

Abstract
This manuscript introduces a versatile system for construction of multimeric proteins to be used as substrates for atomic force microscopy. The construction makes use of a cassette system that allows modules to be cut and ligated in any combination in eight different positions. The modules can be sequenced in situ after construction. A three-module fragment can be produced that is of a size amenable to structural and biophysical analysis to check the effect of placing a protein into a multimeric construct. We show that if the parent titin modules are retained in a construct, they can act both as linkers and as an internal standard for the force measurements. Proteins that cannot be expressed solubly in an eight-module homopolymer have been expressed and subject to force measurements using this system.
Keywords: AFM, titin, polyprotein, mutant, linker, force
The study of the mechanical unfolding of proteins is a relatively new science. The protein generally is attached at one end to a solid substrate, either specifically through gold-sulfur linkages or nonspecifically, and to a cantilever by nonspecific adsorption. To minimize interference from tip-substrate interactions, it is preferable to have a long distance between substrate and tip. This is achieved by using long proteins, either portions of large native multimodular proteins, such as titin, tenascin, or spectrin (Rief et al. 1997, 1999; Oberhauser et al. 1998), or using engineered multiple repeats of a single protein module. The proteins with multiple repeats of a single (or two) domain were an important development because they allowed the peaks in an atomic force microscopy (AFM) trace to be unambiguously assigned to the unfolding of a specific domain (Carrion-Vazquez et al. 1999; Li et al. 2000). In the first such multimodular engineered "polyprotein", the modules were cloned either using a BamH I / Bgl II system (which have compatible cohesive ends) or the nonpalindromic Ava I restriction site (Carrion-Vazquez et al. 1999). This system does not allow in situ sequencing and each new protein, including mutants, has to be constructed "from scratch". Here we present a more versatile vector designed to allow relatively rapid construction and expression of eight individual modules or domains arranged in tandem. The vector is constructed in a cassette fashion that allows individual modules to be mutated, or replaced with different protein domains, at any or all of the eight positions, and in situ sequencing is possible. As with previous constructs, the vector contains a His-tag for rapid affinity purification of the eight-module protein and two cysteine residues for attachment to the gold surface. A three-module fragment can be expressed separately and is of a size that allows standard biophysical and/or structural techniques to be used to evaluate the effect of placing the proteins into a multimodular construct. This method of constructing a multimeric protein avoids the need to synthesize or chemically attach synthetic linkers, but, if some of the TI I27 modules are retained, it provides well-characterized internal standard linkers to any protein domain.
Results and Discussion
Method of construction of wild-type TI I27 multimer
The vector used for the construction of the multimer was a modified version of pRSET A (Invitrogen). Wild-type TI I27, the 27th Ig-domain of the I-band of human cardiac titin, was cloned in at eight positions in the multimer vector. The biophysical properties of isolated domains of TI I27 (wild type and mutants) have been characterized extensively in our laboratory (Fowler and Clarke 2001) and soluble multimers of I27 previously have been expressed and purified for use in AFM (Carrion-Vazquez et al. 1999). The construction is described in Figure 1 ▶.
Fig. 1.

Initial construction of an eight-module expression vector. (a) PCR product of wild-type I27 module 1 is T-cloned then subcloned into pRSET A expression vector. The T-clones can be used for site-directed mutagenesis to construct mutant proteins. (b) Module 2 is subcloned from its T-vector into Sac I – EcoR I site in the pRSET A expression vector containing module 1. Module 3 now can be subcloned into BssH II – EcoRI site. (c) Final eight-module construct in pRSET A, showing position of the two cysteine codons in module 8. (d) The expressed protein showing the position of the histidine tag, (H)6, the linking amino acids encoded by the restriction enzyme site (one letter code), and the C-terminal cysteine residues.
Restriction sites
The multimer was built up in the vector using nine different restriction sites: BamH I, Sac I, BssH II, Kpn I, Nhe I, Xba I, Spe I, Mlu I, and EcoR I. (All enzymes were from MBI Fermentas, Vilnius, LI, or Roche Diagnostics). These restriction enzymes place two amino acids at the boundary between each of the eight domains, the identity of which depends on the six-base recognition site of each restriction enzyme. All the restriction enzymes chosen were common enzymes, readily available, inexpensive, and easy to use. They all cleave DNA to leave cohesive ends and the optimal temperature of each is 37°C except BssH II, which has an optimal temperature of 50°C. Note that as Xba I is subject to overlapping dam methylation, it may be necessary to use a dam− Escherichia coli strain when preparing plasmid DNA. For example, if the Xba I restriction site is followed by the serine codon TCT, digestion of the site will be inhibited.
