

Abstract
Formyltransferase catalyzes the reversible formation of formylmethanofuran from N5-formyltetrahydromethanopterin and methanofuran, a reaction involved in the C1 metabolism of methanogenic and sulfate-reducing archaea. The crystal structure of the homotetrameric enzyme from Methanopyrus kandleri (growth temperature optimum 98°C) has recently been solved at 1.65 Å resolution. We report here the crystal structures of the formyltransferase from Methanosarcina barkeri (growth temperature optimum 37°C) and from Archaeoglobus fulgidus (growth temperature optimum 83°C) at 1.9 Å and 2.0 Å resolution, respectively. Comparison of the structures of the three enzymes revealed very similar folds. The most striking difference found was the negative surface charge, which was −32 for the M. kandleri enzyme, only −8 for the M. barkeri enzyme, and −11 for the A. fulgidus enzyme. The hydrophobic surface fraction was 50% for the M. kandleri enzyme, 56% for the M. barkeri enzyme, and 57% for the A. fulgidus enzyme. These differences most likely reflect the adaptation of the enzyme to different cytoplasmic concentrations of potassium cyclic 2,3-diphosphoglycerate, which are very high in M. kandleri (>1 M) and relatively low in M. barkeri and A. fulgidus. Formyltransferase is in a monomer/dimer/tetramer equilibrium that is dependent on the salt concentration. Only the dimers and tetramers are active, and only the tetramers are thermostable. The enzyme from M. kandleri is a tetramer, which is active and thermostable only at high concentrations of potassium phosphate (>1 M) or potassium cyclic 2,3-diphosphoglycerate. Conversely, the enzyme from M. barkeri and A. fulgidus already showed these properties, activity and stability, at much lower concentrations of these strong salting-out salts.
Keywords: Formyltransferase, crystal structure, monomer/dimer/tetramer association equilibrium, Methanosarcina barkeri, Methanopyrus kandleri, Archaeoglobus fulgidus, methanogenic archaea, sulfate-reducing archaea
Formyltransferase is an enzyme found in all methanogenic archaea, in sulfate-reducing archaea, and in most methylotrophic bacteria (Chistoserdova et al. 1998; Thauer 1998). The enzyme catalyzes the reversible formation of N5-formyltetrahydromethanopterin from N-formylmethanofuran and tetrahydromethanopterin (ΔG°‘ = −5.5 kJ/mole). In methanogenic archaea, this reaction is involved in CO2 reduction to methane, in methanol disproportionation to methane and CO2, and in autotrophic CO2 fixation (Thauer 1998); in sulfate-reducing archaea, the enzyme is involved in the oxidation of lactate to 3 CO2 and in autotrophic CO2 fixation (Thauer and Kunow 1995; Vorholt et al. 1997); and in methylotrophic bacteria, it participates in methanol oxidation to CO2 (Vorholt et al. 1998).
The formyltransferases from all methanogenic and sulfate-reducing archaea investigated to date contain only one type of subunit, which has a molecular mass of ∼35 kD and does not contain any prosthetic groups. The enzyme shows a ternary complex catalytic mechanism (Breitung et al. 1992; Schwörer et al. 1993). The genes encoding formyltransferase from several organisms have been cloned and sequenced; it was found that active enzymes could be heterologously overproduced in Escherichia coli (Donnelly and Wolfe 1986; Shima et al. 1995; Kunow et al. 1996).
Studies of the properties and structure of formyltransferase have mainly concentrated on the enzyme from Methanopyrus kandleri, which is a hyperthermophilic methanogen with a growth temperature optimum of 98°C (Kurr et al. 1991). The enzyme is thermostable and active only at relatively high concentrations of potassium phosphate (>1 M) or of other salts with strongly hydrated anions (strong salting-out salts; Breitung et al. 1992). This unique property is reflected in the internal milieu of this microorganism, where a potassium cyclic 2,3-phosphoglycerate concentration of >1 M was found in the cytoplasm (Kurr et al. 1991; Shima et al. 1998a). Interestingly, M. kandleri lives and thrives in nonhalophilic habitats and copes with the high difference in osmotic pressure by having a thick, multilayered pseudomurein sacculus (Kurr et al. 1991).
Potassium cyclic 2,3-diphosphoglycerate and potassium phosphate were found to be equivalent in activating and stabilizing the enzyme (Shima et al. 1998a). At low concentrations of these salts, the enzyme is inactive and thermolabile. It was shown, by equilibrium sedimentation analysis, that the enzyme from M. kandleri is in a salt-concentration-dependent monomer/dimer/tetramer equilibrium, in which high salt concentrations are required for oligomerization (Shima et al. 1998b). It was found that dimerization is required for activity and tetramerization leads to thermostability (Shima et al. 2000).
The formyltransferase from M. kandleri is not activated or stabilized in 1 M NaCl or KCl, which are salts with only weakly hydrated anions (weak salting-out salts). At concentrations considerably >2 M, however, the two salts were found to substitute for potassium phosphate (Breitung et al. 1992; Shima et al. 1998b). Thus, the properties of the enzyme resemble those of many halophilic enzymes (Madern et al. 2000). The formyltransferase from M. kandleri is, however, additionally adapted to high temperatures (Ermler et al. 1997): The high thermostability of the enzyme at high salt concentrations cannot be explained solely by the observation that the thermostability of proteins generally increases with increasing concentrations of salting-out salts, because this increase is far less pronounced than in the case of the M. kandleri enzyme (Madern et al. 2000).
The crystal structure of the enzyme from M. kandleri was solved to 1.7 Å resolution (Shima et al. 1996; Ermler et al. 1997). It revealed a homotetramer composed essentially of two dimers, the monomer contact within the dimer being much more extensive than the dimer/dimer contact. The structure of the monomer can be subdivided into two tightly connected lobes that are separated by a small cleft. The proximal lobe (the location of the N- and C-terminal ends) consists of a six-stranded mixed β-sheet embedded between two helices and the so-called insertion region. The distal lobe is composed of a central four-stranded β-sheet and a peripheral three-stranded antiparallel β-sheet (β-meander fold), both flanked from the backside by three helices and from the frontside by the proximal lobe. The structure also indicated that the active site is located at the monomer/monomer interface, explaining the requirement of dimerization for activity.
We have now investigated the formyltransferase from Methanosarcina barkeri, which is a methanogen with a growth temperature optimum of 37°C (Boone et al. 1993), and from Archaeoglobus fulgidus, which is a hyperthermophilic sulfate-reducing archaeon with a temperature optimum of 83°C (Stetter 1988). The two enzymes differ from the M. kandleri enzyme by not requiring high concentrations of potassium phosphate or related compounds for activity and thermostability (Breitung and Thauer 1990; Schwörer et al. 1993), thus reflecting that the intracellular concentrations of salts in the two archaea are much lower than in M. kandleri (Tolman et al. 1986; Gorris et al. 1991; Kurr et al. 1991). In this respect, it is consistent that the organisms with similar salt requirements for formyltransferase activity, Methanosarcina and Archaeoglobus, are phylogenetically much more closely related to each other than to Methanopyrus (Fig. 1 ▶; Woese et al. 1991).
