

Abstract
3H-diazirine (3H-DZN), a photoreactive gas similar in size to water, was used to probe the topography of the surface and inner space of proteins. On photolysis 3H-DZN generates 3H-methylene carbene, which reacts unselectively with its molecular cage, inserting even into C-H bonds. Labeling of bovine α-lactalbumin (α-LA, MW: 14,200) with 1 mM 3H-DZN yielded 0.0041 mol CH2/mol of protein, in agreement with the expectation for an unspecific surface-labeling phenomenon. The cooperative urea-induced unfolding of α-LA, as monitored by the extent of 3H-methylene labeling, agrees with that measured by circular dichroism spectroscopy in the far and near ultraviolet regions. At 8 M urea, the unfolded state U was labeled 25–30% more than the native state N primarily because of the increase in the accessible surface area (ASA) of the protein occurring upon unfolding. However, this result lies below the ∼100% increment expected from theoretical estimates of ASA of state U. Among other factors, most likely the existence of a residual structure in U, that involves helices H2 and H4 of the α subdomain, might account for this fact, as shown by a comparative analysis of peptide labeling patterns of N and U samples. In this paper, we demonstrate the usefulness of the 3H-methylene labeling method to monitor conformational transitions and map solvent accessibility along the polypeptide sequence, thus opening the possibility of outlining structural features of nonnative states (i.e., denatured states, molten globule). We anticipate that this technique also would help to identify ligand binding and oligomerization sites in proteins.
Keywords: Diazirine, methylene carbene, bovine α lactalbumin, accessible surface area, conformational transition, protein folding, protein footprinting, urea
The study of the structure of the native state of proteins, as well as that of the denatured state(s) and folding intermediates, is a matter of intense investigation (Creighton 1992; for the current status of the protein folding problem, see Fersht 1999). While X-ray crystallography and nuclear magnetic resonance (NMR) methods have contributed a wealth of knowledge providing a detailed picture of the native state at atomic resolution (the pdb bank: www.rcsb.org holds more than 16,200 entries as of 10/2001), much less is known about the nature and properties of the denatured and intermediate states. The structural analysis of nonnative states remains severely limited by the intrinsically dynamic nature of the molecular ensembles. Witness to this is the current debate on the structure of the denatured state (Dill and Shortle 1991; Baldwin and Zimm 2000; Pappu et al. 2000; Shortle and Ackerman 2001).
Circular dichroism (CD) and fluorescence measurements are widely employed to follow conformational transitions. Although these methods are extremely sensitive to perturbations, most frequently they provide information on changes in either the overall conformation of the protein (CD) or around the molecular environment of either the tryptophan fluorophore (intrinsic fluorescence) or extrinsic fluorophores, such as FITC, ANS, or bisANS, attached to the protein under study. Additional insight into the nature of nonnative ensembles arises from molecular modeling techniques. See Duan and Kollman (1998) for the longest molecular dynamics simulation of folding run to date, and Creamer et al. (1995, 1997) for a critical analysis of the tripeptide model for the denatured state.
Theoretical and thermodynamic arguments have pointed to the importance of minimizing the ASA, particularly that associated with hydrophobic side chains and the peptide backbone, as a main factor conferring stability to the folded state (Lee and Richards 1971; Miller et al. 1987; Pace 2001). Much insight into the protein-folding problem can be gained from the examination of the nature and size of exposed surfaces along a conformational transition. However, we lack a direct experimental method to achieve this goal. In contrast to the richness of detail achievable for the native state, the experimental analysis of the nature and extent of protein surfaces remains almost intractable for the elusive folding intermediates and denatured state(s). Rapid-mixing hydrogen-exchange experiments combined with modern NMR spectroscopic measurements (Roder and Wüthrich 1986; Englander and Krishna 2001) have shed light on the time evolution and structure of folding intermediates (Roder et al. 1988; Udgaonkar and Baldwin 1988), mapping secondary structure formation by estimating differences in exchange rates of the main-chain amide hydrogen atoms with the solvent. However, little information is obtained on the accessibility of amino-acid side chains. Inferences on the extent of change in ASA occurring along a conformational transition could be derived from the correlation observed between theoretical estimates of this parameter and experimentally determined heat capacity changes of denaturation (δCp°), for example, as measured by microcalorimetry (Privalov and Makhatadze 1990; Makhatadze and Privalov 1990; Livingstone et al. 1991; Spolar et al. 1992) or denaturant m values (Myers et al. 1995; Pace and Shaw 2000).
On the other hand, the importance of protein surfaces cannot be overemphasized in oligomerization and ligand-binding phenomena. Specific molecular interactions such as those between protein partners (e.g., between polypeptide hormones and their receptors), proteins and nucleic acids (e.g., between repressors or transcription factors and DNA), or proteins and small ligands, such as lipids or carbohydrates (e.g., fatty acid or retinol-binding proteins and lectins, respectively) mediate all sorts of fundamental biological processes and are typically accompanied by a reduction in ASA.
In many cases, ASA changes occurring upon protein folding or ligand binding can be followed by the differential chemical reactivity of functional groups present on side chains. However, the fact that few of these are amenable to reaction limits the general usefulness of an approach based on classical chemical modification methods (Means and Feeney 1971; Lundblad 1995). In this regard, free radicals, nitrenes, or carbenes overcome this problem, because of their indiscriminate reactivity. These species react even with hydrocarbons, thus making all amino acids suitable targets.
Although still in their developmental stage, two general methods for ASA labeling of proteins recently were introduced. One of them is based on hydroxyl radical (•OH) chemistry (Goshe and Anderson 1999; Maleknia et al. 1999, 2001; Goshe et al. 2000; Nukuna et al. 2001) and the other studies the feasibility of modifying the polypeptide chain with methylene carbene (:CH2) (Richards et al. 2000). Both were proposed as useful methods for differential protein footprinting, for example, in defining ligand-binding areas on proteins and in the characterization of folding intermediates.
Here, we address the use of methylene carbene to analyze a conformational transition and to demonstrate the general usefulness of this approach to gain insights into the structure of the native and nonnative states. The extreme reactivity of the methylene carbene (<50 ps lifetime in solution; Turro et al. 1987) renders it ideal for nonselective chemical modification. This species inserts readily into any X-H bond, that is, where X = C, O, N, or S, or adds to double bonds yielding methylated products by reaction with target groups belonging to its molecular cage (Frey 1966; Turro et al. 1987). Methylene carbene has been shown to insert into C-H bonds in a rather indiscriminate manner (Doering et al. 1956). Unlike nitrenes, which often give rise to products prone to decomposition, methylated products generally are stable. Because of these facts, from a chemical standpoint, methylene carbene becomes a promising reagent to map the differential accessibility of the polypeptide chain.
To serve as a useful labeling reagent, methylene carbene has to be generated in the sample in situ from a suitable stable precursor. Possibly the best source of methylene is the photolysis of DZN, which is essentially an inert compound, unless it is irradiated or heated. A most important consequence of this choice is that the general distribution of this precursor prior to the photolysis event will determine the pattern of labeled sites. In particular, a surface-labeling phenomenon will be most sensitive to the size of the probe. In this regard, DZN, being a small gas molecule comparable in size and shape to water (Scheme 1 ▶), will thus be expected, at least in the geometrical sense, to probe the same surface as a water molecule does.
Scheme 1.

In this paper, we introduce a procedure for labeling proteins with methylene carbene based on a tritiated derivative of DZN (3H-DZN). This novel method allows a straightforward and precise measurement of the extent of modification of the polypeptide chain, thus opening the possibility of characterizing native and nonnative states. In particular, we illustrate the usefulness of this approach for studying the conformational transition of α-LA occurring upon urea denaturation. After fragmentation of the peptide chain, this analysis was extended to map different regions in this structure. Results were interpreted in terms of changes in solvent accessibility and evolution of hydrophobic sites.
Results
Photolysis of DZN
Mapping the solvent-exposed surface of proteins with a chemical probe will depend to a large extent on the nature of the chosen reagent. With the aim at searching for a suitably small precursor for methylene carbene, DZN appears as a natural choice (Scheme 1 ▶). In addition, this molecule offers a number of advantages, not the least of which being its remarkable chemical inertness and its relatively high solubility in aqueous and nonaqueous solvents, that is, we easily achieved 10 mM concentration in water or cyclohexane at atmospheric pressure.
Initially, we characterized the spectral properties of DZN in aqueous solution (Fig. 1 ▶). DZN shows a wide absorption peak between 280 and 330 nm with λmax at 320 nm. This represents the envelope of its fine-structured ultraviolet (UV) spectrum in the gas phase (Graham 1962). The use of this wavelength range allows an easy quantitation of the amount of DZN in the sample, even in the presence of high-protein concentration, and, most importantly, the possibility of avoiding damage to natural protein chromophores if suitably filtered UV light (λ>300 nm) is used for the photolysis.
