

Abstract
Coagulation and complement proteinases are activated in sepsis, and one approach to therapy is to develop proteinase inhibitors that will specifically inhibit these proteinases without inhibiting activated protein C, a proteinase that is beneficial to survival. In this study, we made mutants of the serpin α1-PI, designed to mimic the specificity of C1-inhibitor. The P3-P2-P1 residues of α1-PI were changed from IPM to LGR and PFR, sequences preferred by C1s and kallikrein, respectively. Inhibition of C1s, kallikrein, factor XIIa, and activated protein C was assessed by SDS-PAGE, and by determination of the kapp and SI. α1-PI-LGR inhibited C1s with a rate of 7790 M−1s−1, but only minimal inhibition of C1 in a hemolytic assay was observed. Kallikrein, factor XIIa, and activated protein C were inhibited with rates of 382,180 M−1s−1, 10,400 M−1s−1, and 3500 M−1s−1, respectively. α1-PI-PFR was a poor inhibitor of C1s, factor XIIa, and activated protein C, but had enhanced reactivity with kallikrein. Changing the P4′ residue of α1-PI-LGR Pro to Glu reduced the activity with C1s, consistent with the idea that C1s requires hydrophobic residues in this region of the serpin for optimal interaction. The data provide insight into the requirements for kallikrein and C1s inhibition necessary for designing inhibitors with appropriate properties for further investigation as therapeutic agents.
Keywords: Serpin mutants, kallikrein inhibition, C1 inhibition, serpin reactive center loop
C1-inhibitor is a member of the serpin family of proteinase inhibitors with specificity for plasma kallikrein and factor XIIa of the contact system, and C1 of the classical pathway of complement (Davis 1988). These pathways are activated in sepsis, resulting in C1-inhibitor being found either in complex with proteinases or in an inactive cleaved state (Nuijens et al. 1988, 1989). Numerous studies have shown that C1-inhibitor administration can be of therapeutic use in animal models of sepsis and trauma, and can be beneficial in humans in various inflammatory states, although it is clear that more clinical trials are needed to determine the true efficacy and safety of C1-inhibitor (Kirschfink and Nürnberger 1999; Caliezi et al. 2000; Kirschfink and Mollnes 2001). In general, C1-inhibitor appears to reduce complement and contact system activation, reduce hypoxemia, reduce hypotension, and increase survival. C1-inhibitor replacement therapy is also used successfully to treat C1-inhibitor deficiency (hereditary angioedema) (Waytes et al. 1996; Carugati et al. 2001). However, as with all blood products, the risks of transmission of infectious agents remain (De Filippi et al. 1998). In addition, C1-inhibitor itself has properties that make it less than ideal. It readily converts to an inactive conformation upon even relatively mild denaturing conditions (Patston et al. 1995), and care has to be taken to ensure that activity is not lost during viral inactivation and lyophilization (Williams et al. 1984), such as occurs with the plasma derived serpins antithrombin (Busby et al. 1981; Chang and Harper 1997), and α2-antiplasmin (Mast et al. 1999).
One approach to avoid problems with plasma-derived C1-inhibitor is to create a mutant serpin specifically designed to inhibit kallikrein or C1s, but based on a serpin that has better stability properties, and which can be readily produced in a recombinant expression system. Since the discovery that a naturally occurring mutant of α1-PI was an inhibitor of thrombin rather than elastase (Owen et al. 1983), there has been interest in designing mutant serpins with specificity for particular enzymes. This α1-PI-Pittsburgh mutant had a change of Met to Arg at the P1 residue, hence the change in specificity to Arg-specific proteinases (Schapira et al. 1986; Scott et al. 1986; Travis et al. 1986), and so it was thought that it might be a useful agent in the treatment of sepsis. Although initial animal experiments showed some promise (Colman et al. 1988), this has not turned out to be the case because it also inhibits activated protein C (Heeb et al. 1990; Hermans and Stone 1993; Harper et al. 1998), a proteinase that is beneficial to survival in sepsis (Esmon 2000). This P1-Arg mutant also inhibited kallikrein and factor XIIa (Schapira et al. 1986; Patston et al. 1990), but not C1s (Patston et al. 1990). An additional change of the P2 residue to Ala (as is found in C1-inhibitor) was only able to inhibit C1s very slowly (Patston et al. 1990).
The P3-P2-P1 residues of C1-inhibitor are Val-Ala-Arg, and as such might be expected to be optimal for the main in vivo target proteinases, kallikrein and C1s. However, studies with synthetic peptide substrates and inhibitors indicated that C1s prefers Gly rather than Ala at P2 (McRae et al. 1981; Walker 1987). This is consistent with the cleavage site for C1s in C2 being Leu-Gly-Arg. In contrast, studies with synthetic peptide substrates and inhibitors indicate that kallikrein has a preference for Pro-Phe-Arg in these positions (Levison and Tomalin 1982; Tans et al. 1987). Mutants of C1-inhibitor also suggest that kallikrein favors bulky hydrophobic groups at P3 (Eldering et al. 1993). Together, these results indicate that although the P3 and P2 residues of C1-inhibitor are designed for the inhibition of both kallikrein and C1s in their biological context, they might not be optimal in vitro for inhibition of either kallikrein or C1s, individually. Therefore, we have constructed α1-PI mutants with a P3-P2-P1 sequence of Leu-Gly-Arg, which is predicted to have preference for C1s and with a sequence of Pro-Phe-Arg, which is predicted to have preference for kallikrein. An additional mutant was constructed that contained the P3-P2-P1 sequence of Leu-Gly-Arg with a further change of the P4′ residue from Pro to Glu. Figure 1 ▶ shows a sequence alignment of these mutants and C1-inhibitor. These mutants were tested for their ability to inhibit kallikrein, factor XIIa, C1s, activated protein C, and macromolecular C1.