Fig. 1.

(A) Phylogenetic relationship of 16S-rRNA sequence from Methanosarcina barkeri, Methanopyrus kandleri, Archaeoglobus fulgidus, Methanothermobacter thermoautotrophicus, Methanothermus fervidus, and Methanococcus jannaschii. (B) Phylogenetic relationship of the formyltransferase amino acid sequence from these euryarchaeota. Tree A was constructed from the data given in Boone et al. (1993); Tree B was calculated from formyltransferase sequences using the Clustal method of the Megaligne program (DNA Star).
Results
Formyltransferase from M. barkeri differs from that of M. kandleri in solubility and in salt-dependent oligomerization, activity, and stability. The properties of the A. fulgidus enzyme are somewhere between those of the two methanogenic enzymes. The differences can mainly be explained by differences in negative surface charge and hydrophobic surface fraction.
Solubility
The solubility of the formyltransferases from M. barkeri, A. fulgidus, and M. kandleri in salt solutions was investigated by performing a hanging-drop vapor diffusion experiment at a temperature of 18°C. The drops contained equal volumes of protein solution (5 mg/mL protein, 10 mM potassium phosphate at pH 7.0) and reservoir solutions consisting of different concentrations of potassium phosphate at pH 7.0. Precipitation could be observed at a salt concentration of ∼1.1 M for the M. barkeri enzyme, of 1.6 M for the A. fulgidus enzyme, and of 2.2 M for the M. kandleri enzyme after an incubation time of 1 wk.
Oligomerization
The oligomeric state of formyltransferase from M. barkeri was studied at potassium phosphate concentrations between 20 mM and 100 mM, pH 7.2, using sedimentation equilibrium analysis with an analytical ultracentrifuge. A typical absorbance-versus-radius distribution, A(r) at 280 nm, is shown in Figure 2 ▶. The data could be fitted with high precision according to equation (1) of Shima et al. (1998b, 2000), under the assumption that the enzyme is present as monomer, dimer, and tetramer. The dependence of the oligomeric state of formyltransferase on the potassium phosphate concentration at pH 7.2 is shown in Figure 3 ▶. Under most of the conditions studied, the calculated contribution of the monomer was virtually zero. The relative amounts of the other two species, dimer and tetramer, were ∼20% and ∼80%, respectively, in 20–100 mM potassium phosphate at pH 7.2, and they were independent of the buffer concentration. Oligomers larger than a tetramer could not be detected. According to polyacrylamide gradient gel electrophoresis data, the A. fulgidus enzyme already revealed a tetrameric state at salt concentrations below 0.1 M (Schwörer et al 1993). Therefore, additional ultracentrifugation experiments did not appear to be profitable. The oligomerization behavior of the M. barkeri and A. fulgidus enzymes is clearly in contrast to that of the enzyme from M. kandleri, which is mostly in a monomeric state at a phosphate concentration of 100 mM and which tetramerizes only at potassium phosphate concentrations above 400 mM (Shima et al. 1998b).
Fig. 2.

Sedimentation equilibrium analysis of formyltransferase from Methanosarcina barkeri. (A) Experimental absorbance data A1.2 cm(r) at 280 nm, best fit to the data assuming a monomer/dimer/tetramer model of self-association (—), and calculated local contributions of monomers (---), dimers (- - -), and tetramers (- • - • -). (B) Local difference ΔA1.2 cm at 280 nm between fitted and experimental data. (C) Statistical accuracy of the calculated absorbance contributions of the different oligomers (obtained by integration over the sample volume): changes in the sum of the squared residuals, σ, of fits to the data of A resulting from one nonoptimal absorbance parameter (Schuck 1994). The meaning of the symbols is the same as in A. Initial protein concentration, 0.56 mg/mL; solvent, 0.1 M potassium phosphate at pH 7.2, 0.3 mM DTT; rotor speed, 19,000 rpm; rotor temperature, 4°C.
Fig. 3.

Dependence of the relative amounts of formyltransferase monomers (▪), dimers (□), and tetramers (○) from M. barkeri on the potassium phosphate concentration. Initial protein concentration, 0.35 mg/mL; solvent, potassium phosphate at pH 7.2 at the concentrations indicated, 0.3 mM DTT; rotor speed, 19,000 rpm; rotor temperature, 4°C.
Activity
The activity of the three formyltransferases was dependent on the presence of salts (Table 1). In 5 mM MOPS/KOH at pH 7.2, the activity was essentially zero. Upon addition of potassium phosphate at pH 7.2, the activity increased. Half-maximal activity was observed at a concentration of 0.01 M for the enzyme from M. barkeri, 0.1 M for the enzyme from A. fulgidus, and 1.0 M for the enzyme from M. kandleri.
Table 1.
Dependence of the activity and thermostability of formyltransferase, from a mesophilic and two thermophilic archaea, on the presence of potassium phosphate at pH 7.2
| Formyltransferase from | |||
| Property | Methanosarcina barkeri (37°C)a | Archaeoglobus fulgidus (83°C)a | Methanopyrus kandleri (98°C)a |
| Salt concentration (M) required for 50% activityb | 0.01 | 0.1 | 1.0 |
| Salt concentration (M) required for 100% activityb | 0.1 | 1.0 | 1.5 |
| Salt concentration (M) required to retain 50% activity after 20 min of incubation at 80°Cc | 0.05 | 0.005 | 1.2 |
| Salt concentration (M) required to retain 100% activity after 20 min of incubation at 80°Cc | 0.5 | 0.05 | 1.5 |
a Growth temperature optimum.
b The activity of the M. barkeri enzyme was determined at 37°C and that of the A. fulgidus and M. kandleri enzymes at 65°C.
c 90°C in the case of M. kandleri enzyme.
The effect of the concentration of potassium phosphate at pH7.2 on the activity of formyltransferase from M. barkeri was determined at 4°C, the temperature at which the sedimentation equilibrium data were collected, and at 37°C, the growth temperature optimum of M. barkeri (Fig. 4 ▶). At both temperatures, the maximal activity was reached at a phosphate concentration of 50 mM, the specific activity at 37°C being seven times higher than at 4°C. The results indicate that formyltransferase from M. barkeri is already active at very low salt concentrations, which corresponds to the finding that the enzyme is already in a dimeric and tetrameric state under these conditions (Fig. 3 ▶).
Fig. 4.

Activity of formyltransferase from M. barkeri at 4°C (□) and 37°C (▪) in potassium phosphate at pH 7.2 at the concentrations indicated. The phosphate buffer contained 0.3 mM dithiothreitol. Before starting the reaction, the enzyme (0.4 mg/mL) was incubated at 4°C for 25 h in the same phosphate buffer as used in the respective assay. For assays at 37°C the enzyme solution was diluted 1:20 after incubation. Samples of 2.5 μL were assayed.
