystal structures of the inactive S103A mutant of the CumD enzyme complexed with isobutyrate and acetate ions were determined at 1.6 and 2.0 Å resolution, respectively. The isobutyrate and acetate ions were located at the same position in the active site, and occupied the site for a part of the hydrolysis product with CumD, which has the key determinant group for the substrate specificity of related hydrolases. One of the oxygen atoms of the carboxyl group of the isobutyrate ion was hydrogen bonded with a water molecule and His252. Another oxygen atom of the carboxyl group was situated in an oxyanion hole formed by the two main-chain N atoms. The isopropyl group of the isobutyric acid was recognized by the side-chains of the hydrophobic residues. The substrate-binding pocket of CumD was long, and the inhibition constants of various organic acids corresponded well to it. In comparison with the structure of BphD from Rhodococcus sp. RHA1, the structural basis for the substrate specificity of related hydrolases, is revealed.
Keywords: X-ray crystallography, α/β-hydrolase, substrate specificity, cumene degradation, PCB, structure-function relationship, competitive inhibition, β-ketolase
The monoalkylbenzenes cumene (isopropylbenzene), ethylbenzene, and toluene are important industrially as synthetic intermediates and solvents (Grayson 1985). However, aromatic compounds, especially polychlorinated biphenyls (PCBs) and polycyclic aromatic hydrocarbons, have become some of the most serious environmental pollutants (Blumer and Youngblood 1975; Hites et al. 1977; Keith and Telliard 1979). A number of microorganisms are able to assimilate these compounds, and the responsible catabolic pathways are well established. To improve the biological degradation of these compounds, detailed study for engineering of the catalytic activities and the specificities of enzymes in the degradation pathway is required (Timmis et al. 1994). In the aerobic pathways for bacterial degradation of aromatic carbons, catechol derivatives are cleaved into meta- or ortho-ring fission compounds (Dagley 1986). meta-Cleavage occurs during the degradation of various aromatic compounds such as biphenyls, toluene, ethylbenzene, and cumene (isopropylbenzene; Fig. 1 ▶). The meta-cleavage compounds (2-hydroxy-6-oxohexa-2,4-dienoates and related compounds [HODAs]) are then hydrolyzed, and the resulting 2-hydroxypenta-2,4-dienoates are further converted to tricarboxylic acid cycle intermediates (Dagley 1986; Omori et al. 1986; van der Meer et al. 1992; Zylstra and Gibson 1991). The hydrolases (HODA hydrolases) responsible for the degradation of biphenyls, toluene, ethylbenzene, and cumene are called BphD (EC 3.7.1.8; Furukawa and Miyazaki 1986), TodF (EC 3.7.1.9; Menn et al. 1991), EtbD (Yamada et al. 1998), and CumD/IpbD (Habe et al. 1996a; Eaton et al. 1998), respectively. The strict substrate specificities and the rates of degradation of HODA hydrolases determine the total degradation capacity in many cases (Furukawa et al. 1979, 1993; Bedard and Haberl 1990; Seeger et al. 1995; Seah et al. 1998, 2000, 2001; Cho et al. 2000).
Fig. 1.
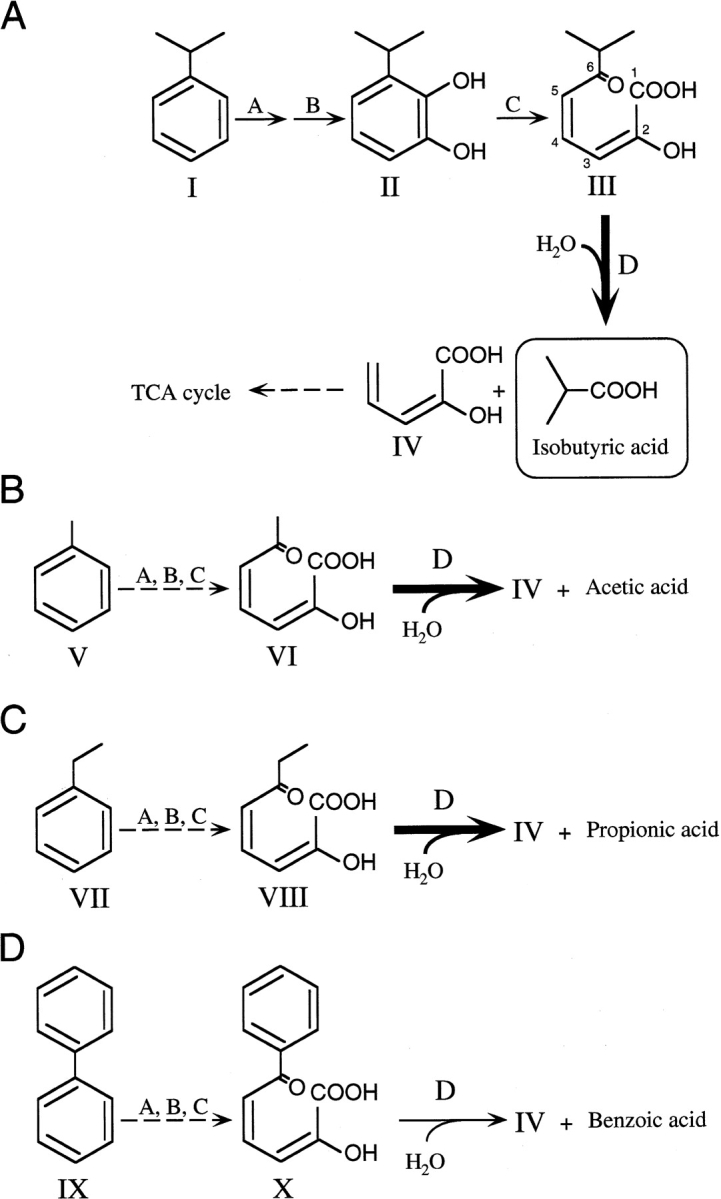
Degradation pathway for cumene (A), toluene (B), ethylbenzene (C), and biphenyl (D). Chemical designations are as follows: compound I, cumene; compound II, 3-isopropylcatechol; compound III, 2-hydroxy-6-oxo-7-methylocta-2,4-dienoate (6-isopropyl-HODA); compound IV, 2-hydroxypenta-2,4-dienoate; compound V, toluene; compound VI, 2-hydroxy-6-oxohepta-2,4-dienoate (6-methyl-HODA); compound VII, ethylbenzene; compound VIII, 2-hydroxy-6-oxoocta-2,4-dienoate (6-ethyl-HODA); compound IX, biphenyl; and compound X, 2-hydroxy-6-oxo-6-phenylhexa-2,4-dienoate (6-phenyl-HODA). Enzyme designations are as follows: A, aromatic ring dioxygenase; B, dihydrodiol dehydrogenase; C, extradiol dioxygenase; and D, meta-cleavage compound hydrolase (HODA hydrolase). Compounds III, VI, and VIII serve as good substrates for CumD, whereas compound X does not.
Most known HODA hydrolases are classified into two major groups. The enzymes in the biphenyl group are involved in the degradation of bicyclic molecules, producing substrates with large C6 side-chains such as biphenyls, carbazole, and tetralin. On the other hand, most enzymes in the monoalkylbenzene group are involved in the degradation of monocyclic compounds with short alkyl side-chains such as toluene, xylene, ethylbenzene, and cumene. These groups were called I and III by Hernáez et al. (2000), and I and II by Nandhagopal et al. (2001), respectively.
The HODA hydrolases belong to the α/β-hydrolase family, similar to lipases and haloalkane dehalogenases (Ollis et al. 1992; Heikinheimo et al. 1999; Nardini and Dijkstra 1999). The α/β-hydrolase family again belongs to a larger group of enzymes showing a unique catalytic reaction, β-ketolases, which cleave the carbon-carbon bonds of 1,3-diketones and 1,5-dioxovinyls (Pokorny et al. 1997).