Fig. 1.

Alignment of the P3-P5′ residues of C1-inhibitor, α1-proteinase inhibitor (α1-PI), and the α1-PI-LGR, α1-PI-PFR, and α1-PI-LGR-P4′E mutants. The mutated residues are highlighted.
Results
Inhibition of kallikrein, C1s, factor XIIa, and activated protein C by α1-PI-LGR and α1-PI-PFR
The inhibition reactions of kallikrein, C1s, factor XIIa, and activated protein C with α1-PI-LGR and α1-PI-PFR mutants were initially assessed by running the reaction mixtures on SDS-PAGE (Fig. 2 ▶). In all cases, complex formation between the α1-PI mutant and the proteinase can be seen as a band of higher molecular weight. kapp and SI are presented in Table 1. The uncorrected rate constants show that α1-PI-LGR is a faster inhibitor of the proteinases than α1-PI-PFR. With α1-PI-LGR, the SI values are close to 1, but with α1-PI-PFR, the SI is elevated with C1s and kallikrein. Correcting the rate constants for the SI shows that in keeping with predictions, α1-PI-LGR is the best serpin inhibitor of C1s known apart from C1-inhibitor, and that α1-PI-PFR shows preference for kallikrein. We did also observe that at longer time points there was some regeneration of enzyme activity (this was particularly prevalent with kallikrein and factor XIIa), which made an accurate determination of the SI difficult. This is also apparent by comparison with the gels, which appear to show that the SI is less than measured. Nevertheless, the conclusion that the SI is markedly elevated with the PFR mutant is valid. This regeneration of activity is consistent with other data showing that residues in this region affect complex stability (Chaillan-Huntington et al. 1997; Plotnick et al. 1997). In regard to complex stability and stoichiometry, it has been shown that mutants of α1-PI-Pittsburgh in which the reactive center loop was shortened by one or two residues had increased complex stability, albeit with an increased SI, and mutants with a longer reactive center loop had reduced stability (Zhou et al. 2001). Although this is different from the mutants investigated here, it does indicate that the composition of the reactive center loop is critical for complex stability.
Fig. 2.

SDS-PAGE (10% acrylamide, nonreduced) of α1-proteinase inhibitor (α1-PI)-LGR, α1-PI-PFR, and α1-PI-LGR-P4′E, and their reactions with C1s, kallikrein, factor XIIa, and activated protein C, as indicated. Each lane contained 5 μg of proteinase and/or inhibitor. The mixtures of proteinase and inhibitor were incubated for 30 min at 37°C prior to stopping the reaction by addition of sample buffer.
Table 1.
The second order rate constants (M−1s−1) and stoichiometries of inhibition (SI) for the reactions of α1-PI-LGR, α1-PI-PFR, and α1-PI-LGR-P4′E with C1s, kallikrein, factor XIIa, and activated protein C
| Rate (M−1 s−1) | SI | Rate × SI (M−1s−1) | |
| α1-PI-LGR with: | |||
| C1s | 7,350 | 1.06 | 7,790 |
| Kallikrein | 357,181 | 1.07 | 382,180 |
| Factor XIIa | 8,673 | 1.2 | 10,400 |
| Activated protein C | 2,713 | 1.3 | 3,530 |
| α1-PI-PFR with: | |||
| C1s | 236 | 2.68 | 630 |
| Kallikrein | 172,045 | 5.05 | 868,530 |
| Factor XIIa | 70 | ND | ND |
| Activated protein C | 3.5 | ND | ND |
| α1-PI-LGR-P4′E with: | |||
| C1s | 220 | 1.2 | 260 |
| Kallikrein | 214,000 | 1.2 | 256,800 |
| Factor XIIa | 1,870 | 1.8 | 3,370 |
| Activated protein C | 2,500 | 2.5 | 6,250 |
ND, not determined due to slow rate of inhibition.
For comparison of the kinetic data measured with the LGR and PFR mutants, C1-inhibitor inhibits C1s at 95,000 M−1s−1 (Lennick et al. 1986), kallikrein at 45,000 M−1s−1 (van der Graff et al. 1983b), factor XIIa at 3600 M−1s−1 (Pixley et al. 1985), and activated protein C at 1 M−1s−1 (Hermans and Stone 1993). Given that the C1s-C1-inhibitor reaction is not particularly fast compared to reactions of other serpins and proteinases, these results suggest that it might be possible to further adapt α1-PI-LGR to make it as good as, or better than, C1-inhibitor as is discussed further below.