Thermostability
The thermostability of formyltransferases was also correlated with the presence of phosphate or related compounds (Table 1). In 5 mM MOPS/KOH at pH 7.2, the three enzymes were completely inactivated at 90°C within a few minutes. Upon addition of potassium phosphate at pH 7.2, the stability increased. Much lower salt concentrations were required to stabilize the enzymes from M. barkeri and A. fulgidus than the enzyme from M. kandleri. Fifty percent inactivation at 80°C after 20 min was observed at a salt concentration of 0.005 M for the A. fulgidus enzyme, of 0.05 M for the M. barkeri enzyme, and of 1.2 M (at 90°C) for the M. kandleri enzyme. The surprisingly high thermostability of the M. barkeri enzyme at low salt concentrations is in agreement with results from differential scanning calorimetry, which indicate that the enzyme in the presence of 0.5 M potassium phosphate has a melting temperature of 95°C (data not shown).
Crystal structures of formyltransferases from M. barkeri and A. fulgidus
The sequence identities between the formyltransferases of M. barkeri and M. kandleri, between A. fulgidus and M. kandleri, and between M. barkeri and A. fulgidus are 59%, 61%, and 68%, respectively, suggesting similar three-dimensional structures (Fig. 5 ▶). Indeed, the corresponding average deviation of the monomers is 0.9 Å, 0.8 Å, and 0.7 Å after superimposing 291 (from 296), 290 (from 296), and 297 (from 297) equivalent Cα-positions. Despite the small overall deviation between the structures, there is a significant conformational change of the lobes relative to each other. The M. barkeri enzyme is present in a more closed form, whereas the M. kandleri enzyme is in a more open form (Fig. 5 ▶). Substantial structural differences were only found in the insertion region, in the β-meander region, and in the loop region between strands 6 and 7 (Fig. 5 ▶). The latter two adjacent regions are shifted in a concerted manner by ∼1.5 Å in the M. kandleri compared with the M. barkeri and A. fulgidus enzymes. The two latter enzymes deviate by only ∼0.6 Å from each other. The temperature factors of the mentioned regions in the three enzymes are significantly higher than those of the core regions. Thus, the regions with the largest conformational changes are also the most flexible ones. Not surprisingly, the mentioned regions, with higher temperature factors, are also involved in substrate binding (Ermler et al. 1997), because some flexibility is required for the participation of the residues. However, a reliable interpretation of the temperature factors is problematic because of the interference between the internal protein flexibility and crystal lattice effects.
Fig. 5.

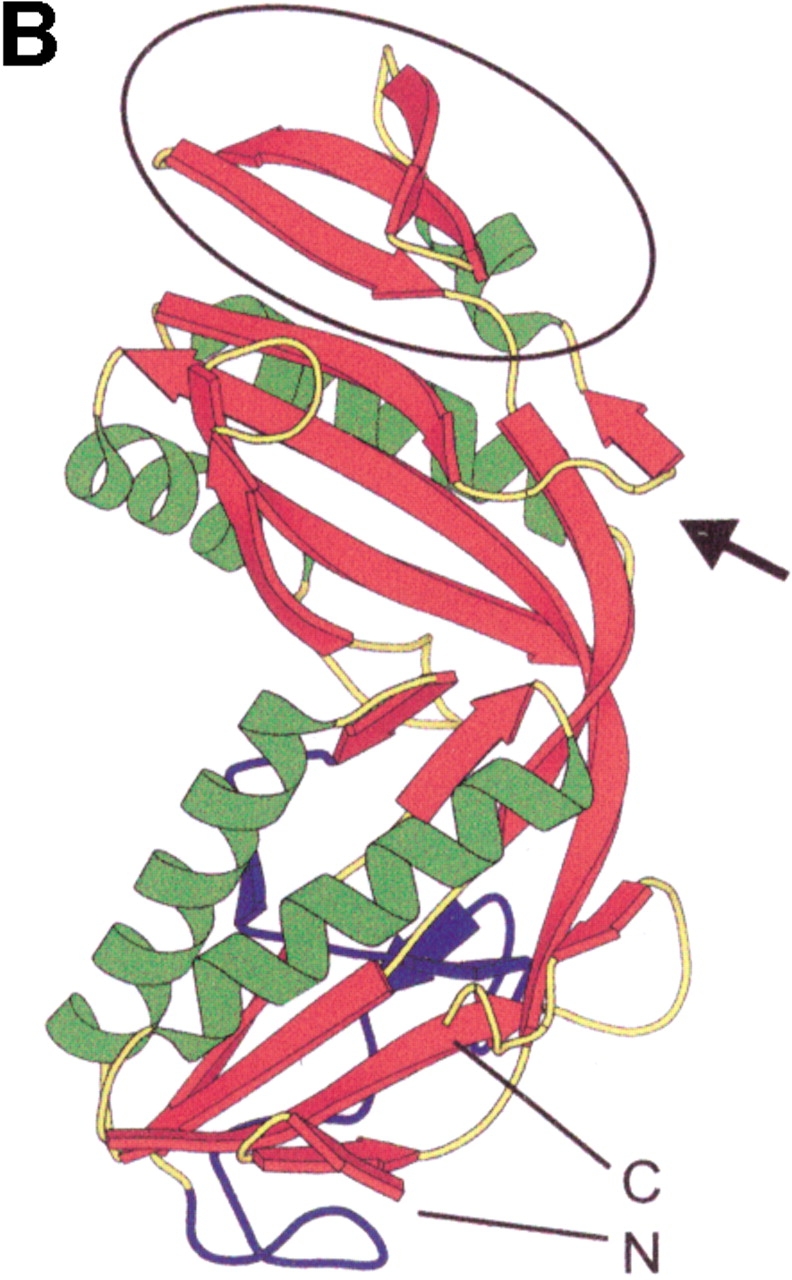

Structure of the formyltransferase. (A) The tetramer presented as a Ribbon diagram indicates a particularly extended contact region between subunits 1 (red) and 2 (green) and the equivalent subunits 3 (blue) and 4 (orange). (B) The Ribbon diagram of the monomer visualizes the location of the insertion region (blue), the meander region (black circle), and the loop between strands 6 and 7 (black arrow). (C) The stereo Cα-plot of the superimposed monomers of the enzymes from M. barkeri (red), A. fulgidus (yellow), and M. kandleri (green) documents their similar fold, in particular, in the core regions of the two lobes. This figure was generated using the program MOLSCRIPT (Kraulis 1991).