Restriction sites were avoided that would encode proline, cysteine, or large amino acids such as phenylalanine or tryptophan and stop codons. In the original construct of Carrion Vasquez et al. (1999), the restriction sites chosen placed either arginine and serine or glycine-leucine-glycine between two TI I27 domains. The protein produced by our method behaved similarly to this (data not shown), showing that the properties of the protein domains in this construct are independent of the linker between them. A longer linker sequence can be added if required (see below).
Cloning
To construct the initial vector, all eight I27 modules were amplified by PCR: eight pairs of primers were designed, each containing the unique restriction site for each module; for example, the forward primer for module one contained a BamH I site and the reverse primer contained a Sac I site. As no commercially available vector existed that contained all of the desired restriction sites, all of the reverse primers for each module also contained an EcoR I site (Fig. 1a ▶). All the PCR products were cloned into a T-vector (Invitrogen), which allowed sequencing of the individual modules. After sequencing, the modules were digested and gel purified (Qiagen) then cloned into the pRSET A parent plasmid (Fig. 1a ▶), one module at a time, until all eight modules were in place (Fig. 1b ▶). All cloning experiments were performed in E. coli XL-1 blue competent cells (Stratagene) made using the standard calcium chloride method. Cloning several copies of an identical gene in tandem array can cause problems with recombination in E. coli. No problems with recombination were encountered in the XL-1 blue strain. E. coli SURE cells (Stratagene) were investigated during the cloning of the wild-type TI I27 but were found to result in greater recombination, and hence, loss of modules, than the XL-1 blue cells.
Protein expression and purification
The expression plasmids were transformed into E. coli C41 cells (Miroux and Walker 1996). The cells were grown in rich medium (2 × TY) at 37°C to an OD600 of 0.4–0.6 cm−1. Expression was induced by addition of IPTG to 0.2 mM. The cells were grown over night at 28°C before harvesting by centrifugation. The cells were resuspended in phosphate buffered saline (PBS, 10 mM sodium phosphate buffer, pH 7.4, 150 mM NaCl), and sonicated on ice. Following centrifugation, the supernatant was incubated with a Ni-affinity matrix (Qiagen), which then was washed thoroughly with PBS (with 20 mM imidazole) followed by 50 mM Tris pH 8.0. It then was eluted with 250 mM imidazole and dialyzed extensively into water. The histidine tag was not removed from the protein.
AFM
All AFM experiments were carried out as previously described (Best et al. 2001) using the molecular force probe from Asylum Research. AFM traces of both TI I27 and an eight-module construct of a fibronectin type III domain of human tenascin (TNfn3) are shown in Figure 2 ▶.
Fig. 2.
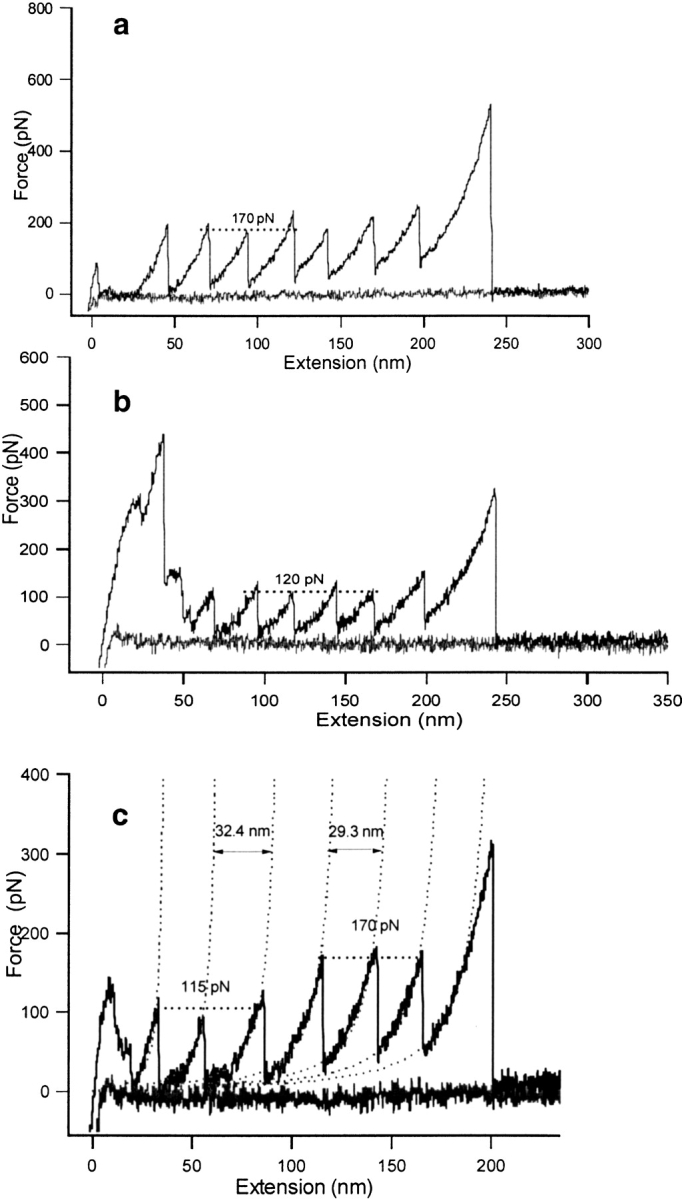
Sample atomic force microscopy traces of homopolymers constructed by this method. All traces collected at 600 nm/sec. (a) Wild-type TI I27. (b) Mutant TI I27 (each module with the mutation V86A). Note that the forces are significantly lower than for wild type. (c) A TI I27 construct with TNfn3 cloned at positions 2, 4, and 6. TNfn3 can be distinguished from TI I27 on the grounds of both peak height and distance between the peaks in the worm-like chain fit (as described in Rief et al. 1997) with the ΔL, increase in contour length from one peak to another, being 324 Å on unfolding of a TNfn3 module and 293Å on unfolding of a TI I27 module. The TI I27 and TNfn3 unfolding forces are the same as in the respective homopolymers.
Construction of mutant multimers