Inhibition of macromolecular C1 by α1-PI-LGR and C1-inhibitor
Although the α1-PI-LGR inhibits C1s, the form of C1s that exists in vivo is not free C1s, but C1s as part of the C1 macromolecule. Therefore, the ability of C1-inhibitor and α1-PI-LGR to inhibit C1 in a hemolytic assay were compared. When C1-inhibitor was preincubated with C1, there was an inhibition of lysis of the EA cells of 11% with 0.5 nM C1-inhibitor, and 80% with 2.5 nM of C1-inhibitor. In contrast, α1-PI-LGR at 25 nM did not inhibit lysis at all, and 250 nM only caused 27% inhibition of lysis. Increasing the α1-PI-LGR concentration to 2.5 μM only caused 32% inhibition. It was not possible to use a higher concentration of α1-PI-LGR in the assay to get a full dose response curve, however from these results we estimate that α1-PI-LGR is ∼500 times less effective than C1-inhibitor at inhibiting C1 than is C1-inhibitor. This is in contrast to the13 times difference in the inhibition rate constants with C1s.
Properties of additional mutants derived from α1-PI-LGR
Even though the rate of reaction of α1-PI-LGR and C1s is enhanced compared to an earlier mutant of α1-PI (Patston 1990), it still remains less than the rate with C1-inhibitor. This difference could be the result of a more extended region of interaction between C1-inhibitor and C1s than just the P3-P1 residues. The rationale for this idea is from studies on exosites for the reaction between t-PA and PAI-1, which concluded that there were electrostatic interactions between residues in the P4′-P9′ region of the serpin with residues in a surface loop of the proteinase (Madison et al. 1989, 1990a,b). This loop is present between residues 36–41 in the aligned trypsin sequence and is called Variable Region 1 (VR1). In C1s, there is an elongated hydrophobic insert at this position (Mackinnon et al. 1987), and in C1-inhibitor the P4′-P9′ region is somewhat hydrophobic in nature (Bock et al. 1986), suggesting that a hydrophobic interaction between these two domains might take place. The corresponding VR1 region of kallikrein has a small insert containing a lysine (Chung et al. 1986). α1-PI-LGR has a glutamic acid at P5′, which might act to enhance the reaction with kallikrein and reduce the reaction with C1s. This is consistent with all α1-PI mutants that contain an arginine at P1 (including α1-PI-Pittsburgh) being effective inhibitors of kallikrein (and better than C1-inhibitor), and with the failure of α1-PI-LGR to be as effective as C1-inhibitor for C1s inhibition. To investigate this, we changed the P5′ Glu of the α1-PI-LGR mutant to Ala. However, the protein was not stable, and spontaneously polymerized (data not shown). Inspection of the structure of native α1-PI (PDB ID: 1 QLP) shows that although the side chain of the P5′ Glu (residue 363) is solvent exposed and has no intramolecular contacts, the carboxyl group forms a hydrogen bond with the amide group of Lys 365. Changing Glu to Ala probably alters the orientation of the residue side chain away from the solvent towards the body of the protein, which in turn causes loss of stabilizing interactions such as the hydrogen bond. The integrity of β-sheet C (of which this region is strand 1), is likely to be critical for the correct folding of the serpin in the native metastable state (Eldering et al. 1995; Patston and Gettins 1996; Chang et al. 1997; Bottomley et al. 2001). Another mutant in which we changed the P9′ residue of the α1-PI- LGR Gln to Glu also formed polymers, consistent with this idea (data not shown).
In view of this, another approach was taken. The P4′ proline was converted to glutamic acid, with the intent of adding another acidic residue that might decrease the reactivity with C1s, to test the validity of the idea that this region is an important exosite for C1s inhibition. This mutant was tested in the same manner as the previous two mutants. The result of analysis by SDS-PAGE of the inhibition of kallikrein, activated protein C, C1s, and factor XIIa are shown in Figure 2 ▶, and the results of assays are shown in Table 1. As predicted, the rate of reaction with C1s was reduced by this mutation. Addition of the extra glutamic acid also reduced the rate with kallikrein, suggesting that the P5′ glutamic acid is optimal for interaction with kallikrein. The importance of P′ residues in directing serpins toward specific serine proteinases is also supported by data with the serpin PI-9, which has a P4′ glutamic acid important for rapid reaction with granzyme B (Sun et al. 2001).
Discussion
In this study, we have constructed mutants of α1-PI with the aim of mimicking the specificity of C1-inhibitor and enhancing selectivity for C1s or kallikrein. These proteinases are inhibited by C1-inhibitor with kapp of <105 M−1s−1, rates that are slow compared to other examples of physiologically important proteinase-serpin interactions, such as t-PA-PAI-1, plasmin-antiplasmin, and antithrombin/heparin-thrombin, which are all about 107 M−1s−1. The structural reasons why the inhibition reactions of C1-inhibitor are slow compared to other serpins are not understood, although clearly C1-inhibitor is designed to operate at this rate and be optimal for the role it plays in vivo. Therefore, to design mutants of α1-PI, which have activity similar to C1-inhibitor and which might be able to substitute for it in vivo, was considered possible, as fast inhibition rates (∼106–107 M−1s−1) would not need to be obtained for the mutant to be able to function. The approach taken was to consider the cleavage sites in substrates of C1s and kallikrein, and modify the reactive center loop of α1-PI accordingly. Thus, we made α1-PI containing the P3-P2-P1 residues of LGR and PFR. The mutants were assayed for inhibition of C1s, kallikrein, factor XIIa, and activated protein C.