The superposition of the tetramers of the three investigated formyltransferases leads to an average deviation of the Cα atoms of 1.1 Å, 0.9 Å, and 0.8 Å, respectively, which indicates a nearly identical arrangement of the subunits. The interfaces between the subunits are basically conserved, although several significant differences are detectable. The character of the interface between subunits 1 and 2, concerning a hydrophobicity of ∼70% and the conservation of about two-thirds of the hydrogen bonds, is maintained. It differs, however, with respect to the size of the contact area and the number of hydrogen bonds. The interface area of the A. fulgidus enzyme is 15% and 6% larger than that of the M. barkeri and M. kandleri enzymes, respectively. Hydrogen bonds between subunits 1 and 2 were found in the corresponding formyltransferases to be 56, 38, and 42. The contact area between subunits 1 and 3 varies between 2% and 5% of the monomer surface and is not well conserved among the three formyltransferases. The bidentate hydrogen bond between Gln 181 of subunit 1 and Gln 176 of subunit 3 (and vice versa) is found in the M. kandleri but not in the M. barkeri and A. fulgidus enzymes. Instead, two new hydrogen bonds between Ser 172 (subunit 1) and Thr 175 (subunit 3) as well as between Ser 172 (subunit 1) and Ser 172 (subunit 3) are formed in the M. barkeri enzyme. In the A. fulgidus enzyme, Ser 172 is exchanged for threonine and Thr 175 for serine, but the hydrogen-bond pattern is maintained. The contact region between subunits 1 and 4 also varies greatly between the enzymes. Although the contact area in all three enzymes is comparable in size, the M. barkeri enzyme contains only two hydrogen bonds, whereas the A. fulgidus and M. kandleri enzymes have six. In the latter enzyme, this contact region is characterized by a strong salt bridge between Glu 64 and Arg 261.
The most pronounced difference between the M. barkeri and A. fulgidus formyltransferases, on one hand, and the M. kandleri enzyme, on the other, is the significantly altered number of negatively charged and hydrophobic residues on the protein surface. The hydrophobic surface fraction, which is 56% (57%) for the M. barkeri (A. fulgidus) enzyme and corresponds well to the average value for mesophilic proteins (Miller et al. 1987), is only 50% for the M. kandleri enzyme. This increased hydrophobicity on the protein surface is predominantly responsible for its increased overall hydrophobicity, as recognized by Kunow et al. (1996) for the M. barkeri enzyme, because the hydrophobic cores of the formyltransferases are highly similar.
The formyltransferases of M. barkeri (A. fulgidus) possess a ratio of 37 (37) negatively to 29 (26) positively charged residues on the surface. This is in drastic contrast to the ratio of 52 to 20 that was found in the M. kandleri enzyme (Fig. 6 ▶). Interestingly, the M. barkeri and the A. fulgidus enzymes contain more positively charged residues than the M. kandleri enzyme. The M. kandleri enzyme contains three sequence clusters (81–88, 111–119, 132–141) with an accumulation of negatively charged residues (Ermler et al. 1997). The M. barkeri and A. fulgidus enzymes also show a dominance of acidic to basic residues in these clusters, but the excess of 7:4 and 9:6, respectively, is by far not as pronounced as that found in the M. kandleri enzyme with 16:5. A more careful analysis of the structure indicates how the negative charges are distributed, or partly neutralized, to prevent protonation or destabilization. For example, the central residue Lys 86 in the segment 81–88 of the M. kandleri enzyme, which stabilizes five surrounding acidic side chains, is replaced by glutamate in the M. barkeri and A. fulgidus enzymes. Interestingly, the solvent-exposed loop in the segment 111–119 is prolonged in the M. kandleri enzyme, most likely as a means to increase the number of negative charges in this region.
Fig. 6.

The electrostatic properties of the formyltransferase tetramer from (A) M. barkeri, (B) A. fulgidus, and (C) M. kandleri. The molecule surface is coated according to the electrostatic potential: The extreme ranges of red and blue represent potentials of −20kBT and 20kBT, respectively (where kB is the Boltzmann constant and T is temperature). The electrostatic surface potential of the enzymes from M. barkeri and A. fulgidus is nearly neutral, and that of the M. kandleri enzyme highly negative, reflecting the dominance of acidic to basic residues. The potentials are calculated under salt-free conditions. The figure was generated using the program GRASP (Nicholls et al. 1993).
Despite the large differences concerning the charged residues, the number of ion pairs are similar, with 0.06, 0.05, and 0.07 per residue for M. barkeri, A. fulgidus, and M. kandleri respectively. Although the numbers are similar, the locations and types of the ion pairs on the surface are not conserved owing to the high number of exchanged amino acids at the protein surface. Conserved ion pairs are only found to link different regions of the structure. Examples include the salt bridge between Lys 207 and Glu 222 within the insertion region and between Glu 152 and Arg 137, which connect the core of the distal lobe with the β-meander region.
Discussion
The structural basis of the different physical and catalytic properties of the M. barkeri, A. fulgidus, and M. kandleri formyltransferases we have discovered is related to the different living conditions of the three organisms.
Solubility
Formyltransferase from M. barkeri is significantly less soluble in salts than the enzyme from M. kandleri; the solubility of the enzyme from A. fulgidus is between that of the methanogenic enzymes. This behavior reflects the low and high intracellular salt concentrations found in the three organisms (Shima et al. 1998a). The structural analysis of the three formyltransferases revealed two features that can be correlated with this difference.
Firstly, the higher percentage of hydrophobic surface area in the M. barkeri and A. fulgidus enzymes compared with the M. kandleri enzyme leads to an increased number of unspecific hydrophobic interactions between the protein molecules in solution, which can induce aggregation. Furthermore, this effect is even stronger at high salt concentrations because the hydrophobic interactions are increased as a result of the salting-out effect.
Secondly, the ratio of acidic to basic residues, in particular in three clusters (see above), on the protein surface is large in the M. kandleri enzyme but significantly reduced in the A. fulgidus enzyme and even more reduced in the M. barkeri enzyme (Fig. 6 ▶). A negatively charged surface raises the solubility, perhaps from the repulsion of approaching protein molecules preventing aggregation and the increased capacity of acidic residues to compete successfully with inorganic ions for water molecules. This interpretation is in agreement with observations in halophilic proteins like malate dehydrogenase (Dym et al. 1995) and ferredoxin (Frolow et al. 1996). The dominance of acidic residues appears to be a general feature for halophilic proteins (Madern et al. 2000).
Oligomerization
In contrast to the strong dependency of the oligomeric state of the formyltransferase from M. kandleri on salt concentration (Shima et al. 1998b), the structurally highly homologous enzymes from M. barkeri and A. fulgidus are mostly tetramers independent of the salt concentration (Fig. 3 ▶). The different oligomerization behavior might be, again, a consequence of the different number of negatively charged residues at the protein surface. In the M. barkeri and A. fulgidus enzymes, repelling forces between negative surface charges of the monomers that approach each other can be compensated at low salt concentration by attractive intersubunit interactions. However, in the M. kandleri enzyme, the repulsion between the negative surface charges can only be compensated at higher salt concentrations. At these concentrations, the charge is partly neutralized (Elcock and McCammon 1998), and the hydrophobic intersubunit interactions are increased because of the salting-out effect. The charged residues adjacent to the subunit interfaces are of special relevance for subunit assembly. According to qualitative analysis, the number of carboxylate groups less than 10 Å from carboxylate groups of a neighboring subunit is only half as many in the M. barkeri and A. fulgidus enzymes as in the M. kandleri enzyme. The repulsive effect of negative charges on tetramerization was recently investigated by exchanging Arg 261 for glutamate in the M. kandleri enzyme. Upon substitution of this amino acid, the dimer/tetramer equilibrium was significantly shifted toward the dimer (Shima et al. 2000).
