

Abstract
A large number of different proteins or protein domains have been investigated as possible scaffolds to engineer antibody-like molecules. We have previously shown that the TEM-1 β-lactamase can accommodate insertions of random sequences in two loops surrounding its active site without compromising its activity. From the libraries that were generated, active enzymes binding with high affinities to monoclonal antibodies raised against prostate-specific antigen, a protein unrelated to β-lactamase, could be isolated. Antibody binding was shown to affect markedly the enzyme activity. As a consequence, these enzymes have the potential to be used as signaling molecules in direct or competitive homogeneous immunoassay. Preliminary results showed that β-lactamase clones binding to streptavidin could also be isolated, indicating that some enzymes in the libraries have the ability to recognize proteins other than antibodies. In this paper, we show that, in addition to β-lactamases binding to streptavidin, β-lactamase clones binding to horse spleen ferritin and β-galactosidase could be isolated. Affinity maturation of a clone binding to ferritin allowed obtaining β-lactamases with affinities comprised between 10 and 20 nM (Kd) for the protein. Contrary to what was observed for β-lactamases issued from selections on antibodies, enzyme complexation induced only a modest effect on enzyme activity, in the three cases studied. This kind of enzyme could prove useful in replacement of enzyme-conjugated antibodies in enzyme-linked immunosorbant assays (ELISA) or in other applications that use antibodies conjugated to an enzyme.
Keywords: β-Lactamase, phage display, protein scaffold, protein assay, combinatorial libraries
Antibody-based molecular receptors have the ability to recognize a large variety of different molecules, from large proteins to small organic molecules, with high affinities and specificities. With the advance of the phage display technology, it has been possible to generate naive, combinatorial, or synthetic libraries of Fab, Fv, and scFv antibodies and to select from them antibodies that bind to virtually any protein antigen or hapten for biotechnological or medical purposes (Hoogenboom 1997; Hoogenboom et al. 1998). However, antibodies have some intrinsic disadvantages related to their size, to the stability of the minor fragments, and to their production. For these reasons, other proteins have also been investigated as potential scaffolds to create receptors for proteins or haptens (Skerra 2000). Exposed loops in these proteins are modified to provide the elements required for antigen binding. To be useful, the protein scaffold must tolerate a large number of different mutations or insertions in localized regions of its surface without compromising folding, to allow the construction of combinatorial libraries from which antibody-like molecules can be selected. Although the strategy has been applied with success in several cases, the results published up to now remain rather scarce, at least for the selection of ligand receptors able to bind globular proteins with high affinities. For instance, selections on libraries based on the Z domain of staphylococcal protein A has allowed the isolation of variants that bind the Taq DNA polymerase, human insulin, human apoliprotein A-1, and RSV G protein with micromolar affinities (Nord et al. 1997; Hansson et al. 1999). Affinity maturation of a Z domain able to bind the Taq DNA polymerase increased the affinity of the domain for the polymerase to 30–50 nM (Kd; Gunneriusson et al. 1999). In two other examples, randomization of some surface-exposed residues of the cellulose-binding domain of the fungal enzyme cellobiohydrolase I and the fibronectin type III domain permitted investigators to isolate mutants that recognize, respectively, alkaline phosphatase and ubiquitin with micromolar affinities (Koide et al. 1998; Smith et al. 1998). A large number of protease inhibitors have also been used as protein scaffolds to display structurally constrained peptides, but they were mainly engineered to generate new protease inhibitors with modified enzyme specificities (Roberts et al. 1992; Dennis and Lazarus 1994; Dennis et al. 1995; Rottgen and Collins 1995; Wang et al. 1995; Markland et al. 1996).