Because there are unique restriction sites at the beginning and end of all eight modules, wild-type modules can be replaced at any or all of the positions in the construct. It therefore was possible to make several different TI I27 mutant constructs, where the same mutation was present in all eight modules. A summary diagram describing the construction is available in supplementary materials.
Four-module sequencing constructs
Because the insertion of mutant modules needs to be confirmed by sequencing, the eight-module construct was split into two halves: one pRSET A construct contained modules one through four (pAFM1–4) and a second pRSET A construct contained modules five through eight (pAFM5–8). Each four-module construct can be sequenced in forward and reverse directions to confirm the identity of each module (requiring accurate sequencing of only two domains in each direction).
Mutation inside the T-vector
All eight wild-type TI I27 modules, each cloned individually in a T-vector, were mutated using the Stratagene Quik-Change mutagenesis method. The presence of the mutation was confirmed by sequencing.
Cloning
The mutated module was digested from the T-vector using the unique restriction sites. The multimeric four-module construct was digested to remove wild-type modules one and two and mutant modules one and two were ligated into the vector. Two modules can be ligated into the vector in the same reaction. Mutant modules three and four were inserted into the multimer in the same fashion, after modules one and two.
It is possible that during a double digest, only one of the restriction enzymes may cut some target plasmid molecules, allowing re-ligation to readily occur, with wild-type modules still in place. Treatment of the cut vector with shrimp alkaline phosphatase (Amersham Pharmacia) greatly reduces re-ligation of vector, which has only been cut by one enzyme.
The restriction sites Nhe I, Xba I, and Spe I all have compatible cohesive ends, therefore modules at positions five and six can be inserted in either direction. Plasmids from minipreps can be screened readily for correct orientation of modules at these positions by restriction digest, as the restriction sites will be lost if the module has ligated into the vector in the reverse direction. We routinely screen four clones and have always found at least one in the correct orientation.
Assembly of mutant construct
Once each four-module construct has been sequenced, the 5–8-module fragment from pAFM5–8 is cloned into pAFM1–4. This has an EcoR I site four bases downstream from the Nhe I site, allowing modules five to eight to be cloned in.
Expression test using a four module construct
It may not be possible to predict if a protein will express solubly in an eight-domain construct. Expression tests with a four-module vector 1–4 that has a stop codon downstream from the Nhe I site (pAFM1–4e) allows expression tests of four modules to be performed. Barnase does not express in such a system but expression of a four-module construct of the first domain of rat CD2 showed that the protein partitioned between the soluble and insoluble fractions (unpubl. data).
Construction of heterologous multimers
As it may not be possible or desirable to express all proteins in a soluble eight-module multimeric form for study by AFM, this system allows the cloning of a single copy or multiple copies of a gene into the TI I27 construct. The presence of flanking wild-type TI I27 domains may assist in producing correctly folded protein at these positions and has the added advantage of acting as an internal standard in AFM experiments. In principle, force peaks from the inserted protein domain can be distinguished from the TI I27 peaks in an AFM trace by either distance between the peaks, or by the different force profile (Fig. 2 ▶).