Fig. 1.

Photolysis of DZN in aqueous solution (ultraviolet [UV] spectra). The sample (3.5 mM DZN) was irradiated for the times in minutes shown in each spectrum with light from a filtered UV source (λ>300 nm), as described in Materials and Methods. The inset shows the decay of the absorbance at 320 nm as a function of time of photolysis in a semilogarithmic representation.
To determine the time course of photolysis of DZN in aqueous solution, a sample of DZN (3.5 mM) was irradiated and the kinetics was monitored by the disappearance of the characteristic absorption at 320 nm (Fig. 1 ▶). This process follows a pseudo first-order kinetics with a half-life of 10.3 min (as measured in our particular setup; see Materials and Methods), which remains invariable under diverse conditions of pH, ionic strength, and concentration of denaturing agents commonly used in the study of proteins (data not shown). On the other hand, upon photolysis, no significant increase in absorption at λ<280 nm was evident, suggesting the lack of significant build-up of chromophoric side products (e.g., diazomethane arising from DZN by photoisomerization; Goldfarb and Pimentel 1960; Amrich and Bell 1964; Moore and Pimentel 1964).
Inner-filter effects were ruled out for the usual range of DZN concentrations used in protein-labeling experiments, as evidenced by the linearity of the decay (inset to Fig. 1 ▶), in agreement with its low extinction coefficient (we measured a value of around 180 M−1 cm−1).
The high quantum yield of photolysis of DZN (Richards et al. 2000, supplementary material) adds to its usefulness as a general photochemical reagent for the polypeptide chain. With protein solutions, the photogenerated methylene carbene would give rise to methylated derivatives of the protein as well as methanol after reaction with water (Scheme 2 ▶).
Scheme 2.

DZN does not perturb the three-dimensional structure of α-LA
We chose α-LA (MW 14,200) as our initial model to test the usefulness of methylene-carbene labeling for probing protein conformation. The availability of a crystallographic structure of this protein (Chrysina et al. 2000), a mutant variant m90v (Pike et al. 1996) and its close structural homology to the extensively studied HEWL, allows the identification of structural features determining the labeling pattern. In this work, we carried out a comparative study of the native state versus the denatured ensemble. On the other hand, α-LA is a classical and extensively studied example of a protein undergoing a transition to the molten-globule state (Dolgikh et al. 1981; Baum et al. 1989; Ptitsyn 1995; Arai and Kuwajima 2000). This opens the possibility of extending this analysis in the future to address structural studies of folding intermediates by means of this technique.
In the development of a methodology for the conformational analysis of proteins, it becomes critically important to prove that the probe does not perturb the three-dimensional (3D) structure of the protein under scrutiny. In this regard, near and far UV-CD spectra are most sensitive to changes in the tertiary and secondary structure, respectively (Fasman 1996). α-LA was shown to fully preserve all the fine structure characteristic of these spectral regions after exposure to DZN and photolysis (Fig. 2 ▶). Thence, we concluded that neither DZN, nor the UV irradiation, causes any significant alteration of protein conformation.
Fig. 2.
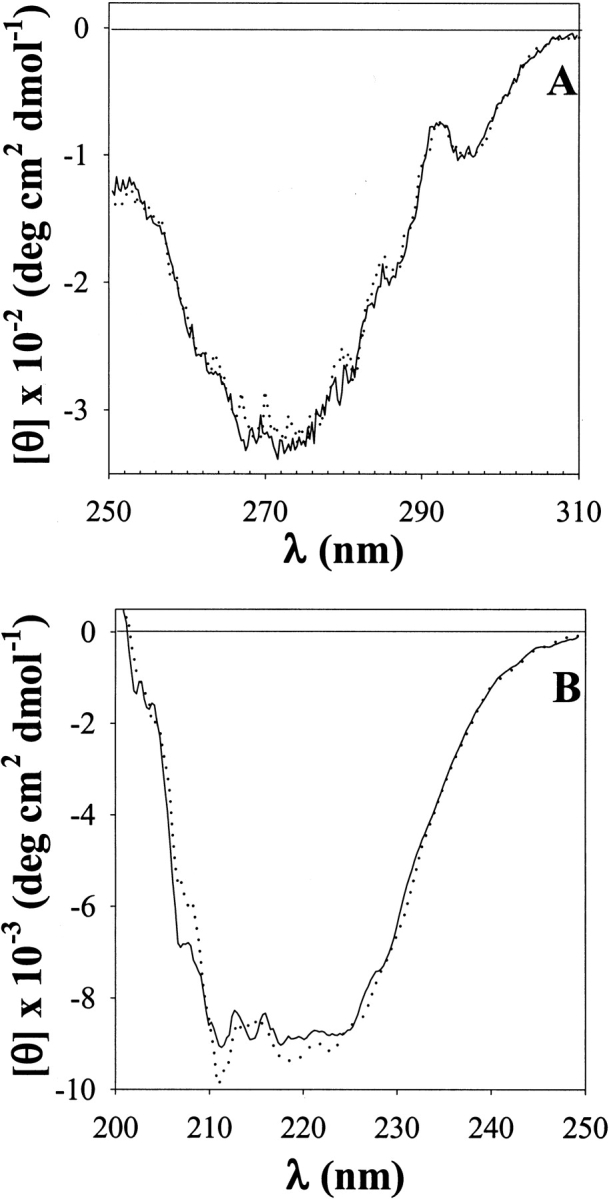
Near (A) and far (B) ultraviolet-circular dichroism (UV-CD) spectra of a 0.7-mM α-LA solution in 20 mM sodium phosphates buffer, pH 7.4 before (solid line) and after (dotted line) the addition of DZN (6.7 mM final concentration) and UV irradiation for 30 min.
Independently, we demonstrated that, under conditions appropriate for photolysis of the reagent, no damage to natural protein chromophores takes place. This was evidenced by the invariance of the absorbance ratio A310/A280, which reflects mainly the photochemical modification of tryptophan residues. By interposing a 300 nm long-pass filter between the light source and the protein solution, no significant increase in this ratio was observed, that is, values of 0.02 and 0.03 were measured for nonirradiated and irradiated samples, respectively, as compared to a value of 0.34 obtained for a sample irradiated with nonfiltered UV light.
Labeling of α-LA with 3H-methylene
The methylene labeling procedure was tested on α-LA samples under various experimental conditions. To evaluate the feasibility of this approach, we carried out the photoreaction of 3H-DZN with α-LA and measured the extent of 3H-methylene incorporation to the polypeptide chain. Initially, 3H-DZN was dissolved in α-LA samples in phosphates buffer and photolyzed (for details, see Materials and Methods). After this, we had to develop a suitable workup procedure to be able to estimate reliably the extent of protein labeling with this reagent. Most importantly, one should remark that the products arising after reaction of 3H-methylene with the polypeptide chain are not expected to exchange the radioactive label with the aqueous solvent, thus allowing a variety of analytical procedures to be carried out without loss of label. To ensure the removal of any noncovalently bound by-product to the protein, we introduced an unfolding step in 8 M urea, followed by dialysis against buffer to eliminate the bulk of radioactivity and subsequent freeze drying. However, after these procedures, we noticed from the HPLC analysis of these samples that small but significant amounts of radioactivity noncovalently associated to the protein still remained, which eluted in the pass-through peak of the chromatogram (data not shown). In connection with this, Richards et al. (2000) observed the presence of a hydrophilic polymer arising after extensive photolysis of DZN. A reversed-phase HPLC separation of each freeze-dried sample then was routinely employed as the final stage of the clean-up procedure to thoroughly eliminate traces of side products (see Materials and Methods). The quality of the labeled protein thus purified was assessed after chromatography on a C4 column (Fig. 3 ▶).
Fig. 3.

Separation of α-LA labeled with 3H-DZN by reversed-phase HPLC on a C4 column. The photolyzed sample contained 0.7 mM α-LA dissolved with 3.1 mM 3H-DZN (1 mCi/mmol) in 20 mM sodium phosphates buffer, pH 7.4. After the clean-up procedure described in Materials and Methods, the labeled protein sample (nearly 0.7 mg) was chromatographed through an HPLC C4 reversed-phase column (Vydac 214TP54, 4.6 mm × 250 mm) eluted isocratically with 0.05% aqueous TFA (20 min) followed by a linear gradient of acetonitrile:water (0 to 60% in 120 min) in 0.05% TFA at a 0.5 mL/min flow rate. α-LA eluted at 44% acetonitrile. Elution was monitored by both ultraviolet absorption at 280 nm (solid line) and by measurement of the radioactivity associated to each collected fraction (gray bars).