Among the different classes of proteins that could be contemplated as possible scaffolds to engineer antibody-like molecules, enzymes might be interesting candidates as they are naturally provided with cavities designed for binding substrates. These could be engineered to accommodate other molecules. For example, random peptide libraries have been inserted into an active-site loop of thioredoxin to design a system for exploring protein–protein interactions (Lu et al. 1995). Several proteins that inhibit cellular functions have been selected from thioredoxin insertion libraries using the two-hybrid system. For instance, inhibitors of cyclin-dependant kinase 2, thymidylate synthase, and E2F transcription factor have been isolated using this approach (Colas et al. 1996; Cohen et al. 1998; Fabbrizio et al. 1999; Blum et al. 2000). Some of the selected aptamers possess affinities in the nanomolar range. The staphylococcal nuclease framework has also been shown to be suitable for the same purpose (Norman et al. 1999). In these cases, the enzymes were used as a passive framework, and the aptamers were inactive.
Enzymes like the Escherichia coli alkaline phosphatase and β-galactosidase have been shown to accommodate linear viral epitopes in their sequence while retaining some activity. Binding of antibodies recognizing the viral epitopes modulates the activity (Brennan et al. 1994, 1995; Benito et al. 1996; Feliu et al. 1998; Ferrer-Miralles et al. 2001). Recently we have shown that the TEM-1 β-lactamase can be engineered to contain insertions of variable sequences in exposed loops (Legendre et al. 1999). Libraries of β-lactamases were constructed in which one or two surface-exposed loops surrounding the active site were modified by insertion of random sequences. Some of the libraries contained a significant percentage of active clones, indicating that the mutations introduced did not interfere with proper folding of the enzyme. Active β-lactamase clones binding with affinities in the micromolar to the nanomolar range to three different monoclonal antibodies directed against prostate-specific antigen (PSA), a protein unrelated to β-lactamase, could be isolated. Binding occurred to mimotopes of the original antigen because no similarity could be detected between the sequences of the inserted peptides and of PSA potential linear epitopes. Antibody binding in many cases induced a large effect on activity. These hybrid enzymes could be used as signaling molecules to detect either antibodies in direct homogeneous immunoassays or the original antigen in competitive immunoassays.
Clones containing an HPQ sequence, characteristic of peptides binding to streptavidin (Devlin et al. 1990), were also incidentally obtained during selections from the β-lactamase libraries. These results indicated that β-lactamase variants binding not only to antibodies but also to various other proteins could be isolated. Again, these enzymes would have the potential to be used as signaling molecules in direct assays. In the present paper, the existing libraries of β-lactamases were selected on three different proteins, which we call antigens. We show that β-lactamase variants binding to streptavidin, ferritin, and β-galactosidase can be isolated from the libraries and that the affinity of the selected enzymes can be increased by affinity maturation. However, the effect of antigen binding on activity was less pronounced than with antibodies.
Results
Selections for affinity on streptavidin, ferritin, and β-galactosidase and characterization of the selected phage-displayed β-lactamases
In an attempt to determine whether the TEM-1 β-lactamase could be used as a scaffold for protein recognition, three different libraries of the enzyme, displayed on fd phages, were selected for affinity on three protein antigens. The libraries used in this work, L1B, L3B and L4, have been described previously (Legendre et al. 1999). They contain one or two insertions of six random codons in substitution for one to three codons of the TEM-1 enzyme, in loops that surround the catalytic site of the enzyme. L1B and L3B libraries contain a single insertion located in loops A and C, respectively, whereas L4 contains one insertion in each loop A and C (Fig. 1 ▶). L3B has been selected on ampicillin to remove inactive clones from the library prior to affinity selection and has a high diversity (Legendre et al. 1999). This library contains only active clones (kcats ≥ 30 sec−1). On the other hand, L1B and L4 have not been selected for activity and contain, respectively, <0.1% and ∼20% of clones whose kcats are higher than 30 sec−1. Streptavidin, ferritin, and β-galactosidase were chosen as model antigens for selection on L1B, L3B, and L4. These proteins were chosen on the basis of their properties. Streptavidin is a tetrameric protein that binds biotin with very high affinity in deep cavities (Weber et al. 1989). It has been shown to bind peptide mimotopes containing an HPQ sequence (Devlin et al. 1990). This could ease the identification of streptavidin binders among the β-lactamase clones selected. The E. coli β-galactosidase is a 440-kD tetrameric enzyme featuring deep substrate-binding cavities (Jacobson et al. 1994). It does not appear to interact with other proteins. Ferritin is an iron storage protein of roughly spherical shape. The horse spleen protein is made up of 24 subunits, ∼22 light chains and 2 heavy chains (Harrison and Arosio 1996). Only one protein, kininogen, has been described as forming a complex with ferritin (Torti and Torti 1998). As such, β-galactosidase and ferritin may appear as challenging targets. These proteins are available in large quantities as purified enzymes.