Cumene is an aromatic compound that is intermediate in size between ethylbenzene and biphenyl. The CumD enzyme isolated from Pseudomonas fluorescens IP01 (Aoki et al. 1996; Habe et al. 1996b)—which can grow on cumene and toluene, as the sole source of carbon, but not on biphenyl—was also shown to be a key enzyme for the assimilation ability (Habe et al. 1996a). A biochemical study on CumD indicated that the enzyme can effectively hydrolyze 2-hydroxy-6-oxo-7-methylocta-2,4-dienoate (6-isopropyl-HODA) but can only slightly hydrolyze 2-hydroxy-6-oxo-6-phenylhexa-2,4-dienoate (6-phenyl-HODA(Saku et al. 2002). Km for 6-phenyl-HODA (0.74 μM) is smaller than that for 6-ispropyl-HODA (7.3 μM), but the kcat for 6-phenyl-HODA is smaller by about 6 × 102–fold than that for 6-isopropyl-HODA. Therefore, its substrate specificity covers larger C6 substituents compared with another well-studied enzyme in the monoalkylbenzene group, TodF. CumD can also effectively hydrolyze 2-hydroxy-6-oxohepta-2,4-dienoate (6-methyl-HODA) and 2-hydroxy-6-oxoocta-2,4-dienoate (6-ethyl-HODA), with the similar Km values for 6-isopropyl-HODA.
The crystal structure of BphD from Rhodococcus sp. strain RHA1 (RHA1 BphD), which belongs to the "biphenyl group", has been reported at 2.4 Å resolution (Nandhagopal et al. 2001). Here, we describe the crystal structure of a member of the "monoalkylbenzene group", CumD from P. fluorescens IP01, complexed with the reaction products, isobutyric acid and acetic acid, at 1.6 and 2.0 Å, respectively. The structures indicate the environment recognizing the C6 side-chain of the substrate, as well as the oxyanion hole, which seems to be catalytically important.
Results
Crystallography
All three crystal structures presented here were obtained using the inactive S103A mutant of CumD (Saku et al. 2002). Despite extensive screening, no crystals of the wild-type CumD enzyme have been obtained, even under the conditions for producing the crystals of the S103A mutant. The statistics for the three crystal structures of CumD described here are summarized in Table 1. At first, pillar-shaped hexagonal crystals (type-I) were used to solve the structure of CumD by means of molecular replacement, using the structure of RHA1 BphD (Protein Data Bank entry 1C4X). The type-I structure was refined at 2.8-Å resolution with a crystallographic R-factor of 19.6%. Under the conditions for producing the type-I crystals, serrated leaf-like crystals (type-II ACT) grew. These crystals belong to a C centered orthorhombic space group and were found to diffract to higher resolution. An acetic acid molecule was found at the active site of the refined type-II ACT structure at 2.0-Å resolution with a crystallographic R-factor of 17.4% (discussed later). The complex structure with isobutyric acid (type-II ISB) was obtained under similar crystallization conditions, using isobutyric acid instead of acetic acid, and refined at 1.6-Å resolution with a crystallographic R-factor of 18.8%. Hereafter, we treat type-II ISB as a representative structure of CumD unless otherwise noted.
Table 1.
X-ray crystallography data statistics
| Type-I | Type-II ACT | Type-II ISB | |
| A. Data collection and processing statistics | |||
| Beamline | BL18B | BL6A | BL18B |
| Wavelength (Å) | 1.0 | 1.0 | 1.0 |
| Method | Weissenberg | Oscillation | Oscillation |
| Space group | P3221 | C2221 | C2221 |
| Cell constant (Å) | a = b = 98.8 | a = 76.5 | a = 77.4 |
| c = 149.3 | b = 116.6 | b = 116.6 | |
| c = 78.0 | c = 78.7 | ||
| Resolution limit (Å) | 2.8 | 2.0 | 1.6 |
| Last shell (Å) | 2.90–2.80 | 2.11–2.00 | 1.69–1.60 |
| No. of total reflections | 128.777 | 169.080 | 187.984 |
| No. of unique reflections | 21.101 | 23.969 | 47.219 |
| Rmergea (%) | 9.5 | 9.6 | 8.2 |
| Of last shell | 29.9 | 29.2 | 41.2 |
| I/sigma (I) | 13.5 | 7.2 | 7.3 |
| Of last shell | 3.2 | 2.5 | 1.8 |
| Completeness (%) | 98.7 | 99.9 | 99.9 |
| Of last shell | 97.1 | 99.9 | 99.9 |
| B. Refinement statistics | |||
| Resolution range (Å) | 49.8–2.8 | 29.6–2.0 | 21.72–1.6 |
| No. of reflections used | 21.088 | 23.950 | 47.196 |
| R-factor (%) | 19.6 | 17.4 | 18.8 |
| Rfreeb | 25.1 | 21.1 | 21.9 |
| No. of monomers | 2 | 1 | 1 |
| No. of non-H atoms | 4444 | 2394 | 2543 |
| No. of residues | 548 | 272 | 271 |
| No. of water molecules | 119 | 238 | 385 |
| No. of ligand molecules | 1 | 2 | 3 |
| Ramachandran plot (%) | (acetate) | (acetate) | (isobutyrate) |
| Most favored | 85.7 | 91.1 | 91.1 |
| Disallowed | 0.6 | 0.4 | 0.4 |
| Root mean square deviation from ideal values | |||
| Bond length (Å) | 0.006 | 0.005 | 0.005 |
| Bond angle (°) | 1.3 | 1.2 | 1.3 |
| Coordinate errorc (Å) | 0.29 | 0.18 | 0.17 |
aRmerge = ΣΣ|I1 − ≤I>|/Σ≤I>.
b Calculated using a test data set: 5% of total data randomly selected from the observed reflections.
c Estimated coordinate error from Luzzati plot.
Dimeric structure
Type-I and type-II crystals contained two subunits and one subunit per asymmetric unit, respectively. The structures of the four subunits presented here (type-I chain A and chain B, type-II ACT, and type-II ISB) were almost identical. The root mean square deviations of Cα atoms between all pairs of these subunits were within 0.36 Å. The two subunits in the asymmetric unit of type-I crystals were related by a noncrystallographic twofold axis corresponding to the 1-to-5 interaction of RHA1 BphD (Nandhagopal et al. 2001). The β8 strands of both of the subunits form an antiparallel β-sheet, and this tight interaction seems to be responsible for the dimeric structure of CumD in solution (Saku et al. 2002). The monomer in the asymmetric unit of the type-II crystal structures showed the same dimeric interaction through a crystallographic twofold axis.
Subunit structure
The subunit structure of the CumD enzyme was very similar to that of RHA1 BphD and had a typical α/β hydrolase fold. Although there are some insertions and deletions (Fig. 2 ▶), the two structures can be aligned throughout the polypeptide (Fig. 3 ▶). The sequence identity between CumD and RHA1 BphD is 34.8%. The secondary structural elements of CumD are named as proposed by Nandhagopal et al. (2001). The subunit of the CumD enzyme is divided into two domains, the core domain (residues 1–133, and 198–282) and the lid domain (residues 134–197). The lid domain of CumD had an obviously deviated conformation compared with that of RHA1 BphD. The mean values of displacement of the core and lid domains were 0.92 Å with 196 Cα atoms, and 1.6 Å with 49 Cα atoms, respectively. The lid domain showed a slightly higher B factor (12.7 Å2 on average as to Cα atoms) compared with the core domain (11.8 Å2). However, the deviation of lid conformation may be caused in part by crystal packing. As to the lid domain, regions involved in the crystal contacts are residues 137–154, 172–181, and 193–198 in CumD and residues 143–164 in RHA1 BphD. The active site of CumD was located between the core and lid domains, deep in the substrate-binding pocket. In the view in Figure 3 ▶, the opening of the pocket is located on the front side. The substrate-binding pocket is divided into two parts by the Ala(Ser)103 residue, proximal and distal to the entrance (P-part and D-part) (Nandhagopal et al. 2001).