The radioactivity associated with the absorption peak of the protein proved that effectively α-LA became labeled covalently with 3H-methylene after photolysis of 3H-DZN. Further analysis of peak shape indicates that increased broadening and a small increase in retention time occurs for the radioactive peak with respect to the mass peak. These effects can be attributed to the slightly more hydrophobic nature of the product and the intrinsic chemical heterogeneity brought about by the methylation reaction.
Labeling was strictly dependent on the photoirradiation of the sample as ascertained by the identity between the rates of 3H-DZN photolysis and 3H-methylene carbene reaction with α-LA (Fig. 4 ▶). In the absence of light, insignificant incorporation of tritium was observed (<1% with respect to a photolyzed sample). It should also be pointed out that no further labeling occurs after 3H-DZN was completely photolyzed, thus excluding the involvement of photolytic reactions with protein chromophores, which could pick up tritium label from the milieu. Another control sample in which a 3H-DZN solution first was photolyzed and immediately thereafter mixed with α-LA showed no incorporation of radioactive label, thus ruling out any contribution of "dark reactions" to the labeling products observed.
Fig. 4.

Time courses of 3H-DZN photolysis and tritium incorporation into α-LA. The photolyzed samples contained 0.6 mM α-LA dissolved with 0.5 mM 3H-DZN (1 mCi/mmol) in 40 mM Tris/HCl buffer, pH 7.4. White dots indicate the decay of the absorbance at 320 nm normalized with respect to the initial amount of 3H-DZN present in the sample. Black dots represent the radioactivity of tritium incorporated to the protein (see Fig. 3 ▶), normalized with respect to the maximal level of modification measured.
The extent of labeling of α-LA is linearly dependent on 3H-DZN concentration (Fig. 5 ▶) in the range assayed (0.7–7.4 mM). Presumably, by applying pressure on the gas phase, one could increase the upper limit further. If need be, this straightforward alternative could serve to enhance the incorporation of label into protein samples. However, literature references (Graham 1962; Richards et al. 2000) warn against explosions or flash burns that may occur whenever manipulation of concentrated samples of DZN is taking place.
Fig. 5.

Labeling of α-LA as a function of 3H-DZN concentration. The photolyzed samples contained 0.7 mM α-LA dissolved in 20 mM sodium phosphates buffer, pH 7.4. The specific radioactivity of 3H-DZN was 0.2 mCi/mmol. The extent of methylene carbene incorporation into α-LA was expressed as the average number of moles of CH2 per mol of protein.
The main advantage of the use of 3H-DZN is that this form of the reagent allows the easy quantitation of products, even at a very low extent of protein modification. This situation arises from the unspecific nature of the reaction where, because of its abundance, the bulk solvent becomes the main target. Under our experimental conditions, between 0.27 and 2.71% of the protein was labeled on a molar basis, thus implying that essentially all modified protein molecules will be monolabeled. A direct consequence of this "single hit modification" is that heterogeneity in the resulting collection of molecular species will be minimized, thus facilitating further analysis of reaction products.
Labeling of α-LA with 3H-methylene depends on the conformational state of the protein
3H-methylene labeling of α-LA as a function of urea concentration shows that this experimental parameter is able to distinguish between different conformational states of this protein. Evidence for this is the ability of this technique to follow the conformational transition, which α-LA undergoes between 4 and 7 M urea (Fig. 6 ▶).
Fig. 6.

(A) Labeling of α-LA with 3H-methylene as a function of urea concentration. α-LA samples (0.055 mM) dissolved in 3.4 mM sodium phosphates buffer, pH 7.4, and 1.4 mM 3H-DZN (1 mCi/mmol) were photolyzed for 45 min in the presence of varying urea concentrations (0–8 M). The extent of methylene carbene incorporation into α-LA was expressed as the average number of moles of CH2 per mol of protein at 1 mM 3H-DZN (solid circles and continuous line). The conformational transition also was followed by changes in the molar ellipticity at 222 nm (open circles and dashed line) and 270 nm (open triangles and dotted line). The inset shows the dependence of 3H-methylene labeling at low urea concentrations. (B) Data shown in panel A was normalized to calculate the denatured fraction at each urea concentration. Values of each experimental series at 2 and 8 M urea were taken as references for the native and the denatured states, respectively.
To attest the validity of our approach, parallel CD measurements were carried out for the sake of comparison. We used the molar ellipticity at 270 nm to check the integrity of the tertiary structure, whereas the value at 222 nm revealed the extent of remaining secondary structure. In the former case, a sharp transition with a midpoint at 5.2 M urea was observed. In addition, this signal becomes nil at 7 M urea. By contrast, the 222 nm signal shows more complexity, that is, in this case the transition appears to be biphasic with a first stage between 4 and 6 M urea followed by a less abrupt decrease above this concentration. Even at 8 M urea, around 20% of the signal still remains, indicating the persistence of a substantial amount of secondary structure in the denatured ensemble. Although less evident than with GuHCl (Kuwajima et al. 1976), the noncoincidence of the transition curves measured at different wavelengths and the existence of residual structure at the highest concentration of denaturant used are features of this complex phenomenon.
In this context, 3H-methylene labeling shows a consistent behavior, namely, a major increase (27% with respect to the baseline prior to the transition) occurs between 4 and 7 M urea. Here, the calculated midpoint is 5.4 M urea. This curve falls in between those measured by CD, consequently its slope does not differ significantly from those calculated from the CD curves (Fig. 6B ▶), pointing to the same unfolding phenomenon.
Far from being a peculiarity of α-LA, the tendency to increase the 3H-methylene labeling upon unfolding appears to be the general behavior observed with other proteins (results not shown). As will be discussed later, in principle, this result can be correlated with the expected increase in the solvent-accessible surface area of the protein. However, other effects also should be taken into consideration. This is illustrated by the small but significant decrease in labeling (6%) that occurs at low urea concentration. This effect also has been observed with at least another protein (Bacillus licheniformis β-lactamase), but its magnitude differs in each case (D.B. Ureta, P.O. Craig, and J.M. Delfino, unpubl.). Undoubtedly, an explanation for this behavior must consider the influence on protein labeling of the interaction of the denaturant with the native state, possibly binding to sites as will be dealt with later. On the other hand, one can safely rule out any effect of urea by itself on the chemistry of 3H-methylene labeling in aqueous solution. Proof of this last statement is the invariance in the labeling yield of α-LA in the range of 0.5–4 M urea, where no significant change in its 3D structure becomes evident by spectroscopic measurements.
Peptide analysis of α-LA labeled with 3H-methylene in the native and unfolded states
To identify sites labeled with 3H-methylene, we measured the radioactivity associated to peptides derived from samples of α-LA modified under native or denaturing conditions (Fig. 7 ▶ and Table 1).
Fig. 7.

Purification of tryptic peptides derived from 3H-methylene labeled α-LA by size exclusion chromatography followed by reversed-phase HPLC. Samples of α-LA (0.14 mM) were photolabeled with 3H-DZN (6.6 mM, 1 mCi/mmol) under native (20 mM sodium phosphates buffer, pH 7.4) or denaturing conditions (in the presence of 8 M urea). After cleanup, the protein samples were reduced, carbamidomethylated, and extensively digested with TPCK-trypsin. The resulting soluble peptide mixtures were subjected to size exclusion chromatography on a Superdex Peptide HR 10/30 column (upper panel). Elution was monitored by both UV absorption at 215 nm (solid line) and by measurement of the radioactivity (dashed line: native sample; dotted line: denatured sample). Panels A through F show the reversed-phase HPLC separation on a C18 column of each fraction indicated in the size exclusion chromatography separation. Elution was monitored by UV absorption at 215 nm. Amino-acid analysis of the peaks was carried out to identify each peptide (Table 1). For further details, see Materials and Methods.
Table 1.