Fig. 1.

Localization of insertion and mutation sites on the three-dimensional structure of TEM-1 β-lactamase. Loops A and C in which insertions were carried out to generate L1B, L3B, and L4 libraries are represented in green. Additional residues mutated in Fer503-1 (T114M, L201P, I287V), Fer503-2 (T114M, N132S, L201Q), and Fer503-5 (T114M) clones are indicated in red. The active site Ser 70 residue is labeled in blue. The model was generated using Molscript V2.1 and Raster 3D V2.5.
Selections were carried out by incubating the libraries with streptavidin-coated beads or with biotinylated ferritin or biotinylated β-galactosidase that were immobilized on streptavidin beads. After washing the unbound phages, clones specifically retained on the beads were eluted at acidic pH and amplified in bacteria. Three to six rounds of selection-amplification were required to isolate β-lactamase clones able to bind the target proteins. In the first place, L1B, L3B, and L4 libraries were individually selected for clones that bind streptavidin. Selections on streptavidin were characterized by a rapid increase in the number of phages eluted along the rounds, indicating that β-lactamase clones able to bind streptavidin were being selected. Between the first and the third round of selection, the number of phages eluted increased by a factor of 150–2800, depending on the library (data not shown). The library that showed the lowest enrichment was L1B. Three rounds of selection were sufficient to enrich the libraries in clones displaying all the HPQ sequence (Table 1).
Table 1.
Characteristics of phage-displayed β-lactamases selected on streptavidin (SV), ferritin, and β-galactosidase
| Selecting agent | Library | Clone | Frq.a | Sequence | Substrate | Specific activity (s−1)b | Relative activity (−/+ ligand)c | Kd (M)d |
| FdBla | wt | Penicillin G | 3790 | |||||
| Nitrocefin | 1428 | |||||||
| SV | L1B | SVL1-01 | 8/12 | V103-Y105 → YHPQNA | Penicillin G | 14.6 | 1/0.83 (+0.33 μM); 0.75 (sat) | 1.0 ± 0.4 × 10−7 |
| SVL1-02 | 2/12 | V103-Y105 → NHPQNI | Penicillin G | 5.8 | 1/0.79 (+0.33 μM) | ND | ||
| SVL1-03 | 1/12 | V103-Y105 → WHPQNS | Penicillin G | 12.4 | 1/0.77 (+0.33 μM); 0.65 (sat) | 2.2 ± 0.3 × 10−7 | ||
| SVL1-04 | 1/12 | V103-Y105 → FENHPQ | Penicillin G | 34.8 | 1/0.86 (+0.33 μM) | ND | ||
| L3B | SVL3-01 | 5/11 | T271 → HPQNDD | Penicillin G | 419 | 1/0.95 (+0.33 μM) | ND | |
| SVL3-02 | 5/11 | T271 → HPQGDN | Penicillin G | 294 | 1/0.90 (+0.33 μM); 0.74 (sat) | 4.6 ± 0.7 × 10−7 | ||
| SVL3-03 | 1/11 | T271 → HPQGDI | Penicillin G | 2271 | ND | ND | ||
| L4 | SVL4-01 | 1/4 | V103-Y105 → QGQGAW | — | ND | ND | ND | |
| T271 → HPQNDY | ||||||||
| SVL4-02 | 1/4 | V103-Y105 → KGKHAW | — | ND | ND | ND | ||
| T271 → HPQGDA | ||||||||
| SVL4-03 | 1/4 | V103-Y105 → FLSGDW | — | ND | ND | ND | ||
| T271 → HPQNDY | ||||||||
| Ferritin | L3B+L4 | Fer503 | 8/8 | V103-Y105 → EWVPFG | Penicillin G | 2613 | 1/1.03 (+0.5 μM) | ND |
| T271 → KWETVT | Nitrocefin | 230 | 1/1.07 (+0.5 μM) | |||||
| β-Gal | L3B+L4 | Gal601 | 1/5 | T271 → ATLEKS | Penicillin G | 209 | 1/1.01 (+0.33 μM) | ND |
| Gal602 | 1/5 | E104Y105 → TGPAAW, T271 → DVRDVG | Penicillin G | 27.4 | ND | ND | ||
| Gal604 | 1/5 | E104Y105 → STGNGW, T271 → CKSVNC | Penicillin G | 8.3 | 1/1.05 (+0.33 μM) | ND | ||
| Gal605 | 1/5 | V103-Y105 → NKSGNF, T271 → KSRIAS | Penicillin G | 84.2 | 1/0.96 (+0.33 μM) | ND | ||
| Gal606 | 1/5 | V103-Y105 → LKAGMW, T271 → PQRNIN | Penicillin G | 1.9 | ND | ND |
ND, not determined.