Activity
The formyltransferases from M. barkeri, A. fulgidus, and M. kandleri display 50% activity at 10 mM, 100 mM, and 1 M phosphate concentrations, respectively (Table 1). The main reason for this difference in behavior is that dimer formation, which is itself a salt-dependent process, is essential for activity.
The three formyltransferases have maximal enzymatic activity at different temperatures: the enzyme from M. barkeri at 65°C (Breitung and Thauer 1990), that from A. fulgidus at 70°C (Schwörer et al. 1993), and that from M. kandleri at 90°C (Breitung et al. 1992). Moreover, at 4°C the enzyme from M. barkeri showed 10% (Fig. 4 ▶) of the optimum activity, but the enzyme from M. kandleri had only 0.2% (Shima et al. 1998b). One explanation might be that a few specific regions around the substrate-binding site of the enzymes from M. barkeri and A. fulgidus have increased flexibility compared with the M. kandleri enzyme. Although the M. kandleri enzyme is well adapted to high temperature, it might be too rigid at low temperatures. This idea is supported by the Q10 value, which was calculated to be 2.0 for the M. kandleri enzyme and 1.5 for the M. barkeri and A. fulgidus enzymes.
Thermostability
A reliable quantitative determination of protein stability on the basis of structural data is not feasible yet. Structural and thermodynamic data can only be linked by comparative and qualitative structural analysis of heat-labile and heat-stable proteins. These studies revealed that a tight network of ion pairs and hydrogen bonds, optimal internal packing, and increased oligomeric interactions are the major stabilization factors for proteins (Szilagyi and Zavodszky 2000).
The formyltransferases from A. fulgidus, M. barkeri, and M. kandleri are highly thermostable, but their stability greatly depends on the salt concentration: 50% activity after incubation at 80°C requires salt concentrations of 0.005 M, 0.05 M, and 1.2 M (at 90°C), respectively. As these values correlate well with the phosphate concentrations necessary for oligomerization, the formation of a tetrameric state is a very important stabilizing factor. Surprisingly, the dimer–dimer interactions are important for stability even though these interactions are few and the participating residues are not well conserved among the three enzymes (Shima et al. 1998b, 2000).
Even after tetramer formation, the stability of the three enzymes increased with increasing salt concentrations owing to the enhancement of the hydrophobic interactions (salting-out effect). The formyltransferase of M. kandleri is soluble and thermostable in 3.0 M potassium phosphate, pH 7.2, up to a temperature of 130°C (Shima et al. 1998b). In contrast, the M. barkeri enzyme becomes insoluble at phosphate concentrations between 1.0 M and 1.5 M (depending primarily on the protein concentration) and loses its activity at ∼80°C in 1 M salt. The stability of the A. fulgidus enzyme dependent on salt concentration was not exactly determined, but the enzyme was stable above 80°C in 1 M potassium phosphate at pH 7.2. Notably, the A. fulgidus enzyme has a larger interface area and a higher number of intersubunit hydrogen bonds than the methanogenic enzymes.
Environmental adaptation
M. kandleri is a hyperthermophilic organism that is adapted to high intracellular salt concentrations. In comparison, the phylogenetically distant methanogen M. barkeri is a mesophilic organism with low intracellular salt concentrations. The sulfate-reducing archaeon A. fulgidus is a hyperthermophilic organism but evolutionarily closely related to M. barkeri. The investigated formyltransferases of these three organisms revealed a nearly identical overall fold (Fig. 5 ▶); however, the structural similarity between the phylogenetically closely related M. barkeri and A. fulgidus is more pronounced than between M. kandleri and M. barkeri and between M. kandleri and A. fulgidus. The molecular adaptation to the highly different living conditions is primarily restricted to the protein surface, where the exchange of specific solvent-exposed residues is primarily responsible for the observed physical and kinetic differences. The structural consequence of molecular adaptation is a significantly different ratio of acidic-to-basic surface residues (Fig. 6 ▶). A similar conclusion was drawn from the structural analysis of three methyl coenzyme M reductases from M. barkeri, Methanothermobacter marburgensis, and M. kandleri (Grabarse et al. 2000).
It has to be emphasized that M. kandleri proteins have not enlarged their field of activity; they operate at high, but no longer at low, salt concentrations and temperatures. In contrast, the M. barkeri proteins are functional at low but not at high salt concentrations owing to decreased solubility. The extreme thermostability of the M. barkeri enzyme was unexpected, as it is not essential in its mesophilic habitat. An explanation for this finding might be that the evolutionary origin of methanogenic archaea is in a high-temperature environment. Interestingly, the M. barkeri enzyme is also significantly active at 4°C, in contrast to other thermostable proteins (e.g., the formyltransferase from M. kandleri), reflecting its presence in a mesophilic organism. The physical properties of the A. fulgidus enzyme lie between those of the methanogenic enzymes, albeit closer to the M. barkeri formyltransferase. It is enzymatically active in a range of higher salt concentrations and temperatures than the M. barkeri enzyme but of lower salt concentrations and temperatures than the M. kandleri enzyme.
Conclusion
The analysis of the formyltransferases from M. barkeri and M. kandleri revealed the astonishing finding that, at low salt concentrations, the protein from the hyperthermophilic organism is thermolabile, whereas the equivalent one from the mesophilic organism is extremely thermostable. This apparent contradiction teaches us two things: Firstly, it is highly precarious to make conclusions from the optimal growth conditions of an organism concerning the potential thermostability of individual proteins. Secondly, the stability of proteins cannot be understood independently of parameters like buffer, pH value, and salt conditions. Only the A. fulgidus enzyme behaved as expected for a protein from a thermophilic organism. Taken together, the formyltransferase system provides an instructive example to illustrate compensatory effects of oligomerization on stability and activity and to investigate the molecular basis for the adaptation strategy of proteins.