Fig. 2.
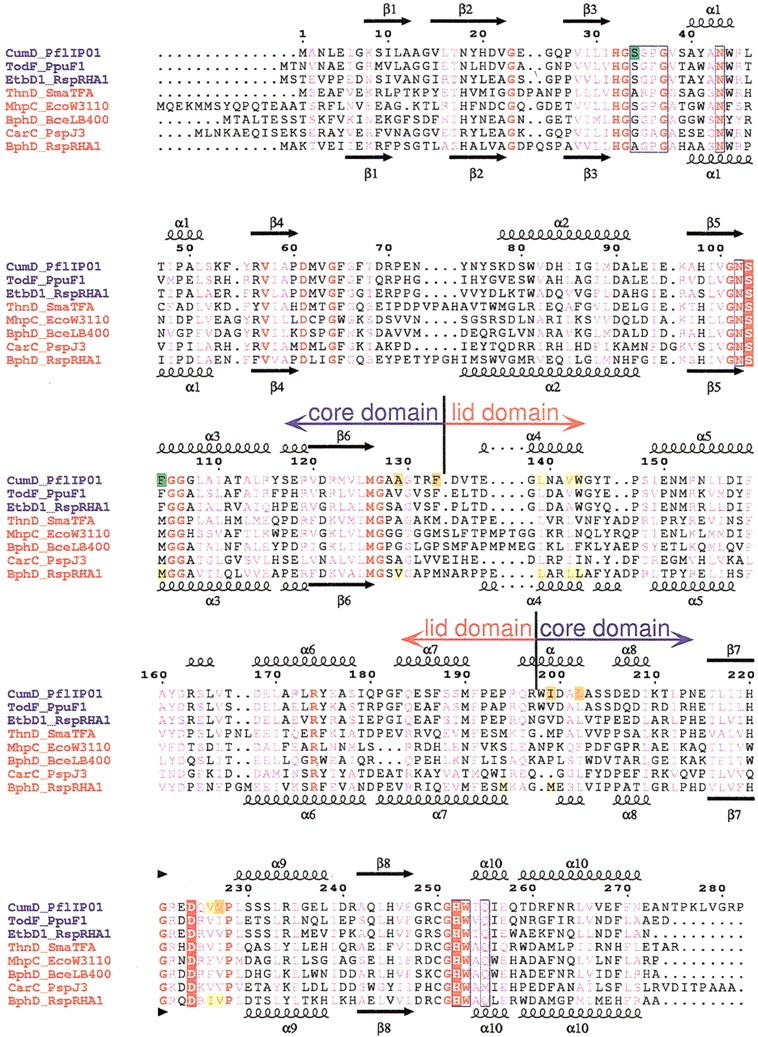
Sequence alignment of HODA hydrolases. Multiple sequence alignment was performed using program ClustalX (Thompson et al. 1997) and then modified on the basis of structural alignment. The sequence names are given in blue and red for members of the monoalkylbenzene and biphenyl groups, respectively. The numbering above the alignment is for the CumD sequence. The secondary structures and their designations are shown above CumD and below RHA1 BphD; arrows, large coils, and small coils represent the β-strands, α-helices, and 310 helices, respectively. Completely conserved residues among the presented sequences are colored red, and conserved residues are shown in pink. The catalytic residues are indicated by red rectangles. The residues involved in the recognition of the 2-hydroxy-6-oxohexa-2,4-dienoate group of the substrate are surrounded by blue frames. The residues involved in the recognition of the carboxylate group and isopropyl group and in the formation of the deeper space of the D-part of CumD are indicated by green, yellow, and orange rectangles, respectively. The residues involved in the recognition of the formation of the D-part of RHA1 BphD are indicated by yellow rectangles. CumD from Pseudomonas fluorescens IP01 (D83955), TodF from P. putida F1 (Y18245), EtbD1 from Rhodococcus sp. RHA1 (AB004320), ThnD from Sphingomonas macrogoltabidus TFA (AF204963), MhpC from Escherichia coli W3110 (D86239), BphD from Burkholderia cepacia LB400 (X66123), CarC from Acrobacterium sp. strain J3 (K. Morii, H. Habe, H. Nojiri, and T. Omori, unpubl.), and BphD from Rhodococcus sp. strain RHA1 (D88016).
Fig. 3.

Superimpositioning of the Cα backbones of CumD and RHA1 BphD. The core and lid domains of CumD are shown in blue and red; those of RHA1 BphD, in light green and green, respectively. The N and C termini are labeled. The isobutyrate ion in the active site (ISB300) and the three catalytic residues of CumD, Ala(Ser)103, Asp224, and His252, are shown as a ball-and-stick model. Root mean square deviation is 1.2 Å, with 245 Cα atoms between type-II ISB and RHA1 BphD (for details, see Materials and Methods).
In the core domain region, all secondary structures of CumD and RHA1 BphD superimposed well, except in several loop regions, such as that between β4 and α2 (residues 70–77). On the other hand, the four α helices in the lid domain region (α4–α7) showed significant disposition. The α4–α5 and C-terminal half of α6 moved to close the D-part of the substrate-binding pocket. The N-terminal half of α6 moved to open the entrance of the P-part. The two regions connecting the core and lid domains formed a hinge between them. The region between α7 and α8 of CumD formed a seven-residue long α helix (Gln196 to Leu202), whereas RHA1 BphD had a short 310 helix here (Fig. 2 ▶).
Active site
The active site residues, Ala(Ser)103, Asp224, and His252, are situated on the core domain facing the lid (Fig. 3 ▶). These residues are clustered and form a hydrogen bond between the Oδ2 atom of Asp224 and Nδ1 of His252 (Fig. 4A ▶). Ala(Ser)103 and His252 are involved in the formation of the inner surface of the substrate-binding pocket in the border region between the P- and D-parts. Trp253, which is a residue adjacent to His252, had different conformations in the type-II ISB, type-II ACT, and type-I structures (Table 2). Moreover, the κ2 dihedral angles of His252 were different in the type-I and type-II structures.
Fig. 4.
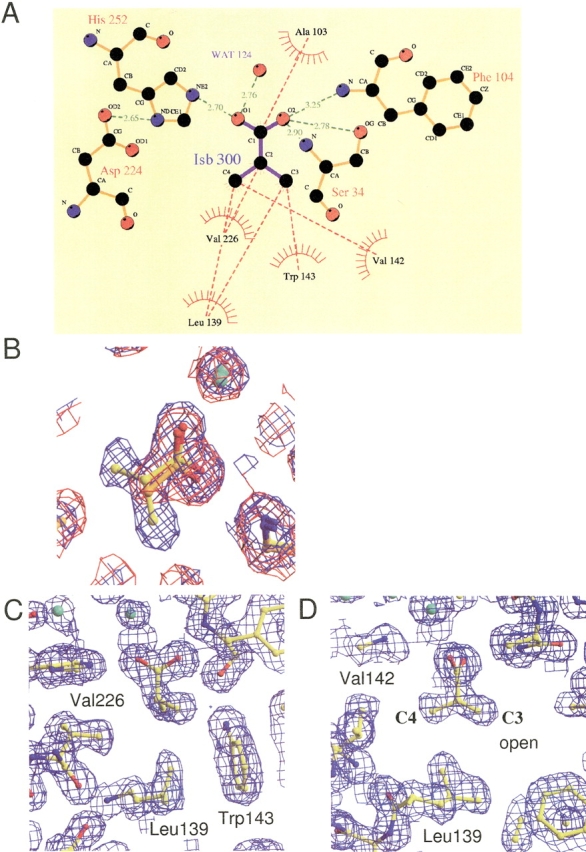
(A) Schematic drawing of the atoms and interactions involved in the recognition of ISB300. (B) Superimpositioning of the sigma-A weighted 2Fo-Fc electron density maps contoured at 2σ and the final model structures of type-II ACT and type-II ISB at the bound acetate and isobutyrate ions. The maps of the type-II ACT and type-II ISB structures are shown in red and blue, respectively. The carbon atoms in the model structures of type-II ACT and type-II ISB are shown in orange and yellow, respectively. Water molecules are represented as green spheres. (C, D) Hydrophobic residues involved in the recognition of the isopropyl group of ISB300. σA-weighted 2Fo-Fc electron density maps contoured at 2σ are shown. The view in D is rotated 90° around the perpendicular axis from that in C.