Tryptic peptides of α-LA separated by size exclusion chromatography

General features of the size exclusion chromatography separation of these peptides are the following. (i) All radioactivity was observed between the boundaries of elution of the peptides and in close association with any given fraction (Fig. 7 ▶), an observation consistent with a general photolabeling phenomenon triggered by a reagent that minimally increases the molecular weight of the product. (ii) In addition, a trend exists to have more labels incorporated as the molecular mass of the peptide increases, in agreement with the indiscriminate reactivity of the methylene carbene with the polypeptide chain. (iii) Although qualitatively similar, the labeling patterns of the native and denatured samples showed clear quantitative differences. In general, most peptides exhibit higher extents of modification when α-LA was labeled in the denatured state. As expected, the total radioactivity eluting in the latter case is 30% higher than that observed for the corresponding pattern of the native state, a result that agrees well with the increment measured for the whole protein. (iv) However, the individual behavior of each peptide (U/N labeling ratio; Table 1) is not uniform, as the yield of the photoreaction with most peptides increases to a different extent and, in a few cases, this value remains invariant or even decreases. The source of these differences is not to be found in the intrinsic chemical nature of the target groups, but rather in the conformational state of the protein, which affects the microenvironment around a given peptide. As will be discussed below, a comparative analysis of the extent of 3H-methylene incorporation constitutes a valuable source of sequential information that is useful for characterizing the status of distinct loci of a protein along a conformational transition.
Discussion
3H-methylene carbene as a general probe of protein surfaces
The procedure described in this paper aims at probing the surface of the polypeptide chain using methylene carbene as a means of addressing conformational features of proteins. For the reasons pointed out in Results, DZN was chosen as the source of methylene carbene. The use of the tritiated form of this reagent (3H-DZN) allowed us to quantitate the extent of labeling of the protein and to identify the sites of reaction. Modification of α-LA was strictly dependent on photoirradiation of the sample. The signal is absolutely dependent on the concentration of 3H-DZN in the sample and on the time of photolysis. In addition, 3H-DZN does not give rise to species that could modify the protein at later stages after photolysis had taken place.
Methylation results as the consequence of the extreme reactivity of the photogenerated carbene species, which reacts with its molecular cage regardless of the chemical nature of the target group. Proof of this is the fact that all peptides derived from labeled α-LA became modified at comparable yields. In addition, the extent of labeling of proteins of different sizes becomes similar once the values (expressed as mol CH2/mol protein) are normalized by their accessible areas, indicating that a general modification phenomenon is taking place (results not shown). In this regard, we also should mention that exhaustive labeling with methylene of HEWL and a 15-residue peptide derived from ribonuclease-S (S-peptide) also was consistent with a nonspecific phenomenon as shown by the mass spectrometric analysis of the distribution of products appearing at 14 mass unit intervals from the unmodified target (Richards et al. 2000). It could be anticipated that the principal products would result from the methylation of side chains of amino acids, not because of a particular chemical selectivity of the reagent, but because of the amount of exposed surface of the polypeptide main chain relative to the side chains.
A simple theoretical analysis that considers the molecular surface of the components of the system yields a reasonably good estimate of the experimentally determined extent of protein modification. Initially, we calculated the ratio R between the total molecular surface area of the protein and the sum of that of the water solvent and protein solute. The former represents the product between the molecular area of α-LA (7236 Å2 based on pdb 1hfz, subunit D) and the number of molecules of this protein present in the system, whereas the latter is essentially the product between the area of water (32.4 Å2) and the number of molecules of this solvent. Although a moderate chemical preference has been demonstrated for the insertion of methylene carbene to polar protic groups (Richards et al. 2000), in principle, one would expect that the yield of the reaction with protein would mainly be determined by the ratio R. Thus, at 1 mM bulk 3H-DZN concentration, ∼0.004 mol CH2/mol α-LA will be expected to be incorporated, a value that agrees very well with that experimentally obtained under these conditions: 0.0041 mol CH2/mol α-LA. Although this extent of reaction is low, it was possible to measure it accurately by the use of 3H-DZN radiolabeled at sufficiently high specific radioactivity (between 0.2 and 1.0 mCi/mmol).
Despite the demonstrated unspecificity of reaction, one cannot rule out the possibility that, to some extent, 3H-methylene labeling also could result from binding of 3H-DZN to sites in the protein. In fact, 3H-DZN shares properties such as comparable size and moderate hydrophobic character with other gases like cyclopropane (Schoenborn 1967; Otting et al. 1997), xenon (Schoenborn 1965; Tilton and Kuntz 1982;Tilton et al. 1984), methane (Otting et al. 1997), or nitrogen (Tilton and Petsko 1988), which have been located into cavities or crevices in proteins. In this regard, preliminary evidence of a site in HEWL occupied by DZN was obtained by pressurizing this gas on a protein crystal (J.J. Caramelo, P.O. Craig, P. Freemont, and J.M. Delfino, unpubl.).
The interaction energy for a molecule the size of 3H-DZN would be expected to be significantly lower than that demonstrated by benzene toward a customized (`Phe-less') mutant of T4 lysozyme (∼1000 M−1; Feher et al. 1996). Typically, the binding affinities measured for small organic molecules to HEWL ranged between 0.1 and 5 M−1 (Liepinsh and Otting 1997). Assuming that 3H-DZN would bind with similar affinity to α-LA, between 0.0001 and 0.0050 mol CH2/mol α-LA will be expected to be incorporated at 1 mM bulk 3H-DZN concentration after photolysis, a range consistent with our experimental observations. Given the weakness of this interaction, a linear behavior would be expected for the dependence of 3H-methylene labeling with reagent concentration, in agreement with results presented here (Fig. 5 ▶). Consistent with this, methane occupancy of sites in HEWL was observed to vary linearly with gas concentration up to the maximum pressure assayed (170 bar; Otting et al. 1997).
3H-methylene labeling is sensitive to conformational changes of the protein
In this paper, we demonstrate the usefulness of 3H-methylene labeling to follow a conformational transition of a protein, as exemplified in the case of α-LA (Fig. 6 ▶). In principle, the observed increment of this signal (27%), occurring concomitantly to protein unfolding, could be correlated with the expected increase in ASA. Nevertheless, given the possible influence of other factors on labeling and the uncertainties associated with the structural models underlying ASA estimates, a strict quantitative correlation between solvent accessibility and 3H-methylene labeling merits further analysis.
In this context, no consensus description exists for the denatured ensemble (Pace and Shaw 2000). A 150% increase in ASA could be calculated between the native state (7236 Å2) and the unfolded state (18143 Å2) if the latter is modeled as a fully extended chain. However, the denatured state of a protein could be equated to a statistical ensemble of conformers, which collectively behaves as a random coil, among which the fully extended chain will be the one with the highest ASA. Thus, a more realistic model would predict an effective accessibility significantly lower than that expected from a purely extended chain. In this regard, Creamer et al. (1995, 1997) developed models that bracket the surface area of the unfolded states between limiting extremes. A Monte Carlo computer simulation of peptides modeled with a hard sphere potential yields the upper bound (ASA max), whereas the surface of fragments excised from known structures renders the lower bound (ASA min). Upon this basis, estimates of ASA for denatured α-LA range between 17,752 and 13,706 Å2, which would yield increments of 145% (very close to the extended model) and 90%, respectively. Independently, a thermodynamic analysis of protein denaturation derived from calorimetric studies implies that the exposure of the polypeptide chain to solvent in the unfolded state would be at most about two-thirds of the value calculated for the fully extended chain model (Lee 1991). Taking into account this result, an upper boundary of about 70% would be set to the increment in ASA associated to α-LA denaturation. Apart from these facts, NMR evidence (Schulman et al. 1997) and CD spectroscopy (see remaining ellipticity at 222 nm in 8 M urea; Fig. 6 ▶) points to the existence of some residual structure in the unfolded state of α-LA, which might account for the difference between theoretical estimates of ASA increment and experimental measurements of 3H-methylene labeling ratios. In addition, the presence of four disulfide bridges might possibly limit the expansion and contribute added stability to the unfolded state. However, the extent of labeling of α-LA in 8 M urea is not greatly enhanced after reduction and blockage of the nascent thiol groups (29% increment relative to the native state).
On the other hand, an additional source of deviation between estimates derived from geometrical calculations and actual labeling ratios can arise from (i) the intrinsic dynamics of the native state, which would allow some extent of permeation of the reagent into the protein; and (ii) the existence of binding sites for 3H-DZN, where the residence time of this molecule would be significantly higher than average.
The effective accessibility of 3H-DZN might be higher than that derived from a purely static model based on the known 3D structure. This picture is consistent with the rapid diffusion of small hydrophobic molecules into the protein interior, as has been demonstrated in HEWL and other proteins by oxygen quenching of tryptophan fluorescence (Lakowicz and Weber 1973) and phosphorescence (Calhoun et al. 1983). Although 3H-DZN will penetrate the folded state at a rate comparable to that observed for other gases, very low local concentration of this molecule is expected to occur in close packed regions as compared to solvent exposed surfaces, or inner cavities and surface pockets that could lodge the reagent. By contrast, the exposure to 3H-DZN will be globally higher and its distribution more homogeneous once the polypeptide chain unfolds (see section below).