Materials and methods
Gene expression and protein purification
Formyltransferases from M. barkeri and M. kandleri were heterologously produced in E. coli and purified as described by Kunow et al. (1996) and Shima et al. (1995). For heterologous overproduction of the A. fulgidus enzyme in E. coli, the ftr gene was amplified by PCR using Pfu DNA polymerase with the following primers: CAT ATG AAA GTG AAT GGT GTT GAG GTT GAG G (sense) and ACC AGA CGG ATC CAG GCC AGA GAT TGT TA (antisense). The primers were constructed using the known A. fulgidus genome sequence (Klenk et al. 1997), which contains the two ftr genes AF2073 and AF2207. The enzyme encoded by AF2207 has been biochemically characterized (Schwörer et al. 1993) and was studied in this work. The PCR product obtained was cloned into the blunt-end vector pPCR-Script SK(+) (Stratagene). After transformation of E. coli XL1 Blue and isolation of the recombinant plasmid, the insert was recloned into the expression vector pET24b(+) (Novagen) via the NdeI and BamHI sites. For expression, E. coli BL21(DE3) pLysS cells (Novagen) were transformed and grown at 30°C in M9 mineral salt medium (Sambrook et al. 1989), supplemented by the antibiotics kanamycin (50 μg/mL) and chloramphenicol (25 μg/mL). Expression of formyltransferase was induced at an OD600 of 0.5–0.6 by adding IPTG to a concentration of 0.5 mM. Cells were harvested 3 h after induction, washed with 50 mM Tricine/KOH (pH 7.0), and stored at −70°C. For purification, the frozen cells were thawed in a small volume of 50 mM Tricine/KOH (pH 7.0) with 200 μg of DNAse I. To complete cell lysis and to degrade large fragments of genomic DNA, the suspension was subjected to ultrasonication. The purification protocol was similar to that previously developed for the enzyme from A. fulgidus cells (Schwörer et al. 1993). Enzyme activity was determined by following the formation of N5-formyltetrahydromethanoperin from formylmethanofuran (100 μM) and tetrahydromethanopterin (65 μM) at 282 nm (Δɛ = 5.1 mM−1 cm−1) in a potassium phosphate (pH 7.0) solution (at the concentrations indicated) containing 0.3 mM dithiothreitol (Schwörer et al. 1993).
Tetrahydromethanopterin and methanofuran were purified from Methanothermobacter marburgensis (formerly Methanobacterium thermoautotrophicum strain Marburg; Wasserfallen et al. 2000) as described (Breitung et al. 1992). Formylmethanofuran was synthesized from methanofuran and 4-nitroformyl formate (Breitung et al. 1992).
Analytical ultracentrifugation
Sedimentation equilibrium experiments were performed as described for the M. kandleri enzyme (Shima et al. 1998b, 2000). The sample volume was 140 μL, the rotor speed 19,000 rpm, and the rotor temperature 4°C. The experiments were carried out in a buffer containing 0.3 mM dithiothreitol and 0–100 mM potassium phosphate at pH 7.2. The loading protein concentration was 0.56 mg/mL. Absorption profiles were recorded at 280 nm. The experimental A(r) data were analyzed according to equation (1) in Shima et al. (1998b, 2000). The statistical accuracy of the calculated parameters was assessed as described (Shima et al. 1998b, 2000). The partial specific volume 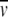 of the enzyme was calculated (Durchschlag 1986) to be 0.736 mL/g, applying the data set of Cohn and Edsall, as tabulated in Durchschlag (1986). In the range of low potassium phosphate concentrations used,
of the enzyme was calculated (Durchschlag 1986) to be 0.736 mL/g, applying the data set of Cohn and Edsall, as tabulated in Durchschlag (1986). In the range of low potassium phosphate concentrations used, 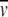 of the protein was assumed to be constant, as reported in Durchschlag and Jaenicke (1982 ,1983).
of the protein was assumed to be constant, as reported in Durchschlag and Jaenicke (1982 ,1983).
Crystallization and data collection
Crystallization experiments were performed with the hanging-drop vapor diffusion method using a sparse matrix crystallization kit (Hampton Research) for initial screening (Jancarik and Kim 1991). Crystals for the M. barkeri enzyme were obtained at 4°C after mixing of equal volumes of enzyme solution (15 mg/mL protein in 10 mM MOPS at pH 7.0) and reservoir solution consisting of 0.1 M sodium citrate, 0.2 M ammonium acetate, 31% MPD, 0.1 M HEPES at pH 6.7, and 5.0 mM formylmethanofuran. The space group of the crystals was C2 and their cell dimensions were a = 127.1 Å, b = 83.8 Å, c = 126.4 Å, and β = 108.4° (at 90 K), which corresponds to one tetramer per asymmetric unit (Vm = 2.5 Å3/D; Matthews 1968). Data at 1.9 Å resolution were collected at the Max-Planck beamline BW6 of the Deutsches Elektronensynchrotron in Hamburg (DESY) using a MAR345 IP detector. The measurement was performed under cryoconditions using the reservoir solution as cryoprotectants. Data were processed with DENZO and SCALEPACK (Otwinowski and Minor 1996). From 215,926 measured reflections, 99,960 were unique. The Rsym value and the completeness were 3.7% (8.1%) and 94% (84.4%) in the resolution range 1.9–30 Å (1.88–1.92 Å).
Rhombohedrally shaped crystals of the A. fulgidus enzyme were grown at 4°C from equal volumes of protein solution (12 mg/mL in 10 mM MOPS at pH 7.0) and a reservoir solution of 0.1 M Imidazol/Malate, 0.2 M KSCN, and 23% PEG3350. The space group was P1 with cell dimensions of a = 77.1 Å, b = 81.8 Å, c = 99.1 Å, α = 90.1°, β = 110.0°, and γ = 93.7° (at 100 K), corresponding to two tetramers per unit cell (Vm = 2.3 Å3/Da; Matthews 1968). The crystals were transferred to a cryogenic buffer consisting of 0.1 M Imidazol/Malate, 0.2 M KSCN, 15% PEG3350, and 25% ethylene glycol. Data were collected at the EMBL beamline BW7B at DESY using a MAR345 IP detector and processed using DENZO (Otwinowski and Minor 1996) and SCALA (CCP4 1994). The total number of reflections was 839,340, with 150,792 being unique. The completeness of the data was 86% (67%), and the Rsym value was 8.0% (22.3%) in the resolution range 2.0–30 Å (2.0–2.1 Å).
Structure determination
The structures of the M. barkeri and A. fulgidus enzymes were determined with the molecular replacement method (AmoRe, Navaza 1994; EPMR, Kissinger et al. 1999) using the structures of the M. kandleri and M. barkeri enzymes, respectively, as search models. After modeling the M. barkeri and A. fulgidus sequences (Kunow et al. 1996; Klenk et al. 1997) into the correctly oriented structures, the coordinates were refined with the program CNS (Brünger et al. 1998), excluding 5% of the data for validation (Rfree). The refinement processes were initiated by a simulated annealing round from 3000 K to 300 K without water molecules included. Model errors, including mostly side-chain conformations and water positions, were removed during several rounds of manual model building using the graphical display program O (Jones et al. 1991) and of positional refinement, B-factor refinement, and automated water picking. The R and Rfree factors converged for the M. barkeri formyltransferase to 19.7% and 24.8%; for the A. fulgidus enzyme to 22.8% and 28.2%.