Table 2.
Torsion angles (°) of the side-chains of His252 and Trp253
| His252 | Trp253 | |||
| κ1 | κ2 | κ1 | κ2 | |
| Type-I A subunit | 177.7 | 74.0 | −81.6 | −35.4 |
| Type-I B subunit | 175.2 | 78.3 | −78.1 | −40.4 |
| Type-II ACT | −168.3 | 50.5 | 54.9 | 11.8 |
| Type-II ISB | −165.3 | 59.4 | −131.3 | 14.1 |
In the type-II ACT structure, the CumD protein bound two acetate ions, one of which (ACT300) was located at the active site and close to Ala(Ser)103. In the type-II ISB structure, three isobutyrate ions were found, one of which (ISB300) was located at the same position as ACT300 (Fig. 4B ▶). One of the other bound isobutyrate ions in the type-II ISB structure (ISB301) was located at the entrance of the substrate-binding pocket. Atoms of ISB301 showed significantly elevated temperature factors (average B-factor = 29.5 Å2) compared with those of ISB300 (13.1 Å2) and showed relatively ambiguous electron density. The other bound ions (ACT301 and ISB302) were located on the surface of the CumD molecule and involved in crystal packing. The organic acids in the active site (ACT300 and ISB300) seemed to occupy the site for a part of the hydrolysis product of CumD, which has the various C6 substituents of the substrates (Fig. 1A, B ▶). A schematic drawing of the interactions around ISB300 is presented in Figure 4A ▶. The carboxyl group of ISB300 was recognized by five hydrogen bonds. The O1 atom of ISB300 was hydrogen-bonded with the Nɛ2 atom of His252 and a water molecule with good electron density (B-factor = 24.5 Å2). The Cβ atom of Ala103 was in van der Waals contact with the C1 atom of ISB300. On the other hand, the O2 atom of ISB300 was hydrogen bonded with the main-chain N atoms of Ser34 and Phe104. These interactions probably correspond to an oxyanion hole, which stabilizes the oxyanion intermediate during catalysis (Fleming et al. 2000). Moreover, the side-chain Oγ atom of Ser34 formed a strong hydrogen bond (distance = 2.78 Å) with the O2 atom of ISB300.
The hydrophobic interactions around the isopropyl group of ISB300 are shown in Figure 4, C and D ▶. Leu139 was located ahead in the C1-to-C2 direction. Of the four side-faces of the hydrophobic cavity, three were occupied by the side-chains of Val226, Trp143, and Val142, and one of them was open with a vacant space. No water molecule was found in this hydrophobic cavity.
Shape of the substrate-binding pocket
The molecular surfaces of CumD and RHA1 BphD are shown in Figure 5 ▶. CumD had a wider entrance to the P-part compared with RHA1 BphD because of the shift of the N-terminal half of α6, as described above. CumD and RHA1 BphD were similar in shape as the molecular surface of the distal half of the P-part, which is an intermediate region of the substrate-binding pocket. The molecular surface of this part is hydrophilic, and a number of waters were bound. The surface of this part is formed by the polar side-chains of Asn43, Asn102, and Gln255 and the main-chain atoms of Ser34 to Gly37, and these residues are highly conserved among HODA hydrolases (Fig. 2 ▶). The D-part was hydrophobic, and the surface of this region differed in shape between CumD and RHA1 BphD. The D-part of CumD was long, but that of RHA1 BphD was somewhat spherical. The vacant hydrophobic space ahead of the C3 atom of the ISB300 molecule in the CumD structure was formed by the side-chains of Ala129, Phe133, Ile199, Leu202, and Val227. The substrate (isopropyl-HODA) and the side-chain of Ser103 were modeled based on the crystal structures of CumD and RHA1 BphD (Fig. 6 ▶). The isobutyrate molecule bound at the entrance of the substrate-binding pocket (ISB301) was far from the modeled substrate.
Fig. 5.
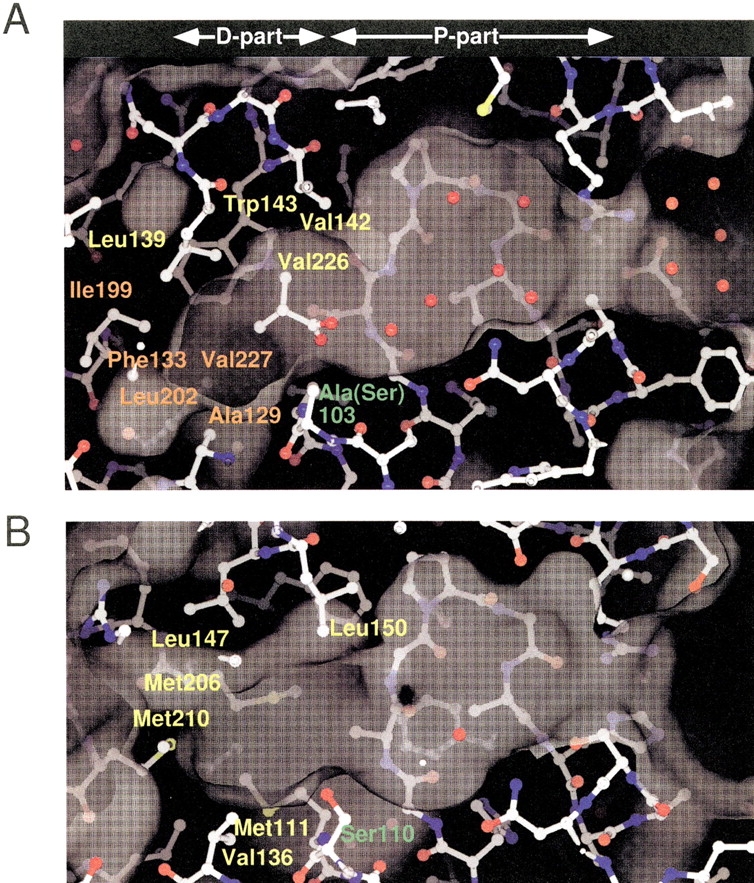
Molecular surfaces of the substrate-binding pockets of CumD (A) and RHA1 BphD (B), calculated using the SPOCK program with probe radius of 1.4 Å. Waters are shown as red spheres. The catalytic serine or mutated alanine residues are labeled in green. The residues involved in the recognition of the isopropyl group of ISB300 and in the formation of the deeper space of the D-part are labeled in yellow and orange, respectively. The residues involved in the formation of the surface of the D-part of RHA1 BphD are labeled in yellow.
Fig. 6.