On the other hand, evidence for the existence of binding sites for 3H-DZN in the native state arises from the small decrease (6%) in labeling observed at very low urea concentration (Fig. 6 ▶). This phenomenon takes place in a millimolar range of the denaturant, comparable to that of 3H-DZN concentration (inset to Fig. 6 ▶), thus ruling out other effects that this denaturant might bring about, for example, loss of flexibility of the structure (Pike and Acharya 1994) or weakening of hydrophobic interactions. Instead, this result could be interpreted as a competition between 3H-DZN and urea for sites present in the native state. As has been mentioned before, small molecules similar in size and shape to 3H-DZN like methane, nitrogen, cyclopropane, or xenon were located by X-ray diffraction or NMR analysis in internal cavities of native proteins with little or no disruption of packing interactions.
Finally, some preference for nonpolar surfaces might be expected from measurements of DZN partition coefficients (Richards et al. 2000), where only moderately higher values for organic than for aqueous solvents were observed, in agreement with the mildly polar character of this molecule (1.59 D dipolar moment; Pierce and Dobyns 1962) and its failure to form hydrogen bonds (Frey 1966). In principle, any attempt to strictly correlate differences in the extent of 3H-methylene labeling with those estimated from ASA also will have to take into account how the distribution of 3H-DZN across the polypeptide interface could be influenced by its hydropathic character and by the environment created by solvent and additives. Notwithstanding the reasons pointed out above, which would merit further investigation, we anticipate that the extent of solvent exposure of the polypeptide chain will primarily influence the degree of 3H-methylene labeling.
Sequence maps of 3H-methylene labeling yield structural features of the native and denatured states
In this paper, we also address a sequential analysis of 3H-methylene–labeled sites to evaluate the usefulness of this technique to map solvent accessibility. In this regard, differences in the extent of reaction of a given peptide derived from protein samples modified under different conditions, and among different peptides derived from the same protein sample, will reveal their differential accessibility to 3H-DZN as a result of changes in microenvironment. This information becomes particularly useful to evaluate aspects of conformational transitions.
As a first approximation, the analysis that follows takes into account the environment of each polypeptide fragment as the only source of variation in the extent of labeling, that is, no major influence is attributed to differences in the intrinsic reactivity of each functional group. Given the extremely low selectivity shown by the methylene carbene (Doering 1956; Turro et al. 1987), the statement above seems well warranted. Notwithstanding this fact, results are expressed as the ratio of 3H-methylene incorporated in the unfolded state relative to the native state, thus minimizing the contribution as a result of any difference in the intrinsic reactivity of individual amino-acid side chains.
In general, the extent of labeling increases for most peptides derived from samples of unfolded α-LA relative to those of the native protein, although the observed increments vary considerably (Fig. 7 ▶). Thus, it becomes interesting to analyze differences in the distribution of label in relation to structural characteristics of the native and unfolded states. One main criterion to start this analysis intends to correlate experimentally determined labeling ratios with the expected increments predicted from simple estimates of ASA (Table 1). ASA increments for each peptide were calculated as the ratio between the exposed surface of the peptide in the native protein (ASA N) and that derived from different models for the unfolded polypeptide chain (ASA U: extended, max and min, as shown in Table 1).
In general, a good correlation was found between the enhancement of 3H-methylene labeling (U/N labeling ratio) and ASA increase (U/N ASA ratio) occurring upon unfolding. However, in absolute value, no peptide ever approaches the theoretically maximal increment of exposure, thus it becomes obvious that estimates derived from the extended model do not realistically represent the nature of the unfolded state. Particularly, the best agreement (both in the trend and in absolute values) was found with the ASA min/N ratio, as exemplified in the case of peptides C and F (shown in red in Fig. 8 ▶). On average, peptides belonging to groups B and D also correlate roughly well with predictions (also shown in red in Fig. 8 ▶). By contrast, other peptides show anomalously low labeling ratios (shown in blue in Fig. 8 ▶), as is the case for peptide E. In addition, after subtracting the contribution of peptide A1 (which is equivalent to the sum of B1 and B2) from group A, it results that peptide A2 also would show an abnormally low labeling ratio.
Fig. 8.
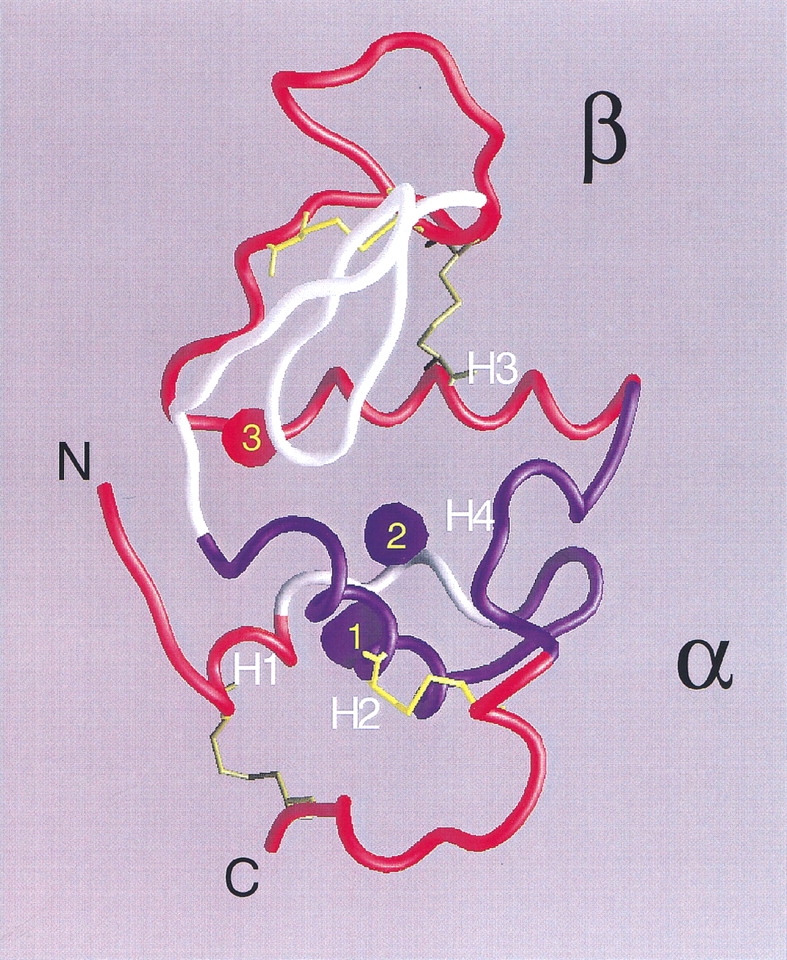
Structure of native α-LA (from pdb 1f6s; Chrysina et al. 2000). Helices H1 (5–11), H2 (23–34), H3 (86–98), and the loop/helix H4 (105–110) are indicated. Peptides (Table 1) having 3H-methylene labeling increments upon unfolding well correlated with theoretical ASA increments are colored in red (B1 and F from the β subdomain; B2 and D3 at the interface between subdomains; D1 and D4 from the N-terminal end, and C and D2 from the C-terminal end, both ends belonging to the α subdomain), whereas those having anomalously low labeling increments are shown in blue (A2 and E from the α subdomain). Peptides not found after tryptic digestion are shown in white. Empty cavities are indicated as blue blobs, whereas in red is that occupied by calcium and water. Cavity 1 (22 Å3) is delimited by residues F9, L12, L23, W26, and V27; cavity 2 (16 Å3) by W26, T29, T30, T33, F53, and W104; and cavity 3 by L52, F80, D82, L85, and D88. Cavity volumes and boundaries were calculated with MSP (Connolly 1993) using a probe radius of 1.4 Å. The final figure was rendered with GRASP (Nicholls et al. 1991).
Preservation of structure in the unfolded state is an issue worth discussing here. The behavior observed for peptides E and A2, contiguous in the 3D structure and belonging to the α subdomain of this protein, could arise from an incomplete unfolding of the polypeptide chain. In this regard, NMR evidence points to the retention of structure at this region even at extreme denaturing conditions in human α-lactalbumin (Schulman et al. 1997). This analysis indicates that helices H2 and H4 (Fig. 8 ▶) are most resistant to urea-induced denaturation. Hence, this region could constitute an organizing core for the whole structure.