As predicted, the LGR mutant had the best inhibition of C1s, in keeping with the limited substrate specificity of this enzyme, and the PFR mutant was a very poor inhibitor of C1s. To determine the true rate for the E + I → EI step, it is necessary to multiply the kapp by the SI (Gettins 1996). Looking at C1s, it can be seen that this step is faster for the LGR mutant than for the PFR mutant. This is consistent with our prediction that the LGR mutation would give specificity for C1s over kallikrein. Similarly with kallikrein, this step is faster with the PFR mutant than with the LGR mutant, again according to predictions. Curiously though, both mutants inhibited kallikrein at rates faster than C1-inhibitor, indicating that C1-inhibitor is not optimal for kallikrein inhibition, when considered in this in vitro context. In vivo however, this rate is optimal in the biological context, especially as kallikrein is also significantly inhibited by α2-macroglobulin, and to a lesser extent antithrombin (Lewin et al. 1983; van der Graaf et al. 1983a; Olson et al. 1993). Although the LGR mutant is slower than C1-inhibitor at inhibiting C1s, it is only the second α1-PI mutant reported to be able to inhibit C1s. An earlier study (Patston et al. 1990) showed that α1-PI containing the P2-P1 residues of Ala-Arg (as found in C1-inhibitor) could weakly inhibit C1s (300 M−1s−1). A study with P2 mutants of C1-inhibitor suggests that Gly at this position does not alter the ability to inhibit C1s, but that bulkier or charged residues (Asn, Thr, Val, or Asp) are not favored (Zahedi et al. 2001). SERP-1, a serpin from myxoma virus, also inhibits C1s with rate constants of 600 M−1s−1 (Nash et al. 1998) or 1300 M−1s−1 (Lomas et al. 1993). This has P3-P2-P1 residues of IPR, the same as in α1-PI-Pittsburgh, but surprisingly does not inhibit kallikrein (Lomas et al. 1993). In fact, compared to α1-PI-Pittsburgh, it has a somewhat limited specificity. The P5′ residue in SERP-1 is alanine and so this might contribute to the lack of activity with kallikrein, however the fact that there is apparently no reaction at all with kallikrein (not even as a substrate), suggests that additional exosites in α1-PI-Pittsburgh or SERP-1 might either enhance or decrease the reactions with kallikrein and other proteinases.
Because C1-inhibitor will not encounter free C1s in vivo, but rather C1s as part of the C1 complex, we carried out hemolytic C1 inhibition assays. α1-PI-LGR was about 500 times less effective at preventing hemolysis than C1-inhibitor, even though it is only 13 times less effective at inhibiting C1s. This finding suggests that C1-inhibitor might have an exosite essential for directing it toward C1s within C1. Where this exosite resides on C1-inhibitor, and what role might be played by C1q, C1r, or C1s itself is not known. One possibility is that the amino-terminal domain of C1-inhibitor, which is a highly glycosylated 100 amino-acid region not found in other serpins, could play a role here, although there is no evidence to support this. The function of this region is unknown (Coutinho et al. 1994).
The inhibition of activated protein C by the three mutants was measured because of the requirement that this enzyme remain active in any proteinase inhibitor-based therapy for sepsis. In this regard, the PFR mutant was a very poor inhibitor of activated protein C, and the LGR mutant inhibited it relatively slowly. However, even this rate is probably too fast for the LGR mutant to be used in sepsis, especially given the likelihood for pharmacological doses to be quite large. It is clear from a number of studies that design of mutant serpins that do not inhibit activated protein C is problematic. In general, a glycine at the P2 position is detrimental to inhibition of activated protein C (Phillips et al. 1994; Hopkins et al. 1995, 2000; Elisen et al. 1998). However, there appear to be cooperative (and not predictable) interactions within the reactive center loop that affect the reaction with activated protein C, thereby making it difficult to design serpins that will not inhibit this enzyme (Hopkins et al. 2000).
In conclusion, we have produced three mutants of α1-PI designed to have specificity for C1s and kallikrein. The α1-PI-LGR mutant was the best inhibitor of C1s, a good inhibitor of kallikrein, and also inhibited activated protein C, but was a poor inhibitor of C1. The inhibition of activated protein C is a drawback in designing inhibitors for use in sepsis. The additional mutation of the P4′ residue reduced C1s inhibition and appeared to offer no benefit. The α1-PI-PFR mutant was a poor inhibitor of C1s and also showed increased SI with all the enzymes. Therefore, the mutant with the most potential is α1-PI-LGR, and future studies will be aimed at enhancing activity toward C1s and C1 and reducing activity toward activated protein C.
Materials and methods
Materials
Oligonucleotides were synthesized by the University of Illinois at Chicago Molecular Biology core facility. The Quick Change mutagenesis system was from Stratagene. The chromogenic substrates S-2302 and S-2366 were from DiaPharma and Spectrozyme C1-E was from American Diagnostica. Plasma kallikrein, active C1s, factor XIIa, and activated protein C were from Enzyme Research Laboratories. Complement reagents (C1-inhibitor, C1, C2, C4, EA cells, human and guinea pig sera) were from Advanced Research Technologies.
Mutagenesis, expression, refolding, and purification of recombinant α1-proteinase inhibitor mutants
α1-PI cDNA with the P1 residue mutated from Met to Arg (α1-PI-Pittsburgh) was provided by Dr. Peter Gettins (Department of Biochemistry and Molecular Biology, University of Illinois at Chicago). Cys 232 was also changed to Ala, which has no effect on the inhibitory activity, but improves the purification, as there is no likelihood of disulfide-bonded dimers forming. Mutation of the P3 and P2 residues to Leu-Gly and Pro-Phe was carried out by PCR essentially as described with other α1-PI mutants (Chaillan-Huntington et al. 1997; Stratikos and Gettins 1998). The recombinant proteins were produced as inclusion bodies in Escherichia coli. The recovery of the active proteins from the inclusion bodies was carried out as described previously (Chaillan-Huntington et al. 1997; Stratikos and Gettins 1998). The purified proteins were stored at −70°C. A further mutation of the α1-PI-LGR mutant with the P4′ residue changed from Pro to Glu and was carried using the same techniques.