a Frequency at which clones appeared after selection on the target protein.
b Specific activities measured on 1mM penicillin G or 1 mM nitrocefin, SD < 10%.
c Effects on enzymes activity at a reference concentration of protein antigen and maximum effects extrapolated from curves in Figure 3 ▶, at antigen saturation.
d Kd extracted from curves in Figure 3 ▶.
Selections on ferritin and β-galactosidase were carried out exclusively on L3B and L4 libraries that were pooled for the occasion. During selection on ferritin, no increase in the number of phages eluted could be observed before the fifth round, in which the number of eluted phages increased by a factor of 60 compared with the first round. Yet, the specific activity of the library on penicillin G increased steadily along the rounds, from ∼100 sec−1 for L3B/L4 to 2300 sec−1 at the end of the selection (data not shown). This indicated that a selection was occurring. Indeed, sequencing of 8 clones from the fifth round revealed that a monoclonal selection had been reached, a single clone being isolated (Table 1).
Six rounds of selection were carried out on β-galactosidase, but in this case, neither an increase in the number of eluted phages nor a significant modification of the library specific activity was observed along the rounds. Five clones from the sixth round were sequenced and proved to be different (Table 1). In view of these results, two hypotheses can be envisaged: either the library diversity was still too large after the sixth round of selection or the selection for binders was not successful. It should be noted that β-galactosidase is an E. coli enzyme whose production in bacteria could interfere with the selection process by sequestrating binders before phage production. However, as the β-lactamase-pIII fusion protein is exported to the periplasmic space, the possibility for the fusion protein to interact with the cytoplasmic β-galactosidase may be limited. The fact that the number of washes applied to eliminate unbound phages was raised along the rounds may make the observation of slight enrichments of phage–enzymes that poorly bind the target protein difficult. Therefore, the lack of increase in elution yield does not necessarily mean that enzymes able to bind to β-galactosidase were not selected. It is interesting to note that four out of the five clones analyzed originated from the L4 library and contained full inserts in both loops A and C (Table 1). It had been shown previously that 50% of the clones in the L4 library do not display a complete insert in loop A but, instead, only point mutations in this region (Legendre et al. 1999). The predominance of clones issued from L4 and featuring two inserts could therefore be indicating that a selection took place. Moreover, four of the inserts sequenced showed the presence of a conserved KS motif (Table1). To find out whether these clones were able to bind to β-galactosidase, an ELISA was carried out on microplates coated with the purified enzyme. Dilutions of three clones displaying the KS motif, Gal601, Gal604, and Gal605, were incubated on β-galactosidase-coated microplates, and bound phages were detected by an anti-M13 antibody conjugated to horseradish peroxidase. Unmodified fd-Bla phages were used as negative control. Figure 2A ▶ shows that all clones tested gave a signal significantly higher than fd-Bla, indicating that these clones really bind the coated protein. Interestingly, Gal604, which contains the KS motif flanked by two cysteines in the C loop, had the apparently highest affinity among the clones analyzed. This could indicate that the two cysteines form a disulfide bridge that imposes a constraint favoring the interaction with β-galactosidase. However, the signals measured by ELISA were quite weak and observed only at high phage concentrations, which probably indicates a low affinity of the phage–enzymes for their target. As a comparison, the ferritin-selected clone Fer503 gave a stronger signal on ferritin-coated plates (Fig. 2B ▶).