Additional considerations should be made in interpreting labeling results. W104 and W26, belonging to peptides E and A2, respectively, and their common environment define the main aromatic cluster of this protein (Chrysina et al. 2000). 3H-DZN could conceivably show a somewhat more favorable partition here, aided by the existence of two small, hollow cavities (blue blobs in Fig. 8 ▶), which potentially would lodge the reagent, thus rendering this region a preferential target for labeling in the native state. In this regard, in the structurally related protein HEWL, Otting et al. (1997) described the binding of small hydrocarbon molecules (such as methane and cyclopropane) in a cavity homologous to cavity two in α-LA. Unlike the former, cavity three (red blob in Fig. 8 ▶) includes structural calcium and water, causing it to be a less likely site for 3H-DZN binding.
On the other hand, the C terminal end of the molecule, as represented by peptide C, exhibits a high extent of exposure to the solvent even in the native state and, consistently, 3H-methylene incorporation does not vary significantly. By contrast, the β subdomain 35–85, mapped by peptides F and B1, and the interfacial helix H3, connecting the β and α subdomains, mapped by peptide B2 and D3, would become disorganized to a large extent upon an increase in urea concentration. This picture is consistent with its higher susceptibility to unfolding as observed by NMR (Schulman et al. 1997).
Conclusions
We introduce a methylene carbene labeling method for proteins and illustrate its usefulness to analyze conformational transitions and to gain insights into the structure of native and nonnative states. The interpretation of our results includes comparisons of the extent of 3H-methylene labeling of the polypeptide chain with overall estimates of ASA and the location of hydrophobic sites. On the other hand, through the analysis of sequential maps, methylene carbene proved to be a reagent useful for discriminating the different accessibility of the polypeptide chain to the solvent. Thus, we anticipate that this would become a valuable method for the characterization of folding intermediates and for differential protein footprinting, for example, in defining ligand-binding areas and protein-protein interfaces. In principle, this approach could be extended to include the structural analysis of other macromolecules such as nucleic acids or complex carbohydrates. In a different vein, with the aid of UV radiation from synchrotron sources, powerful laser beams, or flash photolysis lamps, one could envision taking advantage of the extremely fast reaction of the intermediate methylene carbene species (ps time scale) to address structural issues along many kinetic processes involving changes in protein conformation.
Materials and methods
3H-formaldehyde (1–5 mCi, 90.0 mCi/mmol) was purchased from New England Nuclear; 37 % (wt/vol) formaldehyde was from E. Merck; formamide, urea, and bovine α lactalbumin type I (α-LA) were from Sigma Chemical Co. α-LA was used without further purification, and urea was recrystallized from ethanol before use. TPCK trypsin was from Worthington. Acetonitrile from E. Merck and trifluoroacetic acid (TFA) from Riedel de Haën were of HPLC grade. All other reagents and chemicals used were of analytical grade.
α-LA concentration was determined by its UV absorption at 280 nm on a JASCO 7850 spectrophotometer using an extinction coefficient of E1% = 20.1 (Kronman and Andreotti 1964).
Circular dichroism spectra were recorded on a JASCO J-20 spectropolarimeter, using cuvettes of 1 or 10 mm path length for the far (200–250 nm) and near (250–310 nm) UV regions, respectively. Data were converted to molar ellipticity [θ]M (in units of deg cm2 dmol−1) using a mean residue weight value for α-LA of 115.5 g/mol.
α-LA and their tryptic peptides were separated on a Rainin Dynamax HPLC system.
Synthesis of DZN
DZN was synthesized by a method described earlier (Ohme and Schmitz 1964). The procedure involves four steps starting from (i) vacuum concentration of 37% (wt/vol) formaldehyde to yield paraformaldehyde, (ii) condensation of this solid with formamide at 125°C for 1–2 days, (iii) hydrolysis of the product methylene-bis-formamide with 50% (wt/wt) aqueous H2SO4, and (iv) oxidation of the cation of the resulting salt, methylene diammonium sulfate, with aqueous 1 N NaClO/0.9 M NaOH. 3H-DZN (0.2–1 mCi/mmol) was synthesized in a microscale setup following the above-described procedure starting from 1–5 mCi of 3H-formaldehyde (Scheme 3 ▶).
Scheme 3.

3H-DZN was purified from remaining oxidizing materials that could potentially be deleterious to Trp residues, and from traces of the synthetic intermediate diaziridine by successive passage through sodium bisulphite and citric acid columns, respectively (Richards et al. 2000). The characterization of the reagent was carried out by measuring its UV spectrum in the gas phase (Graham 1962) and its 1H-NMR spectrum in CDCl3 solution measured on a 300 MHz Bruker spectrometer.
3H-DZN concentration measurement
3H-DZN concentration in aqueous solution was estimated both by measuring its absorbance at 320 nm and the concentration of radioactivity of the solution after complete photolysis of 3H-DZN to 3H-methanol had taken place. By these procedures, we were able to calculate an extinction coefficient at 320 nm (E320) for 3H-DZN in aqueous solution of 180 M−1 cm−1.
Photolabeling of α-LA
3H-DZN (1–10 mM, 0.2–1 mCi/mmol) was dissolved in samples of α-LA (0.06–0.7 mM), 20 mM sodium phosphates buffer, pH 7.4, in the presence or in the absence of urea, in quartz cuvettes (2- or 4-mL volume) of 1-cm path length and capped with Teflon stoppers. The buffers used were degassed in advance and kept under an inert atmosphere of nitrogen gas. Photolysis was carried out using a UV light source (Philips HPA 1000 Halogen/Hg lamp) placed at 12 cm from the samples. Light was filtered from the emission below 300 nm (with Oriel 59044 long-pass filter) to prevent photolytic damage to protein chromophores. The cuvettes were immersed in a water bath thermostated at 20°C so that even at the high luminic flux used, no heating occurred. Typically, UV irradiation was extended for 45 min, a time, which corresponds to approximately four half-lives of the reagent in our photolysis setup (t1/2 = 10.3 min).
After photolysis, solid urea was added to all samples (8 M final concentration), the solutions then were dialyzed against 0.07 mM Na2CO3 (pH 7.5–8.0) and freeze dried. Finally, the samples were separated from any remaining radioactive impurity by reversed-phase HPLC on a C4 column (Vydac 214TP510, 10 mm × 250 mm), developed with a linear gradient of acetonitrile:water (0 to 60% in 10 min) in 0.05% TFA, followed by an isocratic elution with 60% acetonitrile in 0.05% TFA (5 min), at a 3 mL/min flow rate. The elution was monitored by UV absorption at 280 nm and the protein was collected manually between 11–14 min. Samples cleaned in this fashion were freeze dried to remove HPLC solvents and redissolved in 20 mM sodium phosphates buffer pH 7.4 before the radioactivity was measured on a Pharmacia 1214 Rackbeta liquid scintillation counter. The extent of 3H-methylene carbene incorporation into α-LA was expressed as the average number of moles of CH2 per mol of protein, as estimated by the radioactivity measured on a sample of known protein concentration by taking into account the specific radioactivity of the reagent.
Reduction, carbamidomethylation, and tryptic digestion of α-LA
Reduction and carbamidomethylation of α-LA were performed according to Waxdal et al. (1968). After the clean-up procedure described before, samples of α-LA (10 mg) labeled with 3H-methylene (5 mM, 1 mCi/mmol) under native (20 mM sodium phosphates buffer, pH 7.4) or denaturing conditions (in the presence of 8 M urea) were dissolved in 1 mL of 0.5 M Tris/HCl buffer, 2 mM EDTA, 6 M guanidinium hydrochloride, pH 8.0, flushed with nitrogen and incubated at 37°C for 30 min. After this time, DTT (22 mg) was added and the samples were further kept for 4 h at 37°C under nitrogen. Then, the solutions were cooled in an ice bath and iodoacetamide (58 mg) was added. After 1 h in the dark at 0–4°C under nitrogen, β-mercaptoethanol (22 μL) was added to quench iodoacetamide in excess and the samples finally were dialyzed at 4°C against 20 mM NH4CO3H, pH 8.0.
Complete enzymatic digestion of these carbamidomethylated samples with TPCK trypsin was achieved in 0.1 M NH4CO3H, pH 8.0, after 12–18 h at 37°C using a 2% (wt/wt) enzyme/substrate ratio.
Peptide mapping
Separation of mixtures of tryptic peptides (∼2 mg) was carried out by size exclusion chromatography on a Superdex Peptide HR 10/30 column (Pharmacia) developed with 50 mM Tris/HCl, 4 M urea, pH 7.0 buffer at a flow rate of 0.5 mL/min. Elution was monitored by both UV absorption at 215 nm and by measurement of the radioactivity.