Assay of proteinase inhibition by the recombinant α1-proteinase inhibitor mutants
SDS-PAGE was carried out using the Laemmli method (Laemmli 1970) with a Bio-Rad mini gel system. Samples were nonreduced. Gels were stained with Gel-Code Blue (Pierce). Kallikrein and factor XIIa were assayed using 0.6 mM S-2302, C1s was assayed with 0.75 mM Spectrozyme C1E, and activated protein C was assayed using 0.56 mM S-2366, all at 37°C in 50 mM Tris-HCl, pH 7.4, 150 mM NaCl, essentially as described previously (Patston et al. 1990, 1991). The concentration of each enzyme was taken to be that provided by the supplier. The kapp for the reactions of kallikrein (at 0.14 μM) with the three α1-PI mutants were determined under pseudo-first–order conditions by a discontinuous assay by standard techniques as described previously (Patston et al. 1990). Similarly, the stoichiometry of inhibition (SI) of each reaction at 37°C was determined by titrations as described previously (Patston et al. 1991). For the reaction to reach completion, α1-PI-LGR was incubated with C1s for 1 hr, with kallikrein for 1 hr, with factor XIIa for 2 hr, and with activated protein C for 24 hr; α1-PI-PFR was incubated with C1s for 24 hr, and with kallikrein for 1 hr; and α1-PI-LGR-P4′E was incubated with C1s for 24 hr, with kallikrein for 1 hr, with factor XIIa for 3 hr, and with activated protein C for 24 hr. In each case, the enzyme activity in the absence of inhibitor was stable under the conditions used. Serpins inhibit proteinases by the suicide substrate mechanism shown in Scheme 1.
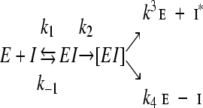 |
Scheme 1 |
The measured kapp rate kapp is defined as kapp = (k2/Ks) × (k4/k4 + k3). This term is dependent on the amount of partitioning between the noninhibitory substrate pathway and the inhibitory pathway as indicated by the (k4/k4 + k3) term. The SI is defined as (k4 + k3/k4), therefore multiplying kapp by the SI will give the rate constant k′app, which represents the true rate of inhibition (Gettins et al. 1996).
Assay of C1 inhibition by hemolytic assay
C1 was assayed using the procedure described by supplier of the reagents (Advanced Research Technologies). C1-inhibitor (1 nM or 5nM), or α1-PI-LGR (50 nM, 500 nM, or 5 μM) (both proteins in 100 μL) were incubated with 100 μL of C1 (0.6 μg/mL or 0.75 μM) at 30°C for 30 min in SGVB++. This amount of C1 was determined to give ∼60% lysis of the EA cells in the absence of any inhibitor. Next, 100 μL of SGVB++ containing 1.5 μg C4 and 0.2 μg C2, and 200 μL of EA cells at 3 × 108 cells/mL were added, and this mixture was incubated at 30°C for 12 min. The samples were then put on ice and 1 mL of guinea pig serum diluted 1:50 in GVB0/40 mM EDTA, pH 7.2 was added, and then incubated at 30° for 30 min. Samples were then centrifuged at 1000 × g for 5 min, and the absorbance of the supernatant at 412 nm determined. Total cell lysis was determined by addition of 1 mL of water to a mix of 200 μL EA cells at 3 × 108 cells/mL, and 100 μL of SGVB++.
Acknowledgments
We thank Scott Suda, Betsy Brown, and Inder Chand for their assistance with the mutagenesis, expression, and purification of the α1-PI mutants. This work was supported by National Institutes of Health grant HL-49242. The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
α1-PI, α1-proteinase inhibitor
P1, the amino acid at the N-terminal side of the scissile bond in the reactive center loop of the serpin
P2, the amino acid at the N-terminal side of the P1 residue
P3 the amino acid at the N-terminal side of the P2 residue
SI, stoichiometry of inhibition
kapp, measured apparent second order rate constant
k′app, kapp × SI
SGVB++, sucrose gelatin veronal buffer with 0.15 mM Ca++ and 1 mM Mg++
GVB0, gelatin veronal buffer without divalent cations
EA cells, antibody sensitized sheep erythrocytes
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0207302.