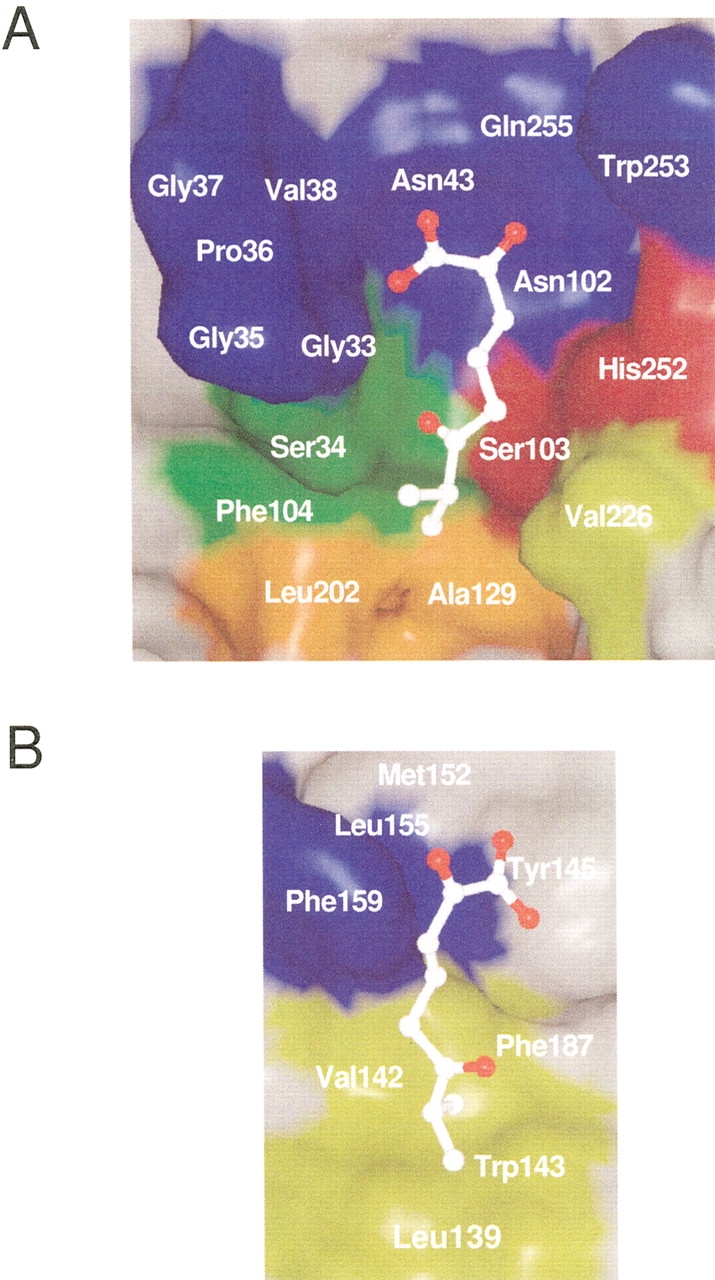
Modeled substrate in the substrate-binding pocket. The inner surface of the core-domain side (A) and that of the lid-domain side (B) are shown. The side-chain of Ser103 was modeled and energy-minimized with the substrate (see Materials and Methods). The modeled substrate is shown with a ball-and-stick model. The inner surface formed by the residues involved in the catalysis, in the recognition of the carboxylate group and isopropyl group of isobutyric acid, and in the formation of the deeper space of the D-part are colored in red, green, yellow, and orange, respectively. Other conserved residues, which are shown in pink in Fig. 2 ▶, are shown in blue.
Inhibition constants of various organic acids
The activity of CumD was shown to be inhibited by acetate buffer (Saku et al. 2002). Because an acetate or isobutyrate ion was bound in the D-part of the active site, the inhibition constants (Ki) for various organic acids are considered to indicate the affinity between these molecules and the D-part. Although there is another binding site for isobutyrate in the substrate-binding pocket, the bound molecule (ISB301) showed significantly higher B-factor, and the affinity is speculated to be weaker than that of the D-part. All organic acids investigated here showed competitive inhibition toward the CumD activity (data not shown). The Ki values of these organic acids are shown in Table 3. As a general tendency, organic acids with longer, and thus hydrophobic, side-chains showed smaller Ki values. Isomers as to the alkyl group, such as n-butyric acid and isobutyric acid, showed different Ki values, indicating that the shape of the hydrophobic pocket of the D-part is responsible for the degree of inhibition. The Ki of isobutyric acid was smaller than that of n-butyric acid, and for the isomers of n-valeric acid, the Ki value was the smallest for n-valeric acid and the largest for trimethylacetic acid. As for the two 2-methylbutyric acid reagents tested here, one of the two optical isomers, (S)-(+)-2-methylbutyric acid, showed a smaller Ki compared with the racemic compound (±)-2-methylbutyric acid, indicating that the stereo-specific shape of the D-part affects the Ki value.
Table 3.
Inhibition constants of various organic acids for the CumD enzyme
| Compound name | Structural formula | Ki (mM) |
| Acetic acid | CH3CO2H | 8.0 (0.32)a |
| Propionic acid | CH3CH2CO2H | 4.8 (0.54) |
| n-Butyric acid | CH3(CH2)2CO2H | 3.6 (1.1) |
| Isobutyric acid | (CH3)2CHCO2H | 2.9 (0.64) |
| n-Valeric acid | CH3(CH2)3CO2H | 0.47 (0.048) |
| Isovaleric acid | (CH3)2CH2CHCO2H | 3.6 (0.69) |
| (±)-2-methylbutyric acid | 1.6 (0.41) | |
| (S)-(+)-2-methylbutyric acid | C2H5C*H(CH3)CO2H | 1.2 (0.13) |
| Trimethylacetic acid | (CH3)3CCO2H | 8.6 (0.35) |
| Hexanoic acid | CH3(CH2)4CO2H | 0.22 (0.016) |
| Benzoic acid | C6H5CO2H | 0.65 (0.036) |
a Numerical values in parentheses indicate standard errors.
Discussion
Active site structure and catalytic mechanism
The acetate and isobutyrate ions bound to the D-part of the active site of CumD are considered to represent the interaction of a part of the hydrolysis product with CumD, because both organic acids were similarly bound in a reasonable manner. The alkyl side-chains of these molecules faced on the hydrophobic D-part, and the carboxyl group interacted with catalytically important elements. The catalytic mechanism of a HODA hydrolase, MhpC, has been investigated in detail (Henderson and Bugg 1997; Lam and Bugg 1997; Fleming et al. 2000). A dienol substrate is initially ketonized, followed by base-catalyzed attack by water, rather than nucleophilic attack of the active site serine. The active site serine is suggested to act as a base for the deprotonation of water. Through formation of a gem-diol intermediate, stereo-specific fragmentation through C-C cleavage proceeds.
The water atom attacking the keto-intermediate would be situated near the O1 atom of ISB300, because the Oγ atom of Ser103 in the wild-type CumD enzyme could interact with a putative water atom around this position. Moreover, the interactions with the Nɛ2 atom of His252 and a water molecule observed in this study may support the activation of the water attacking the keto-intermediate. In the crystal structure of RHA1 BphD, extra electron density around the side-chain of the active site Ser110 was observed (Nandhagopal et al. 2001). The extra density seems to represent a possible chemical modification, but no precise interpretation has been performed yet. The crystal structures of the S103A mutant of CumD reported here showed no such extra density. Unfortunately, because no crystals of the wild-type CumD enzyme were obtained, it is unclear whether or not the side-chain of Ser103 was modified.
We speculate that the interactions around the O2 atom of ISB300, hydrogen bonded with the main-chain N atoms, might correspond to those around the oxygen atom of the keto-intermediate in a nonplanar conformation, and the negatively charged oxygen atom of the following tetrahedral gem-diol intermediate proposed by Henderson and Bugg (1997). Interestingly, the side-chain of the Oγ atom of Ser34 formed a strong hydrogen bond with the O2 atom of ISB300. This serine residue is not conserved among HODA hydrolases (Fig. 2 ▶), and an alanine or glycine residue replaces it in some enzymes. This residue could have some effect on the catalysis by HODA hydrolases.
The complex structure with a substrate, that is, 6-isopropyl-HODA, has not been obtained yet, despite some efforts. The molecular surface of the deeper half of the P-part of CumD was similar with that of RHA1 BphD in shape and hydrophilic nature. This strongly supports the suggestion that this part binds to the 2-hydroxy-6-oxohexa-2,4-dienoate moiety of a substrate (Fig. 6 ▶). However, the conformations of the side-chains of Trp253 and His252 were different among the crystal structures reported here, indicating the mobility of these residues, perhaps in some steps of the catalytic cycle.