The globular protein barnase was expressed successfully using this method. Barnase was inserted at positions two, four, and six, with a six amino-acid linker (Gly-Ser-Gly-Ser-Gly-Ser) at the N and C termini of each of the barnase modules. It was not possible to solubly express a multimer of barnase, but this method allowed the study of the forced unfolding of barnase by expressing it alternately with wild-type I27. Barnase unfolded at much lower forces than wild-type I27 (Best et al. 2001). In some cases, no barnase peaks could be identified, only the internal standard peaks of wild-type I27. The internal control of the TI I27 multimers made interpretation of these traces significantly easier. As proof of principle, TNfn3 also was cloned into positions 2, 4, and 6 of the TI I27 construct. In this case, TNfn3 force peaks could be distinguished by the longer contour length (Fig. 2 ▶). Both TI I27 and TNfn3 behaved exactly as in a homopolymer construct. In principle, not just single domains but domain pairs or several successive domains can be cloned into the TI I27 vector.
Three-module construct as a control system
It is important to establish that the proteins maintain structure and biophysical properties (thermodynamic stability and kinetic properties) in the multimodular construct. The first three modules can be cloned directly into the BamH I – Kpn I sites of pRSET A, and expressed using the stop codon located downstream. We have demonstrated, for a TI I27–barnase–TI I27 construct that this three-module protein is of an ideal size for standard biophysical measurements and also for nuclear magnetic resonance (NMR) (Best et al. 2001). We were able to demonstrate that both barnase and TI I27 had the same structure, stability, and kinetic properties in the multimer construct. However, the small protein chymotrypsin inhibitor 2 (CI2) had a lower stability (and possibly different structure) in a similar TI I27–CI2–TI I27 construct (R. Best and J. Clarke, unpubl.).
Conclusion
We have developed a versatile set of vectors for studying proteins that are to be used in AFM experiments. These vectors allow preliminary expression tests and biophysical studies to be performed on the protein of interest to help determine if it will be suitable for study by AFM, and rapid reliable production of protein wild-type and mutant multimers. The use of the extensively characterized TI I27 as a linker provides an internal standard and may allow expression of protein that could otherwise not be cloned into an AFM construct.
Acknowledgments
This work was supported by the Wellcome Trust. J.C. is a Wellcome Trust Senior Research Fellow.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0212702.
References
- Best, R.B., Li, B., Steward, A., Daggett, V., and Clarke, J. 2001. Can non-mechanical proteins withstand force? Stretching barnase by atomic force microscopy and molecular dynamics simulation. Biophys. J. 81 2344–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrion-Vazquez, M., Oberhauser, A. F., Fowler, S.B., Marszalek, P.E., Broedel, S.E., Clarke, J., and Fernandez, J.M. 1999. Mechanical and chemical unfolding of a single protein: A comparison. Proc. Natl. Acad. Sci. 96 3694–3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler, S.B., and Clarke, J. 2001. Mapping the folding pathway of an immunoglobulin domain: Structural detail from phi value analysis and movement of the transition state. Structure 9 355–366. [DOI] [PubMed] [Google Scholar]
- Li, H., Oberhauser, A.F., Fowler, S.B., Clarke, J., and Fernandez, J.M. 2000. Atomic force microscopy reveals the mechanical design of a modular protein. Proc. Natl. Acad. Sci. 92 6527–6531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miroux, B. and Walker, J.E. 1996. Over-production of proteins in Escherichia coli: Mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J. Mol. Biol. 260 289–298. [DOI] [PubMed] [Google Scholar]
- Oberhauser, A.F., Marszalek, P.E., Erickson, H.P., and Fernandez, J.M. 1998. The molecular elasticity of the extracellular matrix protein tenascin. Nature 393 181–185. [DOI] [PubMed] [Google Scholar]
- Rief, M., Gautel, M., Oesterhelt, F., Fernandez, J.M., and Gaub, H.E. 1997. Reversible unfolding of individual titin immunoglobulin domains by AFM. Science 276 1109–1112. [DOI] [PubMed] [Google Scholar]
- Rief, M., Pascual, J., Saraste, M., and Gaub, H.E. 1999. Single molecule force spectroscopy of spectrin repeats: Low unfolding forces in helix bundles. J. Mol. Biol. 286 553–561. [DOI] [PubMed] [Google Scholar]