References
- Bock, S.C., Skriver, K., Nielsen, E., Thogerson, H.C., Wiman, B., Donaldson, V.H., Eddy, R.L., Marrinan, J., Radziejewska, E., Huber, R., Shows, T.B., and Magnusson S. 1986. Human C1 inhibitor: Primary structure, cDNA cloning, and chromosomal localization.Biochemistry 25 4292–4301. [DOI] [PubMed] [Google Scholar]
- Bottomley, S.P., Lawrenson, I.D., Tew, D., Dai, W., Whisstock, J.C., and Pike, R.N. 2001. The role of strand 1 of the C β-sheet in the structure and function of α(1)-antitrypsin. 2001.Protein Sci. 10 2518–2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busby, T.F., Atha, D.H., and Ingham, K.C. 1981. Thermal denaturation of antithrombin III. Stabilization by heparin and lyotropic anions.J. Biol. Chem. 256 12140–12147. [PubMed] [Google Scholar]
- Caliezi, C., Wuillemin, W.A., Zeerleder, S., Redondo, M., Eisele, B., and Hack, C.E. 2000. C1-Esterase inhibitor: An anti-inflammatory agent and its potential use in the treatment of diseases other than hereditary angioedema.Pharmacol. Rev. 52 91–112. [PubMed] [Google Scholar]
- Carugati, A., Pappalardo, E., Zingale, L.C., and Cicardi, M. 2001. C1-inhibitor deficiency and angioedema.Mol. Immunol. 38 161–1739. [DOI] [PubMed] [Google Scholar]
- Chaillan-Huntington, C.E., Gettins, P.G.W., Huntington, J.A., and Patston, P.A. 1997. The P6-P2 region of serpins is critical for proteinase inhibition and complex stability.Biochemistry 36 9562–9570. [DOI] [PubMed] [Google Scholar]
- Chang, W.S. and Harper PL. 1997. Commercial antithrombin concentrate contains inactive L-forms of antithrombin.Thromb. Haemost. 77 323–328. [PubMed] [Google Scholar]
- Chang, W.S., Whisstock, J., Hopkins, P.C., Lesk, A.M., Carrell, R.W., and Wardell, M.R. 1997. Importance of the release of strand 1C to the polymerization mechanism of inhibitory serpins.Protein Sci. 6 89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung, D.W., Fujikawa, K., McMullen, B.A., and Davie, E.W. 1986. Human plasma prekallikrein, a zymogen to a serine protease that contains four tandem repeats.Biochemistry 25 2410–2417. [DOI] [PubMed] [Google Scholar]
- Colman, R.W., Flores, D.N., De La Cadena, R., Scott, C.F., Cousens, L., Barr, P.J., Hoffman, I.B., Kueppers, F., Fisher, D., Idell, S., and Pisarello, J. 1988. Recombinant α 1-antitrypsin Pittsburgh attenuates experimental Gram-negative septicemia.Am. J. Pathol. 130 418–426. [PMC free article] [PubMed] [Google Scholar]
- Coutinho, M., Aulak, K.S., and Davis III, A.E. 1994. Functional analysis of the serpin domain of C1 inhibitor.J. Immunol. 153 3648–3654. [PubMed] [Google Scholar]
- Davis, III, A.E. 1988. C1 inhibitor and hereditary angioneurotic edema.Ann. Rev. Immunol. 6 595–628. [DOI] [PubMed] [Google Scholar]
- De Filippi, F., Castelli, R., Cicardi, M., Soffredini, R., Rumi, M.G., Silini, E., Mannuci, P.M., and Colombo, M. 1998. Transmission of hepatitis G virus in patients with angioedema treated with steam-heated plasma concentrates of C1 inhibitor.Transfusion 38 307–311. [DOI] [PubMed] [Google Scholar]
- Eldering, E., Huijbregts, C.C., Nuijens, J.H., Verhoeven, A.J., and Hack, C.E. 1993. Recombinant C1 inhibitor P5/P3 variants display resistance to catalytic inactivation by stimulated neutrophils.J. Clin. Invest. 91 1035–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldering, E., Verpy, E., Roem, D., Meo, T., and Tosi, M. 1995. COOH-terminal substitutions in the serpin C1 inhibitor that cause loop overinsertion and subsequent multimerization.J. Biol. Chem. 270 2579–2587. [DOI] [PubMed] [Google Scholar]
- Elisen, M.G.L.M., Bouma, B.N., Church, F.C., and Meijers, J.C.M. 1998. Inhibition of serine proteases by reactive site mutants of protein C inhibitor (plasminogen activator inhibitor 3).Fibrinol. Proteol. 12 283–291. [Google Scholar]
- Esmon, C.T. 2000. Regulation of blood coagulation.Biochim. Biophys. Acta. 1477 349–360. [DOI] [PubMed] [Google Scholar]
- Gettins, P.G.W., Patston, P.A., and Olson, S.T. 1996. Serpins: Structure, function and biology. Molecular Biology Intelligence Unit series, R.G. Landes, Austin, TX.