Peaks from the size exclusion chromatography were collected manually and further separated by reversed-phase HPLC on a C18 column (Vydac 218TP54, 4.6 mm × 250 mm) eluted with 0.05% aqueous TFA (10 min), followed by a linear gradient of acetonitrile:water (0 to 60% in 60 min) in 0.05% TFA at a 1 mL/min flow rate. Elution was monitored by UV absorption at 215 nm. Peptides eluted between 10 and 44% acetonitrile. Their identification was achieved by amino-acid analysis on an Applied Biosystems 420 A amino-acid analyzer.
Molecular modeling
Calculations of the ASA of α-LA and its peptides in the native state (ASA N) were based on the crystallographic structures available for this protein (1f6s; Chrysina et al. 2000) and for a m90v mutant variant (1hfz; Pike et al. 1996), with the program MacroModel (Mohamadi et al. 1990) using a probe radius of 1.4 Å. Considering DZN as an idealized sphere of radius 2.1 Å did not produce significant differences in the estimates of ASA. The interactive module of this program was used for constructing the protein and its tryptic peptides in an extended chain conformation with the aim of calculating ASA extended.
Additional ASA estimates of the unfolded protein and its tryptic peptides were obtained through a web resource (http://grserv.med.jhmi.edu/folded.html;Creamer et al. 1995, 1997) that makes use of models that bracket the surface area of the unfolded state between limiting extremes (ASA max and ASA min).
The analysis of cavities present in native α-LA was based on coordinates of structure 1f6s devoid of calcium and water molecules, and performed with the program MSP (Connolly 1993) using a probe radius of 1.4 Å.
Figure 8 ▶ was rendered with the program Grasp (Nicholls et al. 1991).
MacroModel, MSP, and Grasp were run on SGI workstations (Indigo R4000 XS24Z and O2 R10000).
Acknowledgments
For their encouragement and helpful comments along the various stages of this work, we are indebted to Dr. Julio Caramelo, Dr. Cecilia Arighi, Dr. Frederic M. Richards, Dr. Gerard Olack, Ms. Claudia Kleinman, Dr. Ana Cauerhff, Dr. Rodolfo Gonzalez Lebrero, Dr. Pablo Castello, Dr. Luis Gonzalez Flecha, Dr. Juan Pablo Rossi, Dr. Rolando Rossi, and Dr. Alejandro C. Paladini. We thank the expert technical assistance with amino-acid analysis of Ms. Dora Beatti, Ms. Susana Linskens, and Ms. Evangelina Dacci.
This research has been supported by grants to JMD from the National Research Council of Argentina (CONICET), the University of Buenos Aires (UBA), the Agencia Nacional de Promoción Científica y Tecnológica (ANPCYT), Fundación Antorchas, and the European Union. POC is the recipient of a graduate student fellowship from CONICET.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
DZN, diazirine
3H-DZN, tritiated derivative of diazirine
α-LA, bovine α lactalbumin
ASA, accessible surface area
HEWL, hen egg white lysozyme
HPLC, high performance liquid chromatography
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.4710102
References
- Amrich, M.J. and Bell, J.A. 1964. Photoisomerization of diazirine. J. Am. Chem. Soc. 86 292–293. [Google Scholar]
- Arai, M., and Kuwajima, K. 2000. Role of the molten globule state in protein folding. Adv. Protein Chem. 53 209–282. [DOI] [PubMed] [Google Scholar]
- Baldwin, R.L. and Zimm, B.H. 2000. Are denatured proteins ever random coils? Proc. Natl. Acad. Sci. 97 12391–12392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum, J., Dobson, C.M., Evans, P.A., and Hanley, C. 1989. Characterization of a partly folded protein by NMR methods: Studies on the molten globule state of guinea pig α-lactalbumin. Biochemistry 28 7–13. [DOI] [PubMed] [Google Scholar]
- Calhoun, D.B., Vanderkooi, J.M., Woodrow, G.V. III, and Englander, S.W. 1983. Penetration of dyoxigen into proteins studied by quenching of phosphorescence and fluorescence. Biochemistry 22 1526–1532. [DOI] [PubMed] [Google Scholar]
- Chrysina, E.D., Brew, K., and Acharya, K.R. 2000. Crystal structures of apo- and holo-bovine α-lactalbumin at 2.2-Å resolution reveal an effect of calcium on inter-lobe interactions. J. Biol. Chem. 275 37021–37029. [DOI] [PubMed] [Google Scholar]
- Connolly, M.L. 1993. The molecular surface package. J. Mol. Graph. 11 139–141. [DOI] [PubMed] [Google Scholar]
- Creamer, T.P., Srinivasan, R., and Rose, G.D. 1995. Modeling unfolded states of peptides and proteins. Biochemistry 34 16245–16250. [DOI] [PubMed] [Google Scholar]
- Creamer, T.P., Srinivasan, R., and Rose, G.D. 1997. Modeling unfolded states of proteins and peptides. II. Backbone solvent accessibility. Biochemistry 36 2832–2835. [DOI] [PubMed] [Google Scholar]
- Creighton, T.E. 1992. Protein folding, 1st ed. W.H. Freeman and Company, New York.
- Dill, K.A. and Shortle, D. 1991. Denatured states of proteins. Annu. Rev. Biochem. 60 795–825. [DOI] [PubMed] [Google Scholar]
- Doering, W.E., Buttery, R.G., Laughlin, R.G., and Chaudhuri, N. 1956. Indiscriminate reaction of methylene with the carbon-hydrogen bond. J. Am. Chem. Soc. 78 3224. [Google Scholar]
- Dolgikh, D.A., Gilmashin, R.I., Brazhnikov, E.V., Bychkova, V.E., Semisotnov, G.V., Venyaminov, S.Y., and Ptitsyn, O.B. 1981. α-lactalbumin: Compact state with fluctuating tertiary structure? FEBS Lett. 136 311–315. [DOI] [PubMed] [Google Scholar]
- Duan, Y. and Kollman, P.A. 1998. Pathways to a protein folding intermediate observed in a 1-microsecond simulation in aqueous solution. Science 282 740–744. [DOI] [PubMed] [Google Scholar]
- Englander, S.W. and Krishna, M.M.G. 2001. Hydrogen exchange. Nat. Struct. Biol. 8 741–742. [DOI] [PubMed] [Google Scholar]
- Fasman, G.D. 1996. Circular dichroism and the conformational analysis of biomolecules. Plenum Press, New York.
- Feher, V.A., Baldwin, E.P., and Dahlquist, F.W. 1996. Access of ligands to cavities within the core of a protein is rapid. Nat. Struct. Biol. 3 516–521. [DOI] [PubMed] [Google Scholar]
- Fersht, A. 1999. Structure and mechanism in protein science. A guide to enzyme catalysis and protein folding, 3rd ed. W.H. Freeman and Company, New York.
- Frey, H.M. 1966. The photolysis of diazirines. Adv. Photochem. 4 225–256. [Google Scholar]
- Goldfarb, T.D. and Pimentel, G.C. 1960. Spectroscopic study of the photolysis of diazomethane in solid nitrogen. J. Am. Chem. Soc. 82 1865–1868. [Google Scholar]
- Goshe, M.B. and Anderson, V.E. 1999. Hydroxyl radical-induced hydrogen/deuterium exchange in amino acid carbon-hydrogen bonds. Radiat. Res. 151 50–58. [PubMed] [Google Scholar]
- Goshe, M.B., Chen, Y.H., Anderson, V.E. 2000. Identification of the sites of hydroxyl radical reaction with peptides by hydrogen/deuterium exchange: Prevalence of reactions with the side chains. Biochemistry 39 1761–1770. [DOI] [PubMed] [Google Scholar]
- Graham, W.H. 1962. A direct method of preparation of diazirine. J. Am. Chem. Soc. 84 1063–1064. [Google Scholar]
- Kronman, M.J. and Andreotti, R.E. 1964. Inter and intramolecular interactions of α-lactalbumin. I. The apparent heterogeneity at acid pH. Biochemistry 3 1145–1151. [DOI] [PubMed] [Google Scholar]
- Kuwajima, K., Nitta, K., Yoneyama, M., and Sugai, S. 1976. Three-state denaturation of α-lactalbumin by guanidine hydrochloride. J. Mol. Biol. 106 359–373. [DOI] [PubMed] [Google Scholar]
- Lakowicz, J.R. and Weber, G. 1973. Quenching of protein fluorescence by oxygen. Detection of structural fluctuations in proteins on the nanosecond time scale. Biochemistry 12 4171–4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, B. and Richards, F.M. 1971. The interpretation of protein structures: Estimation of static accessibility. J. Mol. Biol. 55 379–400. [DOI] [PubMed] [Google Scholar]
- Lee, B. 1991. Isoenthalpic and isoentropic temperatures and the thermodynamics of protein denaturation. Proc. Natl. Acad. Sci. 88 5154–5158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liepinsh, E. and Otting, G. 1997. Organic solvents identify specific ligand binding sites on protein surfaces. Nat. Biotechnol. 15 264–268. [DOI] [PubMed] [Google Scholar]
- Livingstone, J.R., Spolar, R.S., and Record, M.T. Jr. 1991. Contribution to the thermodynamics of protein folding from the reduction in water-accessible nonpolar surface area. Biochemistry 30 4237–4244. [DOI] [PubMed] [Google Scholar]
- Lundblad, R.L. 1995. Techniques in protein modification. CRC Press LLC, Boca Raton, Florida.