Fig. 2.

Characterization of binding properties of (A) β-galactosidase- and (B) ferritin-selected β-lactamases. 107 to 1013 phages were incubated per well on microplates coated with β-galactosidase or ferritin. Bound phages were detected with an anti-M13 antibody conjugated to horseradish peroxidase. Readings were carried out at 405 nm every minute for ∼1 h. Readings taken at 60 min (β-galactosidase) or 15 min (ferritin) were plotted as a function of phage concentration. (A) (♦) FdBla; (○) Gal601; (▪) Gal604; (▴) Gal605. (B) (♦) FdBla; (□) Fer503; (▪) Fer503-1; (•) Fer503-2; (○) Fer503-3.
Specific activity measurements on penicillin G or nitrocefin of phage–enzymes issued from the selection showed that, with the exception of clone Fer503, clones isolated from L3B were very active, whereas clones issued from L1 and L4 had a lower activity (Table 1). This is consistent with the fact that the L3B library has been previously selected for activity and has a higher average activity than L1B and L4 (Legendre et al. 1999). The activity of Fer503 was very high, close to the activity of fd-Bla. Antigen binding had only a weak effect on enzyme activity of clones selected on streptavidin and no effect at all on clones selected on ferritin or β-galactosidase, as attested by kinetic measurements in the presence of a relatively high (0.3–0.5 μM) antigen concentration (Table 1). The effect of streptavidin binding on individual clones isolated from L4 (SVL4-01 and SVL4-02) was not determined because the activity of the library after the third round of selection was quite low and remained unaffected by streptavidin (data not shown). The absence of major effect of protein binding on enzyme activity is in striking contrast with results obtained previously with monoclonal antibodies, wherein most of the β-lactamases issued from L3B and L4 had their activity strongly modulated by antibody binding (Legendre et al. 1999). To examine whether the lack of effect could result from a low affinity of the selected enzymes for their target protein, inhibition curves were determined for clones SVL1-01, SVL1-03, and SVL3-02 (Fig. 3A ▶). This allowed extracting the dissociation constants of the streptavidin–enzyme complexes as well as the maximal inhibitory effect of streptavidin binding. All clones examined proved to have a relatively high affinity for streptavidin, with Kd between 1 × 10−7 M and 3.5 × 10−7 M, but had their activity decreased by <35% in the presence of a saturating amount of streptavidin (Table 1). The specificity of the inhibition was confirmed by competition with biotin, which prevented the inhibition of the clones by streptavidin.
Fig. 3.
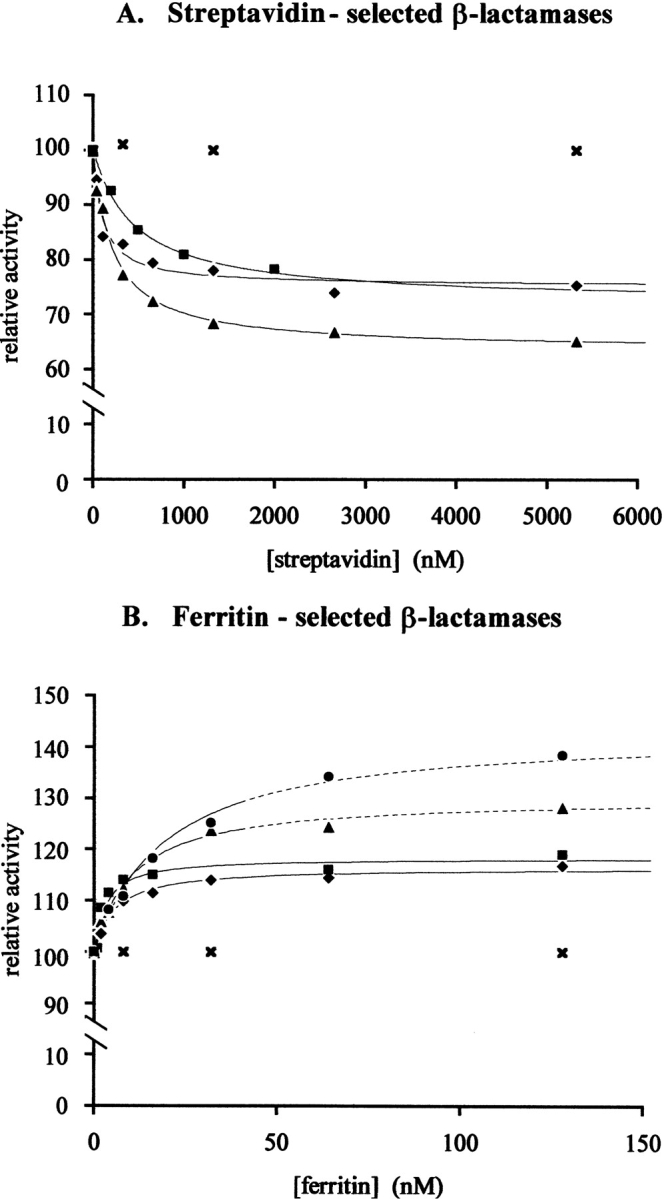
Activity modulation of streptavidin- and ferritin-selected β-lactamases as a function of streptavidin or ferritin concentration. Measurements were carried out on 1 mM penicillin G at 20°C. (A) (♦) SVL1-01; (▴) SVL1-03; (▪) SVL3-02; (|qX) FdBla. (B) (▪) Fer503-1; (♦) Fer503-3; (•) purified Fer503-1; (▴) purified Fer503-3; (×) FdBla. (Solid lines) phage–enzymes; (dotted lines) purified enzymes.