- Harper, P.L., Taylor, F.B., De La Cadena, R.A., Courtney, M., Colman, R.W., and Carrell, R.W. 1998. Recombinant antitrypsin Pittsburgh undergoes proteolytic cleavage during E. coli sepsis and fails to prevent the associated coagulopathy in a primate model.Thromb. Haemost. 80 816–821. [PubMed] [Google Scholar]
- Heeb, M.J., Bischoff, R., Courtney, M., and Griffin, J.H. 1990. Inhibition of activated protein C by recombinant α 1-antitrypsin variants with substitution of arginine or leucine for methionine358.J. Biol. Chem. 265 2365–2369. [PubMed] [Google Scholar]
- Hermans, J.M. and Stone, S.R. 1993. Interaction of activated protein C with serpins.Biochem. J. 295 239–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins, P.C.R., Crowther, D.C., Carrell, R.W., and Stone, S.R. 1995. Development of a novel recombinant serpin with potential antithrombotic properties.J. Biol. Chem. 270 11866–11871. [DOI] [PubMed] [Google Scholar]
- Hopkins, P.C.R., Pike, R.N., and Stone, R.N. 2000. Evolution of serpin specificity: Cooperative interactions in the reactive-site loop sequence of antithrombin specifically restrict the inhibition of activated protein C.J. Mol. Evol. 51 507–515. [DOI] [PubMed] [Google Scholar]
- Kirschfink, M. and Mollnes, T.M. 2001. C1-inhibitor: An anti-inflammatory reagent with therapeutic potential.Expert Opin. Pharmacother. 2 1073–1083. [DOI] [PubMed] [Google Scholar]
- Kirschfink, M. and Nürnberger, W. 1999. C1 inhibitor in anti-inflammatory therapy: From animal experiment to clinical application.Mol. Immunol. 36 225–232. [DOI] [PubMed] [Google Scholar]
- Laemmli, U.K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4.Nature 227 680–685. [DOI] [PubMed] [Google Scholar]
- Lennick, M., Brew, S.A., and Ingham, K.C. 1986. Kinetics of interaction of C1 inhibitor with complement C1s.Biochemistry 25 3890–3898. [DOI] [PubMed] [Google Scholar]
- Levison, P.R. and Tomalin, G. 1982. The kinetics of hydrolysis of some extended N-aminoacyl-L-arginine methyl esters by human plasma kallikrein. Evidence for subsites S2 and S3.Biochem. J. 203 149–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewin, M.F., Kaplan, A.P., and Harpel, P.C. 1983. Studies of C1 inactivator-plasma kallikrein complexes in purified systems and in plasma.J. Biol. Chem. 258 6415–6421. [PubMed] [Google Scholar]
- Lomas, D.A., Evans, D.L., Upton, C., McFadden, G., and Carrell, R.W. 1993. Inhibition of plasmin, urokinase, tissue plasminogen activator, and C1s by a myxoma virus serine protease inhibitor.J. Biol. Chem. 268 516–521. [PubMed] [Google Scholar]
- Mackinnon, C.M., Carter, P.E., Smyth, S.I., Dunbar, B., and Fothergill, J.E. 1987. Molecular cloning of cDNA for human complement component C1s. The complete amino acid sequence.Eur. J. Biochem. 169 547–553. [DOI] [PubMed] [Google Scholar]
- Madison, E.L., Goldsmith, E.J., Gerard, R.D., Gething, M.J.H., and Sambrook, J.F. 1989. Serpin-resistant mutants of human tissue-type plasminogen activator.Nature 339 721–724. [DOI] [PubMed] [Google Scholar]
- Madison, E.L., Goldsmith, E.J., Gerard, R.D., Gething, M.J.H., Sambrook, J.F., and Bassel-Duby, R.S. 1990a. Amino acid residues that affect interaction of tissue-type plasminogen activator with plasminogen activator inhibitor 1.Proc. Natl. Acad. Sci. 87 3530–3533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madison, E.L., Goldsmith, E.J., Gething, M.J.H., Sambrook, J.F., and Gerard, R.D. 1990b. Restoration of serine protease-inhibitor interaction by protein engineering.J. Biol. Chem. 265 21423–21426. [PubMed] [Google Scholar]
- Mast, A.E., Stadanlick, J.E., Lockett, J.M., and Dietzen, D.J. 1999. Solvent/detergent-treated plasma has decreased antitrypsin activity and absent antiplasmin activity.Blood 94 3922–3927. [PubMed] [Google Scholar]
- McRae, B.J., Lin, T.Y., and Powers, J.C. 1981. Mapping the substrate binding site of human C1r and C1s with peptide thioesters. Development of new sensitive substrates.J. Biol. Chem. 25612362–12366. [PubMed] [Google Scholar]
- Nash, P., Whitty, A., Handwerker, J., Macen, J., and McFadden, G. 1998. Inhibitory specificity of the anti-inflammatory myxoma virus, SERP-1.J. Biol. Chem. 273 20982–20991. [DOI] [PubMed] [Google Scholar]
- Nuijens, J.H., Huijbregts, C.C.M., Eerenberg-Belmer, A.J.M., Abbink, J.J., Strack van Schijndel, R.J.M., Felt-Bersma, R.J.F., Thijs, L.G., and Hack, C.E. 1988. Quantification of plasma factor XIIa-Cl-inhibitor and kallikrein-Cl-inhibitor complexes in sepsis.Blood 721841–1848. [PubMed] [Google Scholar]
- Nuijens, J.H., Eerenberg-Belmer, A.J.M., Huijbregts, C.C.M., Schreuder, W.O., Felt-Bersma, R.J.F., Abbink, J.J., Thijs, L.G., and Hack, C.E. 1989. Proteolytic inactivation of plasma C1-inhibitor in sepsis.J. Clin. Invest. 84 443–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson, S.T., Sheffer, R., and Francis, A.M. 1993. High molecular weight kininogen potentiates the heparin-accelerated inhibition of plasma kallikrein by antithrombin: Role for antithrombin in the regulation of kallikrein.Biochemistry 32 12136–12147. [DOI] [PubMed] [Google Scholar]