Model errors were detected with the program PROCHECK (Laskowski et al. 1993), which analyzes deviations of geometric parameters from their standard values. Superpositions of the structures were performed with the program MULTI_GA_FIT (May and Johnson 1994). The accessible surface of the protein was calculated with the program NACCESS (Hubbard et al. 1991) and the classification of a surface region as hydrophobic versus polar was performed according to the method of Miller et al. (1987).
Accession numbers
The coordinates of the M. barkeri and A. fulgidus formyltransferases have been deposited to the Protein Data Bank (PDB). The PDB ID codes are 1M5S and 1M5H, respectively.
Acknowledgments
We thank Melanie Sordel-Klippert and Michaela Wicke for excellent technical assistance, Eberhard Warkentin for discussion, and Hartmut Michel for generous support. We also thank Harumi Fukada and Katsutada Takahashi for analysis using differential scanning calorimetry. The staff of the Max-Planck beamline BW6 and the EMBL beamline BW7B at DESY, Hamburg are gratefully acknowledged for their help during data collection. We are indebted to Erica J. Lyon for corrections of the text.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0211002.
References
- Boone, D.R., Whitman, W.B., and Rouviére, P. 1993. Diversity and taxonomy of methanogens. In Methanogenesis (ed. J.G. Ferry), pp. 35–80. Chapman & Hall, New York.
- Breitung, J. and Thauer, R.K. 1990. Formylmethanofuran:tetrahydromethanopterin formyltransferase from Methanosarcina barkeri: Identification of N5-formyltetrahydromethanopterin as the product.FEBS Lett. 275 226–230. [DOI] [PubMed] [Google Scholar]
- Breitung, J., Börner, G., Scholz, S., Linder, D., Stetter, K.O., and Thauer, R.K. 1992. Salt dependence, kinetic properties and catalytic mechanism of N-formylmethano-furan:tetrahydromethanopterin formyltransferase from the extreme thermophile Methanopyrus kandleri.Eur. J. Biochem. 210 971–981. [DOI] [PubMed] [Google Scholar]
- Brünger, A., Adams, P.D., Clore, G.M., Delano, W.L., Gros, P., GrosseKunstleve, R., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography and NMR system: A new software suite for macromolecular structure determinations.Acta Crystallogr. D 54 905–921. [DOI] [PubMed] [Google Scholar]
- CCP4. 1994. The CCP4 Suite: Programs for protein crystallography.Acta Crystallogr. D 50 760–763. [DOI] [PubMed] [Google Scholar]
- Chistoserdova, L., Vorholt, J.A., Thauer, R.K., and Lidstrom, M.E. 1998. C1 transfer enzymes and coenzymes linking methylotrophic bacteria and methanogenic archaea.Science 281 99–102. [DOI] [PubMed] [Google Scholar]
- Donnelly, M.I. and Wolfe, R.S. 1986. The role of formylmethanofuran:tetrahydromethanopterin formyltransferase in methanogenesis from carbon dioxide.J. Biol. Chem. 261 16653–16659. [PubMed] [Google Scholar]
- Durchschlag, H. 1986. Specific volumes of biological macromolecules and some other molecules of biological interest. In Thermodynamic data for biochemistry and biotechnology (ed. H.-J. Hinz), pp. 45–128. Springer-Verlag, Berlin.
- Durchschlag, H. and Jaenicke, R. 1982. Partial specific volume changes of proteins: Densimetric studies.Biochem. Biophys. Res. Commun. 108 1074–1079. [DOI] [PubMed] [Google Scholar]
- —. 1983. Partial specific volume changes of proteins: Ultracentrifugal and viscometric studies.Int. J. Biol. Macromol. 5 143–148. [Google Scholar]
- Dym, O., Mevarech, M., and Sussman, J.L. 1995. Structural features that stabilize halophilic malate dehydrogenase from an archaebacterium.Science 267 1344–1346. [DOI] [PubMed] [Google Scholar]
- Elcock, A. and McCammon, J.A. 1998. Electrostatic contributions to the stability of halophilic proteins.J. Mol. Biol. 280 731–748. [DOI] [PubMed] [Google Scholar]
- Ermler, U., Merckel, M.C., Thauer, R.K., and Shima, S. 1997. Formylmethanofuran:tetrahydromethanopterin formyltransferase from Methanopyrus kandleri—New insights into salt-dependence and thermostability.Structure 5 635–646. [DOI] [PubMed] [Google Scholar]
- Frolow, F., Harel, M., Sussman, J.L., Mevarech, M., and Shoham, M. 1996. Insights into protein adaption to a saturated salt environment from the crystal structure of a halophilic 2Fe–2S ferredoxin.Nat. Struct. Biol. 3 452–458. [DOI] [PubMed] [Google Scholar]
- Gorris, L.G.M., Voet, A.C.W.A., and Van der Drift, C. 1991. Structural characteristics of methanogenic cofactors in the non-methanogenic archaebacteriumArchaeoglobus fulgidus. Biofactors 3 29–35. [PubMed] [Google Scholar]
- Grabarse, W., Mahlert, F., Shima, S., Thauer, R.K., and Ermler, U. 2000. Comparison of three methyl coenzyme M reductases from phylogenetically distant organisms: Unusual amino acid modification, conservation and adaptation.J. Mol. Biol. 303 329–344. [DOI] [PubMed] [Google Scholar]
- Hubbard, S.J., Campbell, S.F., and Thornton, J.M. 1991. Molecular recognition. Conformational analysis of limited proteolytic sites and serine proteinase protein inhibitors.J. Mol. Biol. 220 507–530. [DOI] [PubMed] [Google Scholar]
- Jancarik, J. and Kim, S.H. 1991. Sparse-matrix sampling: A screening method for crystallization of proteins.J. Appl. Crystallogr. 24 409–411. [Google Scholar]
- Jones, T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models.Acta Crystallogr. A 47 110–119. [DOI] [PubMed] [Google Scholar]
- Kissinger, C.R., Gehlhaar, D.K., and Fogel, D.B. 1999. Rapid automated molecular replacement by evolutionary search.Acta Crystallogr. D 55 484–491. [DOI] [PubMed] [Google Scholar]
- Klenk, H.P., Clayton, R.A., Tomb, J.F., White, O., Nelson, K.E., Ketchum, K.A., Dodson, R.J., Gwinn, M., Hickey, E.K., Peterson, J.D., et al. 1997. The complete genome sequence of the hyperthermophilic, sulphate-reducing archaeon Archaeoglobus fulgidus.Nature 390 364–370. [DOI] [PubMed] [Google Scholar]
- Kraulis, P.J. 1991. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures.J. Appl. Crystallogr. 24 946–950. [Google Scholar]