Affinity maturation of a ferritin-selected β-lactamase
Because the presence of ferritin or β-galactosidase had no effect on the activity of β-lactamases selected on ferritin or β-galactosidase, the dissociation constants of the complexes could not be determined through inhibition curves. This led us to wonder whether the lack of effect of antigen binding was the result of low affinity or, as it appears for streptavidin-selected clones, of an absence of effect of antigen complexation on the enzyme ability to catalyze the conversion of its substrate into product. Notwithstanding the second possibility, a low affinity of the enzyme–antigen complexes is indicated by the low enrichments observed during the selection on ferritin and β-galactosidase, compared with streptavidin. To answer this question, an attempt was made to increase the affinity of clone Fer503 for its target by subjecting its gene to error-prone PCR and by selecting the library of mutants generated on ferritin. A library of 5.3 × 106 mutants of Fer503 was generated with an average of six point mutations per β-lactamase gene. The library, Fer503L1, conserved a relatively high specific activity (200 sec−1) on penicillin G, indicating that the mutagenesis was not too disruptive. Four rounds of selection were carried out on the library, ending up with a 5000-fold increase in the number of phages eluted compared with the first round of selection on L3B + L4. The average activity of the selected phages also increased along the rounds, to recover, at the end of the fourth round, the activity of clone Fer503. Three individual clones of the fourth round, Fer503-1, -2, and -3, were characterized. Sequencing of these clones indicated that both inserts present in Fer503 were conserved but revealed the presence of additional mutations on the surface of the enzyme (Table 2; Fig. 1 ▶). Among the mutations, the T114M was observed in the three clones, suggesting that this mutation is directly involved in ferritin binding. Analysis of Fer503-1, -2, and -3 binding properties by ELISA showed that the affinity of Fer503 was improved by about three orders of magnitude (Fig. 2 ▶). All clones showed a high activity on penicillin G and nitrocefin and had their activity slightly affected by 0.5 μM ferritin (Table 2). Ferritin stimulated activity of Fer503–1, 2 and 3 clones on penicillin G and slowed down activity of Fer503-1 and -2 on nitrocefin. Kds determined from activation curves measured on penicillin G for clones Fer503-1 and -3 (Fig. 3B ▶) were in the nanomolar range (Table 2), showing that the engineering of a strong interaction between the TEM-1 β-lactamase and the horse spleen ferritin had succeeded. As in the case of streptavidin, the interaction between β-lactamase and ferritin was not able to disrupt or significantly affect the enzyme activity. Ferritin binding affected the enzyme activity on phage by <20% (Table 2).