- Owen, M.C., Brennan, S.O., Lewis, J.H., and Carrell, R.W. 1983. Mutation of antitrypsin to antithrombin. α 1-antitrypsin Pittsburgh (358 Met to Arg), a fatal bleeding disorder.N. Eng. J. Med. 309 694–698. [DOI] [PubMed] [Google Scholar]
- Patston, P.A., Roodi, N., Schifferli, J.A., Bischoff, R., Courtney, M., and Schapira, M. 1990. Reactivity of α 1-antitrypsin mutants against proteolytic enzymes of the kallikrein-kinin, complement, and fibrinolytic systems.J. Biol. Chem. 265 10786–10791. [PubMed] [Google Scholar]
- Patston, P.A., Gettins, P., Beechem, J., and Schapira M. 1991. Mechanism of serpin action: Evidence that C1 inhibitor functions as a suicide substrate.Biochemistry 30 8876–8882. [DOI] [PubMed] [Google Scholar]
- Patston, P.A., Hauert, J., Michaud, M., and Schapira, M. 1995. Formation and properties of C1-inhibitor polymers.FEBS Lett. 368 401–404. [DOI] [PubMed] [Google Scholar]
- Patston, P.A. and Gettins, P.G.W. 1996. Significance of secondary structure predictions on the reactive center loop region of serpins: A model for the folding of serpins into a metastable state.FEBS Lett. 373 87–92. [DOI] [PubMed] [Google Scholar]
- Phillips, J.E., Cooper, S.T., Potter, E.E., and Church, F.C. 1994. Mutagenesis of recombinant protein C inhibitor reactive site residues alters target proteinase specificity.J. Biol. Chem. 269 16696–16700. [PubMed] [Google Scholar]
- Pixley, R.A., Schapira,. M., and Colman, R.W. 1985. The regulation of human factor XIIa by plasma proteinase inhibitors.J. Biol. Chem. 260 1723–1729. [PubMed] [Google Scholar]
- Plotnick, M.I., Schechter, N.M., Wang, Z.M., Liu, X., and Rubin, H. 1997. Role of the P6-P3′ region of the serpin reactive loop in the formation and breakdown of the inhibitory complex.Biochemistry 3614601–14608. [DOI] [PubMed] [Google Scholar]
- Schapira, M., Ramus, M.A., Jallat, S., Carvallo, D., and Courtney, M. 1986. Recombinant α 1-antitrypsin Pittsburgh (Met 358 Arg) is a potent inhibitor of plasma kallikrein and activated factor XII fragment.J. Clin. Invest. 77 635–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott, C.F., Carrell, R.W., Glaser, C.B., Kueppers, F., Lewis, J.H., and Colman, R.W. 1986. &alpha-1-antitrypsin-Pittsburgh. A potent inhibitor of human plasma factor XIa, kallikrein, and factor XIIf.J. Clin. Invest. 77 631–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratikos, E. and Gettins, P.G.W. 1998. Mapping the serpin-proteinase complex using single cysteine variants of α 1-proteinase inhibitor Pittsburgh.J. Biol. Chem. 273 15582–15589. [DOI] [PubMed] [Google Scholar]
- Sun, J., Whisstock, J.C., Harriott, P., Walker, B., Novak, A., Thompson, P.E., Smith, A.I., and Bird, P.I. 2001. Importance of the P4′ residue in human granzyme B inhibitors and substrates revealed by scanning mutagenesis of the proteinase inhibitor 9 reactive center loop.J. Biol. Chem. 276 15177–15184. [DOI] [PubMed] [Google Scholar]
- Tans, G., Janssen-Claessen, T., Rosing, J., and Griffin, J.H. 1987. Studies on the effect of serine protease inhibitors on activated contact factors. Application in amidolytic assays for factor XIIa, plasma kallikrein and factor XIa.Eur. J. Biochem. 164 637–642. [DOI] [PubMed] [Google Scholar]
- Travis, J., Matheson, N.R., George, P.M., and Carrell, R.W. 1986. Kinetic studies on the interaction of α 1-proteinase inhibitor (Pittsburgh) with trypsin-like serine proteinases.Biol. Chem. Hoppe-Seyler. 367 853–859. [DOI] [PubMed] [Google Scholar]
- van der Graaf, F., Koedam, J.A., and Bouma, B.N. 1983a. Inactivation of kallikrein in human plasma.J. Clin. Invest. 71 149–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Graaf, F., Koedam, J.A., Griffin, J.H., and Bouma, B.N. 1983b. Interaction of human plasma kallikrein and its light chain with C1 inhibitor.Biochemistry 22 4860–4866. [DOI] [PubMed] [Google Scholar]
- Walker, B. 1987. A kinetic study of the inhibition of C1s by peptides bearing C-terminal arginyl chloromethane.Biochem. Soc. Trans. 15 657. [Google Scholar]
- Waytes, A.T., Rosen, F.S., and Frank, M.M. 1996. Treatment of hereditary angioedema with a vapor-heated C1 inhibitor concentrate.New Engl. J. Med. 334 1630–1634. [DOI] [PubMed] [Google Scholar]
- Williams, C., Wickerhauser, M., Busby, T.F., and Ingham, K.C. 1984. Pasteurization of C1 inactivator in the presence of citrate salts.Vox Sang. 46 260–269. [DOI] [PubMed] [Google Scholar]
- Zahedi, R., MacFarlane, R.C., Wisnieski, J.J., and Davis III, A.E. 2001. C1 inhibitor: Analysis of the role of amino acid residues within the reactive center loop in target protease recognition.J. Immunol. 167 1500–1506. [DOI] [PubMed] [Google Scholar]
- Zhou, A., Carrell, R.W., and Huntington, J.A. 2001. The serpin inhibitory mechanism is critically dependent on the length of the reactive center loop.J. Biol. Chem. 276 27541–27547. [DOI] [PubMed] [Google Scholar]