Substrate specificity of HODA hydrolases
The D-parts of CumD and RHA1 BphD have different shapes, and the residues involved in their formation are also different (Fig. 5 ▶). Compared with RHA1 BphD, the substrate-binding pocket of CumD is wider at the entrance and narrower at the D-part, because of the movement of the α helices in the lid domain. The long and spherial shapes of the D-parts of CumD and BphD correspond well to their cognate substrates (Saku et al. 2002; Seah et al. 1998). The residues forming the hydrophobic pocket of the D-part can be divided into four regions based on the amino acid sequence (Ala129 to Phe133, Leu139 to Trp143, Ile199 to Leu202, and Val226 to Val227; Fig. 2 ▶). The former three regions are situated around the two hinge regions between the lid and core domains. These residues of CumD and RHA1 BphD are superimposed in Figure 7 ▶. The main-chain trace between β6 and α4 (Thr131 to Thr136) of CumD is largely shifted, and the N terminus of α4 approaches the substrate. The residues at the N terminus of α4, Leu139, Leu142, and Trp143, significantly affect the formation of the narrower shape of the D-part of CumD.
Fig. 7.

Superimpositioning of CumD and RHA1 BphD at the D-part. Viewed from a similar direction as in Fig. 3 ▶. Backbone traces are colored as in Fig. 3 ▶. The catalytic residues and the residues involved in the formation of the D-part of CumD are shown as a ball-and-stick model and are labeled in red and black, respectively. The residues of RHA1 BphD are shown and labeled in green.
There is a change in secondary structure in the other hinge region (Pro192 to Leu202). Probably because of Pro192 and Pro194 of CumD, the C terminus of α7 is broken, and a new α helix (Gln196 to Leu202) is formed. In the C terminus of the longer α7 helix of BphD, Met206 forms a hydrophobic surface at a similar position to Trp143 of CumD. Ile199 of CumD and corresponding Met210 of BphD are also involved in the formation of the hydrophobic pocket.
A residue corresponding to Phe104 of CumD and Met111 of BphD is located immediately after the active site serine and is completely conserved as phenylalanine and methionine in the monoalkylbenzene and biphenyl groups, respectively (Fig. 2 ▶). Met111 of BphD is involved in the formation of the substrate-binding pocket, but the side-chain of Phe104 of CumD does not point into the pocket (Fig. 7 ▶). Met111 and Val136 of BphD obstruct the deeper space, which is present in CumD, and thereby reduce the depth of the pocket.
CumD and TodF from P. putida F1 show high similarity in amino acid sequence (identity = 63.8%), but their substrate specificities are significantly different. CumD can efficiently hydrolyze 6-isopropyl-HODA, but TodF cannot. Moreover, CumD can slightly hydrolyze 6-phenyl-HODA. The difference in the residues forming the surface of the D-part between CumD and TodF was only three mutations. Ala129, Ile199, and Val227 of CumD are substituted by valine, valine, and isoleucine in TodF, respectively. As for TodF, the difference in the three residues probably reduces the depth of the pocket and the activities toward 6-isopropyl-HODA and 6-phenyl-HODA.
The Ki values of CumD corresponded well to the shape and nature of the D-part revealed by crystallographic analysis. Because the surface of the D-part is hydrophobic, substrates with longer alkyl chains strongly inhibited the activity. As for the isomers of n-valeric acid, isovaleric acid with a branch at the tip of the alkyl chain showed weaker inhibition compared with that with a straight chain, corresponding to the narrow and long pocket of the D-part. Moreover, trimethylacetic acid showed the weakest inhibition of the isomers, indicating that the D-part is not wide in the direction of Val226 (Fig. 4C ▶). Interestingly, (S)-(+)-2-methylbutyric acid showed significantly stronger inhibition than (±)-2-metnylbutyric acid, which exactly corresponded to the vacant space ahead of the C3 atom of ISB300. Hexanoic acid with the longest alkyl chain showed the strongest inhibition among all inhibitors tested. The hydrophobic pocket of the D-part seems to be deep enough for binding hexanoic acid, almost extending to the back surface of the molecule (Fig. 5A ▶). However, we could not completely exclude the possibility that the Ki values indicate the affinity for other binding site in the substrate-binding pocket than the D-part. Because high concentration of isobutyriate (200 mM) was used for crystallization, the molecules tend to bind to weaker sites. Crystallographic analysis of complex structures with other organic acids strongly bound at a similar position with ISB300, such as propionic acid, n-butyric acid, n-valeric acid, (S)-(+)-2-methylbutyric acid, and benzoic acid, is in progress. Of these complex structures, a weak binding site only for benzoate is observed at a similar position with ISB301 but not for others (data not shown).
The Km values of CumD for 6-methyl-, 6-ethyl-, and 6-isopropyl-HODA are not greatly different from each other (Saku et al. 2002) and do not correlate with the Ki values for acetic, propionic, and isobutyric acid. The Km values for these substrates may be largely dependent on the 2-hydroxy-6-oxohexa-2,4-dienoate group of the substrate. On the other hand, the Ki for benzoic acid was significantly smaller, similar to the small Km for 6-phenyl-HODA. Substrates or inhibitors containing a phenyl ring are bound strongly to the D-part, but the manner of binding may not be proper for efficient catalysis, that is, base-catalyzed attack by water.
Materials and methods
Crystallography
The S103A mutant of the CumD enzyme was expressed in Escherichia coli, and purified as described previously (Saku et al. 2002). The type-I crystals were grown at 5°C by the hanging-drop vapor-diffusion method (protein concentration, 5.0 mg/mL), with a reservoir solution comprising 15% PEG 4000, 0.2 M ammonium acetate, and 0.1 M sodium acetate (pH 3.8). The type-II ACT crystals were grown by the same method, with a reservoir solution comprising 20% PEG 4000, 0.2 M ammonium acetate, and 0.1 M sodium acetate (pH 3.8). The type-II ISB crystals were also grown by the same method, with a reservoir solution comprising 20% PEG 4000, 0.1 M ammonium isobutyrate, and 0.1 M sodium isobutyrate (pH 3.9). The concentrations of acetate and isobutyrate are ∼38 and 69 times Ki of the wild-type CumD, but the binding affinity of the ligands to the S103A mutant is not known. Before data collection, the type-I crystals were transferred to a solution containing 20% glycerol in addition to the reservoir solution. Instead of glycerol, 15% 2-methyl-2,4-pentanediol was used as a cryoprotectant for type-II crystals. The concentration of cryoprotencant was increased by 5% in each step and equilibrated for 10 min between them. The data for the type-I, type-II ACT, and type-II ISB crystals were collected with a Weissenberg camera and CCD cameras on the BL6A and BL18B stations of the Photon Factory, High Energy Accelerator Research Organization (KEK), at 100 K. Diffraction images of type-I and type-II crystals were indexed, integrated, and scaled using the HKL program suite (Otwinowski and Minor 1997) and DPS/MOSFLM program suite (Steller et al. 1997; Powell 1999), respectively. The data collection and processing statistics are given in Table 1a. Phase calculation was performed by molecular replacement with programs AMORE (Navaza 1994) and MOLREP (Vagin and Teplyakov 1997) in the CCP4 program suite. The intact crystal structure of RHA1 BphD (Protein Data Bank entry 1C4X) was used as a search model for the type-I crystals. The initial phases of the type-II crystals were calculated using the type-I structure as a search model. Refinement was performed using CNS 1.1 (Brunger et al. 1998). Model building in the electron density map was performed using programs O (Jones et al. 1991) and Xtalview (McRee 1999). The refinement statistics are given in Table 1b. The φ,ξ angles of Ala103 occur in the disallowed regions of the Ramachandran plot. It is known that the strained turn structure, the so-called nucleophile elbow, is conserved well in all the α/β-hydrolases (Heikinheimo et al. 1999). The coordinates and structure factors have been deposited in the Protein Data Bank (accession nos. 1IUN, 1IUO, and 1IUP for the type-I, type-II ACT, and type-II ISB structures, respectively). Assignment of the secondary structures was performed using program DSSP (Kabsh and Sander 1983). Least-square fit between CumD and RHA1 BphD was performed in the following regions; residues 7–24, 25–73, 78–133, 135–146, 148–164, 171–179, 182–192, and 200–272 for CumD and residues 8–25, 28–76, 85–140, 143–154, 158–174, 183–191, 194–204, and 211–283 for RHA1 BpdhD. The figures were generated with ESPript (Gouet et al. 1999), LIGPLOT (Wallace et al. 1995), XFIT in the Xtalview program suite, MOLSCRIPT (Kraulis 1991), RASTER3D (Merritt and Bacon 1997), RASMOL 2.7.1 (Bernstein 2000), and SPOCK (Christopher and Baldwin 1998). The molecular surfaces shown in Figures 5 and 6 ▶ ▶ were calculated using SPOCK.