- Kunow, J., Shima, S., Vorholt, J.A., and Thauer, R.K. 1996. Primary structure and properties of the formyltransferase from the mesophilic Methanosarcina barkeri: Comparison with the enzymes from thermophilic and hyperthermophilic methanogens.Arch. Microbiol. 165 97–105. [DOI] [PubMed] [Google Scholar]
- Kurr, M., Huber, R., König, H., Jannasch, H.W., Fricke, H., Trincone, A., Kristjansson, J.K., and Stetter, K.O. 1991. Methanopyrus kandleri, gen. and sp. nov. represents a novel group of hyperthermophilic methanogens, growing at 110°C.Arch. Microbiol. 156 239–247. [Google Scholar]
- Laskowski, R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures.J. Appl. Crystallogr. 26 283–291. [Google Scholar]
- Madern, D., Ebel, C. and Zaccai, G. 2000. Halophilic adaptation of enzymes.Extremophiles 4 91–98. [DOI] [PubMed] [Google Scholar]
- Matthews, B.W. 1968. Solvent content of protein crystals.J. Mol. Biol. 33 491–497. [DOI] [PubMed] [Google Scholar]
- May, A.C.W. and Johnson, M.S. 1994. Protein structure comparisons using a combination of a genetic algorithm, dynamic programming and least-squares minimization.Protein Eng. 7 475–485. [DOI] [PubMed] [Google Scholar]
- Miller, S., Lesk, A.M., Janin, J., and Chothia, C. 1987. The accessible surface area and stability of oligomeric proteins.Nature 328 834–836. [DOI] [PubMed] [Google Scholar]
- Nicholls, A., Bharadwaj, R., and Honig, B. 1993. GRASP: Graphical representation and analysis of surface properties.Biophys. J. 64 166–170. [Google Scholar]
- Otwinowski, Z. and Minor, W. 1996. Processing of X-ray diffraction data collected in oscillation mode.Methods Enzymol. 276 307–326. [DOI] [PubMed] [Google Scholar]
- Sambrook, J., Fritsch, E.F., and Maniatis, T.L. 1989. Molecular cloning: A laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Schuck, P. 1994. Simultaneous radial and wavelength analysis with the Optima XL-A analytical ultracentrifuge.Prog. Colloid. Polym. Sci 86 1–13. [Google Scholar]
- Schwörer, B., Breitung, J., Klein, A.R., Stetter, K.O., and Thauer, R.K. 1993. Formylmethanofuran:tetrahydromethanopterin formyltransferase and N5,N10-methylenetetrahydromethanopterin dehydrogenase from the sulfate-reducing Archaeoglobus fulgidus: Similarities with the enzymes from methanogenic archaea.Arch. Microbiol. 159 225–232. [DOI] [PubMed] [Google Scholar]
- Shima, S., Weiss, D.S., and Thauer, R.K. 1995. Formylmethanofuran:tetrahydromethanopterin formyltransferase (Ftr) from the hyperthermophilic Methanopyrus kandleri: Cloning, sequencing and functional expression of the ftr gene and one step purification of the enzyme overproduced in Escherichia coli.Eur. J. Biochem. 230 906–913. [DOI] [PubMed] [Google Scholar]
- Shima, S., Thauer, R.K., Michel, H., and Ermler, U. 1996. Crystallization and preliminary X-ray diffraction studies of formylmethanofuran:tetrahydromethanopterin formyltransferase from Methanopyrus kandleri.Prot. Struct. Funct. Genet. 26 118–120. [DOI] [PubMed] [Google Scholar]
- Shima, S., Hérault, D.A., Berkessel, A., and Thauer, R.K. 1998a. Activation and thermostabilization effects of cyclic 2,3-diphosphoglycerate on enzymes from the hyperthermophilic Methanopyrus kandleri.Arch. Microbiol. 170 469–472. [DOI] [PubMed] [Google Scholar]
- Shima, S., Tziatzios, C., Schubert, D., Fukada, H., Takahashi, K., Ermler, U., and Thauer, R.K. 1998b. Lyotropic-salt-induced changes in monomer/dimer/tetramer association equilibrium of formyltransferase from the hyperthermophilic Methanopyrus kandleri in relation to the activity and thermostability of the enzyme.Eur. J. Biochem. 258 85–92. [DOI] [PubMed] [Google Scholar]
- Shima, S., Thauer, R.K., Ermler, U., Durchschlag, H., Tziatzios, C., and Schubert, D. 2000. A mutation affecting the association equilibrium of formyltransferase from the hyperthermophilic Methanopyrus kandleri and its influence on the enzyme’s activity and thermostability.Eur. J. Biochem 267 6619–6623. [DOI] [PubMed] [Google Scholar]
- Stetter, K.O. 1988. Archaeoglobus fulgidus gen. nov. sp. nov.—A new taxon of extremely thermophilic archaebacteria.Syst. Appl. Microbiol. 10 172–173. [Google Scholar]
- Szilagyi, A. and Zavodszky, P. 2000. Structural differences between mesophilic, thermophilic and extremely thermophilic protein subunits: Results of a comprehensive survey.Structure 8 493–504. [DOI] [PubMed] [Google Scholar]
- Thauer, R.K. 1998. Biochemistry of methanogenesis: A tribute to Marjory Stephenson.Microbiology 144 2377–2406. [DOI] [PubMed] [Google Scholar]
- Thauer, R.K. and Kunow, J. 1995. Sulfate reducing archaea. In Biotechnology handbook (ed. N. Clark), pp. 33–48. Plenum, London.
- Tolman, C.J., Kanodia, S., Roberts, M.F., and Daniels, L. 1986. 31P-NMR spectra of methanogens: 2,3-Cyclopyrosphoglycerate is detectable only in methanogenic strains.Biochim. Biophys. Acta 886 345–352. [DOI] [PubMed] [Google Scholar]
- Vorholt, J.A., Hafenbradl, D., Stetter, K.O., and Thauer, R.K. 1997. Pathways of autotrophic CO2 fixation and of dissimilatory nitrate reduction to N2O in Ferroglobus placidus.Arch. Microbiol. 167 19–23. [DOI] [PubMed] [Google Scholar]
- Vorholt, J.A., Chistoserdova, L., Lidstrom, M.E., and Thauer, R.K. 1998. The NADP-dependent methylene tetrahydromethanopterin dehydrogenase in Methylobacterium extorquens AM1.J. Bacteriol. 180 5351–5356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserfallen, A., Nölling, J., Pfister, P., Reeve, J., and de Macario, E.C. 2000. Phylogenetic analysis of 18 thermophilic Methanobacterium isolates supports the proposals to create a new genus, Methanothermobacter gen. nov., and to reclassify several isolates in three species, Methanothermobacter thermautotrophicus comb. nov., Methanothermobacter wolfeii comb. nov., and Methanothermobacter marburgensis sp. nov.Int. J. Syst. Evol. Microbiol. 50 43–53. [DOI] [PubMed] [Google Scholar]
- Woese, C.R., Achenbach, L., Rouviére, P., and Mandelco, L. 1991. Archaeal phylogeny—Reexamination of the phylogenetic position of Archaeoglobus fulgidus in light of certain composition-induced artifacts.Syst. Appl. Microbiol. 14 364–371. [DOI] [PubMed] [Google Scholar]