- Makhatadze, G.I. and Privalov, P.L. 1990. Heat capacity of proteins. I. Partial molar heat capacity of individual amino acid residues in aqueous solution: Hydration effect. J. Mol. Biol. 213 375–384. [DOI] [PubMed] [Google Scholar]
- Maleknia, S.D., Brenowitz, M., and Chance, M.R. 1999. Millisecond radiolytic modification of peptides by synchrotron X-rays identified by mass spectrometry. Anal. Chem. 71 3965–3973. [DOI] [PubMed] [Google Scholar]
- Maleknia, S.D., Ralston, C.Y., Brenowitz, M.D., Downard, K.M., and Chance, M.R. 2001. Determination of macromolecular folding and structure by synchrotron X-ray radiolysis techniques. Anal. Biochem. 289 103–115. [DOI] [PubMed] [Google Scholar]
- Means, G.E. and Feeney, R.E. 1971. Chemical modification of proteins. Holden-Day, San Francisco.
- Miller, S., Janin, J., Lesk, A.M., and Chothia, C. 1987. Interior and surface of monomeric proteins. J. Mol. Biol. 196 641–656. [DOI] [PubMed] [Google Scholar]
- Mohamadi, F., Richards, N.G.J., Guida, W.C., Liskamp, R., Lipton, M., Caufield, C., Chang, G., Hendrickson, T., and Still, W.C. 1990. MacroModel—An integrated software system for modeling organic and bioorganic molecules using molecular mechanics. J. Comput. Chem. 11 440–467. [Google Scholar]
- Moore, C.B. and Pimentel, G.C. 1964. Matrix reaction of methylene with nitrogen to form diazomethane. J. Chem. Phys. 41 3504–3509. [Google Scholar]
- Myers, J.K., Pace, C.N., and Scholtz, J.M. 1995. Denaturant m values and heat capacity changes: Relation to changes in accessible surface areas of protein unfolding. Protein Sci. 4 2138–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls, A., Sharp, K.A., and Honig, B. 1991. Protein folding and association: Insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins 11 281–296. [DOI] [PubMed] [Google Scholar]
- Nukuna, B.N., Goshe, M.B., and Anderson, V.E. 2001. Sites of hydroxyl radical reaction with amino acids identified by (2)H NMR detection of induced (1)H/(2)H exchange. J. Am. Chem. Soc. 123 1208–1214. [DOI] [PubMed] [Google Scholar]
- Ohme, R. and Schmitz, E. 1964. Notiz über eine einfache synthese des cyclo-diazomethans. Chem. Ber. 97 297–298. [Google Scholar]
- Otting, G., Liepinsh, E., Halle, B. and Frey, U. 1997. NMR identification of hydrophobic cavities with low water occupancies in protein structures using small gas molecules. Nat. Struct. Biol. 4 396–404. [DOI] [PubMed] [Google Scholar]
- Pace, C.N. and Shaw, K.L. 2000. Linear extrapolation method of analysing solvent denaturation curves. Proteins 4 1–7. [DOI] [PubMed] [Google Scholar]
- Pace, C.N. 2001. Polar group burial contributes more to protein stability than nonpolar group burial. Biochemistry 40 310–313. [DOI] [PubMed] [Google Scholar]
- Pappu, R.V., Srinivasan, R., and Rose, G.D. 2000. The Flory isolated-pair hypothesis is not valid for polypeptide chains: Implications for protein folding. Proc. Natl. Acad. Sci. 7 12565–12570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce, L. and Dobyns, V. 1962. Molecular structure, dipole moment, and quadrupole coupling constants of diazirine. J. Am. Chem. Soc. 84 2651–2652. [Google Scholar]
- Pike, A.C.W. and Acharya, K.R. 1994. A structural basis for the interaction of urea with lysozyme. Protein Sci. 3 706–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike, A.C.W., Brew, K., and Acharya, K.R. 1996. Crystal structures of guinea-pig, goat and bovine α-lactalbumin highlight the enhanced conformational flexibility of regions that are significant for its action in lactose synthase. Structure 4 691–703. [DOI] [PubMed] [Google Scholar]
- Privalov, P.L. and Makhatadze G.I. 1990. Heat capacity of proteins. II. Partial molar heat capacity of the unfolded polypeptide chain of proteins: Protein unfolding effects. J. Mol. Biol. 213 385–391. [DOI] [PubMed] [Google Scholar]
- Ptitsyn, O.B. 1995. Molten globule and protein folding. Adv. Protein Chem. 47 83–229. [DOI] [PubMed] [Google Scholar]
- Richards, F.M., Lamed, R., Wynn, R., Patel, D., and Olack, G. 2000. Methylene as a possible universal footprinting reagent that will include hydrophobic surface areas: Overview and feasibility: Properties of diazirine as a precursor. Protein Sci. 9 2506–2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roder, H. and Wüthrich, K. 1986. Protein folding kinetics by combined use of rapid mixing techniques and NMR observation of individual amide protons. Proteins 1 34–42. [DOI] [PubMed] [Google Scholar]
- Roder, H., Elove, G.A., and Englander, S.W. 1988. Structural characterization of folding intermediates in cytochrome c by H-exchange labeling and proton NMR. Nature 335 700–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenborn, B.P. 1965. Binding of xenon to horse haemoglobin. Nature 208 760–762. [DOI] [PubMed] [Google Scholar]
- Schoenborn, B.P. 1967. Binding of cyclopropane to sperm whale myoglobin. Nature 214 1120–1122. [DOI] [PubMed] [Google Scholar]
- Schulman, B.A., Kim, P.S., Dobson, C.M., and Redfield, C. 1997. A residue-specific NMR view of the non-cooperative unfolding of a molten globule. Nat. Struct. Biol. 4 630–634. [DOI] [PubMed] [Google Scholar]
- Shewale, J.G., Sinha, S.K., and Brew, K. 1984. Evolution of α-lactalbumins. J. Biol. Chem. 259 4947–4956. [PubMed] [Google Scholar]
- Shortle, D. and Ackerman, M.S. 2001. Persistence of native-like topology in a denatured protein in 8 M urea. Science 293 487–489. [DOI] [PubMed] [Google Scholar]
- Spolar, R.S., Livingstone, J.R., and Record, M.T. Jr. 1992. Use of liquid hydrocarbon and amide transfer data to estimate contributions to thermodynamic functions of protein folding from the removal of nonpolar and polar surface from water. Biochemistry 31 3947–3955. [DOI] [PubMed] [Google Scholar]
- Tilton, R.F. Jr. and Kuntz, I.D. Jr. 1982. Nuclear magnetic resonance studies of xenon-129 with myoglobin and hemoglobin. Biochemistry 21 6850–6857. [DOI] [PubMed] [Google Scholar]
- Tilton, R.F. Jr., Kuntz, I.D. Jr., and Petsko, G.A. 1984. Cavities in proteins: Structure of a metmyoglobin-xenon complex solved to 1.9 Å. Biochemistry 23 2849–2857. [DOI] [PubMed] [Google Scholar]
- Tilton, R.F. Jr. and Petsko, G.A. 1988. A structure of sperm whale myoglobin at a nitrogen gas pressure of 145 atmospheres. Biochemistry 27 6574–6582. [DOI] [PubMed] [Google Scholar]
- Turro, N.J., Cha, Y., and Gould, I.R. 1987. Reactivity and intersystem crossing of singlet methylene in solution. J. Am. Chem. Soc. 109 2101–2107. [Google Scholar]
- Udgaonkar, J.B. and Baldwin, R.L. 1988. NMR evidence for an early framework intermediate on the folding pathway of ribonuclease A. Nature 335 694–699. [DOI] [PubMed] [Google Scholar]
- Waxdal, M.J., Konigsberg, W.H., Henley, W.L., and Edelman, G.M. 1968. Biochemistry 7 1959–1966. [DOI] [PubMed] [Google Scholar]