Modeling study
The modeling study was performed based on the similar modeling study of RHA1 BphD (Nandhagopal et al. 2001). A ketonised 6-isopropyl-HODA (Fleming et al. 2000) was placed manually to satisfy the binding features of epoxide hydrolase (1CR6) and lipase (1EX9). The side-chain of Ser103 was modeled based on the conformation of Ser110 of RHA1 BphD. An energy minimization of the bound substrate, Ser103, His252, and Trp253 residues was performed with CNS 1.1 for 500 cycles.
Measurement of inhibition constants
(±)-2-Methylbutyric acid and (S)-(+)-2-methylbutyric acid were purchased from Sigma-Aldrich. Other chemicals were purchased from Wako Pure Chemical Industries or Nacalai Tesque, unless otherwise noted. Preparation of the substrate (6-isopropyl-HODA) and kinetic measurement of the activity of CumD were performed as described previously (Saku et al. 2002). Because a large error in measurement with lower concentrations of the substrate than the Km value (7.3 μM) was inevitable, reliable Ki values could not be obtained from Dixon plots (Dixon and Webb 1979). Therefore, saturation curves were measured with various concentrations of inhibitors, and then Ki values were determined by plotting Km/Vmax versus [I] (secondary plot or "replot"). At least three saturation curves with different inhibitor concentrations between 0.45 to 13.5 mM were measured to obtain linear secondary plots. The enzyme concentration of a CumD subunit in the reaction mixtures was 2.4 nM.
Acknowledgments
We thank the staff of the Photon Factory for their assistance with the data collection; Dr. H. Taguchi, Dr. A. Ibuka, and K. Arai for donation of the beam time and their help in data collection; and Dr. T. Senda for offering the coordinates of RHA1 BphD before disclosure at Protein Data Bank. The data collection was approved by the Photon Factory Advisory Committee (proposal 00G127). This work was supported in part by PROBRAIN (Program for Promotion of Basic Research Activities for Innovative Biosciences in Japan), and the TARA (Tsukuba Advanced Research Alliance) Sakabe Project.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0209602.
References
- Aoki, H., Kimura, T., Habe, H., Yamane, H., Kodama, T., and Omori, T. 1996. Cloning, nucleotide sequence, and characterization of the genes encoding enzymes involved in the degradation of cumene to 2-hydroxy-6-oxo-7-methylocta-2,4-dienoic acid in Pseudomonas fluorescens IP01.J. Ferment. Bioeng. 81 187–196. [Google Scholar]
- Bedard, D.L. and Haberl, M.L. 1990. Influence of chlorine substitution pattern on the degradation of polychlorinated biphenyls by eight bacterial strains.Microbiol. Ecol. 20 87–102. [DOI] [PubMed] [Google Scholar]
- Bernstein, H.J. 2000. Recent changes to RasMol, recombining the variants.Trends Biochem. Sci. 25 453–455. [DOI] [PubMed] [Google Scholar]
- Blumer, M. and Youngblood, W.W. 1975. Polycyclic aromatic hydrocarbons in soils and recent sediments.Science 188 53–55. [DOI] [PubMed] [Google Scholar]
- Brunger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N.S., Read, R.J., Rice, L.M., Simonson, T., and Warren, G.L. 1998. Crystallography and NMR system: A new software suite for macromolecular structure determination.Acta Crystallogr. D Biol. Crystallogr. D54 905–921. [DOI] [PubMed] [Google Scholar]
- Cho, M.C., Kang, D.O., Yoon, B.D., and Lee, K. 2000. Toluene degradation pathway from Pseudomonas putida F1: Substrate specificity and gene induction by 1-substituted benzenes.J. Indust. Microbiol. Biotechnol. 25 163–170. [Google Scholar]
- Christopher, J.A. and Baldwin, T.O. 1998. SPOCK: Real-time collaborative molecular modeling.J. Mol. Graphics Model. 16 285. [Google Scholar]
- Dagley, S. 1986. Biochemistry of aromatic hydrocarbon degradation in Pseudomonads. In The bacteria (eds. J.R. Sokatch and L.N. Ornston), Vol. 10, pp. 527–555. Academic Press, New York.
- Dixon, M. and Webb, E.S. 1979. Enzymes, 3rd ed. Academic Press, New York.
- Eaton, R.W., Selifonova, O.V., and Gedney, R.M. 1998. Isopropylbenzene catabolic pathway in Pseudomonas putida RE204: nucleotide sequence analysis of the ipb operon and neighboring DNA from pRE4.Biodegradation 9 119–132. [DOI] [PubMed] [Google Scholar]
- Fleming, S.M., Robertson, T.A., Langley, G.J., and Bugg, T.D.H. 2000. Catalytic mechanism of a C-C hydrolase enzyme: Evidence for a gem-diol intermediate, not an acyl enzyme.Biochemistry 39 1522–1531. [DOI] [PubMed] [Google Scholar]
- Furukawa, K. and Miyazaki, T. 1986. Cloning of a gene cluster encoding biphenyl and chlorobiphenyl degradation in Pseudomonas pseudoalcaligenes.J. Bacteriol. 166 392–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa, K., Tomizuka, N., and Kamibayashi, A. 1979. Effect of chlorine substitution on the bacterial metabolism of various polychlorinated biphenyls.Appl. Environ. Microbiol. 38 301–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa, K., Hirose, J., Suyama, A., Zaiki, T., and Hayashida, S. 1993. Gene components responsible for discrete substrate specificity in the metabolism of biphenyl (bph operon) and toluene (tod operon).J. Bacteriol. 175 5224–5232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouet, P., Courcelle, E., Stuart, D.I., and Metoz, F. 1999. ESPript: Multiple sequence alignments in PostScript.Bioinformatics 15 305–308. [DOI] [PubMed] [Google Scholar]
- Grayson, M. 1985. Kirk-Othmer concise encyclopedia of chemical technology. John Wiley and Sons, Inc., New York.
- Habe, H., Kasuga, K., Nojiri, H., Yamane, H., and Omori, T. 1996a. Analysis of cumene (isopropylbenzene) degradation genes from Pseudomonas fluorescens IP01.Appl. Environ. Microbiol. 62 4471–4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habe, H., Kimura, T., Nojiri, H., Yamane, H., and Omori, T. 1996b. Cloning and nucleotide sequences of the genes involved in the meta-cleavage pathway of cumene degradation in Pseudomonas fluorescens IP01.J. Ferment. Bioeng. 81 247–254. [Google Scholar]
- Heikinheimo, P., Goldman, A., Jeffries, C., and Ollis, D.L. 1999. Of barn owls and bankers: A lush variety of α/β hydrolases.Structure 7 R141–R146. [DOI] [PubMed] [Google Scholar]
- Henderson, I.M.J. and Bugg, T.D.H. 1997. Pre-steady-state kinetic analysis of 2-hydroxy-6-keto-nona-2,4-diene-1,9-dioic acid 5,6-hydrolase: Kinetic evidence for enol/keto tautomerization.Biochemistry 36 12252–12258. [DOI] [PubMed] [Google Scholar]
- Hernáez, M.J., Andújar, E., Ríos, J.L., Kaschabek, S.R., Reineke, W., and Santero, E. 2000. Identification of a serine hydrolase which cleaves the alicyclic ring of tetralin.J. Bacteriol. 182 5448–5453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hites, R.A., Laflamme, R.E., and Farrington, J.W. 1977. Sedimentary polycyclic aromatic hydrocarbons: The historical record.Science 198 829–831. [DOI] [PubMed] [Google Scholar]
- Jones, T.A., Zou, J.-Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models.Acta Crystallogr. A 47 110–119. [DOI] [PubMed] [Google Scholar]
- Kabsh, W. and Sander, C. 1983. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features.Biopolymers 22 2577–2637. [DOI] [PubMed] [Google Scholar]
- Keith, L.H. and Telliard, W.A. 1979. Priority pollutants, I: A perspective view.Environ. Sci. Technol. 13 416–423. [Google Scholar]
- Kraulis, P.J. 1991. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures.J. Appl. Crystallogr. 24 946–950. [Google Scholar]
- Lam, W.W.Y. and Bugg, T.D.H. 1997. Purification, characterization, and stereochemical analysis of a C-C hydrolase: 2-Hydroxy-6-keto-nona-2,4-diene-1,9-dioic acid 5,6-hydrolase.Biochemistry 36 12242–12251. [DOI] [PubMed] [Google Scholar]
- McRee, D.E. 1999. XtalView/Xfit: A versatile program for manipulating atomic coordinates and electron density.J. Struct. Biol. 125 156–165. [DOI] [PubMed] [Google Scholar]
- Menn, F.M., Zylstra, G.J., and Gibson, D.T. 1991. Location and sequence of the todF gene encoding 2-hydroxy-6-oxohepta-2,4-dienoate hydrolase in Pseudomonas putida F1.Gene 104 91–94. [DOI] [PubMed] [Google Scholar]
- Merritt, E.A. and Bacon, D.J. 1997. Raster3D: Photorealistic molecular graphics.Methods Enzymol. 277 505–524. [DOI] [PubMed] [Google Scholar]
- Nandhagopal, N., Yamada, A., Hatta, T., Masai, E., Fukuda, M., Mitsui, Y., and Senda, T. 2001. Crystal structure of 2-hydroxy-6-oxo-6-phenylhexa-2,4-dienoic acid (HPDA) hydrolase (BphD enzyme) from the Rhodococcus sp. strain RHA1 of the PCB degradation pathway.J. Mol. Biol. 309 1139–1151. [DOI] [PubMed] [Google Scholar]
- Nardini, M. and Dijkstra, B.W. 1999. α/β-Hydrolase fold enzymes: The family keeps growing.Curr. Opin. Struct. Biol. 9732–373. [DOI] [PubMed] [Google Scholar]
- Ollis, D. L., Cheah, E., Cygler, M., Dijkstra, B., Frolow, F., Franken, S.M., Harel, M., Remington, S.J., Silman, I. and Schrag, J. 1992. The α/β hydrolase fold.Protein Eng. 5 197–211. [DOI] [PubMed] [Google Scholar]
- Omori, T., Sugimura, K., Ishigooka, H., and Minoda, Y. 1986. Purification and some properties of a 2-hydroxy-6-oxo-6-phenylhexa-2,4-dienoic acid hydrolyzing enzyme from Pseudomonas cruciviae S93B1 involved in the degradation of biphenyl.Agric. Biol. Chem. 50 931–937. [Google Scholar]
- Otwinowski, Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscilation mode.Methods Enzymol. 276 307–326. [DOI] [PubMed] [Google Scholar]
- Pokorny, D., Steiner, W., and Ribbons, D.W. 1997. β-Ketolases: Forgotten hydrophobic enzymes?Trends Biotechnol. 15 291–296. [Google Scholar]
- Powell, H.R. 1999. The Rossmann Fourier autoindexing algorithm inMOSFLM. Acta Cryst. D5:5 1690–1695. [DOI] [PubMed] [Google Scholar]
- Saku, T., Fushinobu, S., Jun, S.-Y., Ikeda, N., Nojiri, H., Yamane, H., Omori, T., and Wakagi, T. 2002. Purification, characterization, and steady-state kinetics of a meta-cleavage compound hydrolase from Pseudomonas fluorescens IP01.J. Biosci. Bioeng. 93 568–574. [DOI] [PubMed] [Google Scholar]
- Seah, S.Y.K., Labbe, G., Nerdinger, S., Johnson, M.R., Snieckus, V., and Eltis, L.D. 2000. Identification of a serine hydrolase as a key determinant in the microbial degradation of polychlorinated biphenyls.J. Biol. Chem. 275 15701–15708. [DOI] [PubMed] [Google Scholar]
- Seah, S.Y.K., Terracina, G., Bolin, J.T., Riebel, P., Snieckus, V., and Eltis, L.D. 1998. Purification and preliminary characterization of a serine hydrolase involved in the microbial degradation of polychlorinated biphenyls.J. Biol. Chem. 273 22943–22949. [DOI] [PubMed] [Google Scholar]
- Seah, S.Y.K., Labbe, G., Kaschabek, S.R., Reifenrath, F., Reineke, W., and Eltis, L. D. 2001. Comparative specificities of two evolutionarily divergent hydrolases involved in microbial degradation of polychlorinated biphenyls.J. Bacteriol. 183 1511–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeger, M., Timmis, K.N., and Hofer, B. 1995. Conversion of chlorobiphenyls into phenylhexadienoates and benzoates by the enzymes of the upper pathway for polychlorobiphenyl degradation encoded by the bph locus of Pseudomonas sp. strain LB400.Appl. Environ. Microbiol. 61 2654–2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steller, I., Bolotovsky, R., and Rossmann, M.G. 1997. An algorithm for automatic indexing of oscillation images using Fourier analysis.J. Appl. Cryst. 30 1036–1040. [Google Scholar]
- Thompson, J.D., Gibson, T.J., Plewniak, F., Jeanmougin, F., and Higgins, D.G. 1997. The CLUSTAL_X windows interface: Flexible strategies for multiple alignment aided by quality analysis tools.Nucleic Acids Res. 25 4876–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmis, K.N., Steffan, R.J., and Unterman, R. 1994. Designing microorganisms for the treatment of toxic wastes.Annu. Rev. Microbiol. 48 525–557. [DOI] [PubMed] [Google Scholar]
- Vagin, A. and Teplyakov, A. 1997. MOLREP: An automated program for molecular replacement.J. Appl. Cryst. 30 1022–1025. [Google Scholar]
- van der Meer, J.R., de Vos, W.M., Harayama, S., and Zehnder, A.J.B. 1992. Molecular mechanisms of genetic adaptation to xenobiotic compounds.Microbiol. Rev. 56 677–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace, A.C., Lakowski, R.A., and Thornton, J.M. 1995. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions.Protein Eng. 8 127–134. [DOI] [PubMed] [Google Scholar]
- Yamada, A., Kishi, H., Sugiyama, K., Hatta, T., Nakamura, K., Masai, E., and Fukuda, M. 1998. Two nearly identical aromatic compound hydrolase genes in a strong polychlorinated biphenyl degrader, Rhodococcus sp. strain RHA1.Appl. Environ. Microbiol. 64 2006–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zylstra, G.J. and Gibson, D.T. 1991. Aromatic hydrocarbon degradation: A molecular approach. In Genetic engineering (ed. J.K. Setlow), Vol. 13, pp. 183–203. Plenum Press, New York. [DOI] [PubMed]