Table 2.
Affinity maturation of phage-displayed β-lactamases selected on ferritin
| Selecting agent | Library | Clone | Frq.a | Sequence | Substrate | Specific activity (s−1)b | Relative activity (−/+ ligand)c | Kdd |
| Ferritin | Fer503L1 | Fer503-1 | 1/3 | = Fer503 + T114M, | Penicillin G | 2733 | 1/1.18 (+0.5 μM); 1.18 (sat) | 2.8 ± 1.1 × 10−9 |
| L201P, I287V | Nitrocefin | 372 | 1/0.88 (+0.5 μM) | |||||
| Fer503-2 | 1/3 | = Fer503 + T114M, | Penicillin G | 498 | 1/1.07 (+0.5 μM) | ND | ||
| N132S, L201Q | Nitrocefin | 56 | 1/0.84 (+0.5 μM) | |||||
| Fer503-3 | 1/3 | = Fer503 + T114M | Penicillin G | 3132 | 1/1.18 (+0.5 μM); 1.18 (sat) | 5.5 ± 1.3 × 10−9 | ||
| Nitrocefin | 373 | 1/1.01 (+0.5 μM) |
ND, not determined.
a Frequency at which clones appeared after selection on the target protein.
b Specific activities measured on 1mM penicillin G or 1 mM nitrocefin, SD < 10%.
c Effects on enzymes activity at a reference concentration of protein antigen and maximum effects extrapolated from curves in Figure 3 ▶, at ferritin saturation.
d Kd extracted from curves in Figure 3 ▶.
Purification and characterization of free enzymes
From the genetic construction, it can be expected that three to five copies of β-lactamase are displayed per phage. However, the number of enzymes displayed can be lower as a result of proteolysis during phage morphogenesis and production (Legendre et al. 1999). Comparison of the specific activity measured for fd-Bla and of the value of kcat for purified TEM-1 on penicillin G (Tables 1 and 3) indicates that phages displaying the wild-type TEM-1 β-lactamase carry on average three active enzymes on their surface. Therefore, measurements carried out on phages may be subject to errors linked to avidity phenomena (streptavidin, ferritin, and β-galactosidase are all multimeric proteins) or steric hindrance.
Table 3.
Parameters of purified enzymes
| Kd (M)b | |||||
| Clone | Substrate | kcat (s−1) | Relative activity (−/+ ferritin)a | Activation curve | ELISA |
| TEM-1 | Penicillin G | 1290 ± 90 | |||
| Nitrocefin | 300 ± 43 | ||||
| Fer503-1 | Penicillin G | 947 ± 25 | 1/1.43 | 2.0 ± 0.3 × 10 −8 | 1.8 ± 0.2 × 10 −8 |
| Nitrocefin | 172 ± 12 | ND | |||
| Fer503-3 | Penicillin G | 786 ± 46 | 1/1.30 | 1.0 ± 0.2 × 10−8 | 1.6 ± 0.2 × 10−8 |
| Nitrocefin | 116 ± 20 | ND | |||
To address these problems, β-lactamases from clones Fer503-1 and Fer503-3 were purified as free enzymes and characterized. The corresponding β-lactamase genes were amplified by PCR and recloned in the pET26b(+) expression vector. The proteins were recovered from the periplasmic fraction of bacteria induced to express β-lactamase from the pET vector and were purified by chromatography on Q sepharose and Ni2+ columns. The enzyme purity was checked by SDS-PAGE (Fig. 4 ▶).
Fig. 4.

Fer503-1 and Fer503-3 purification as free enzymes. The purity of Fer503-1 and Fer503-3 was analyzed on a 10% acrylamide gel. Protein bands were revealed by Coomassie blue staining. As control, several different amounts of purified TEM-1 enzymes were loaded. Concentrations of purified enzymes were determined by the bicinconinic assay.
As expected, kcat measurements showed that the enzyme activity was lower than the specific activity measured on phage but remained close to that of the wild-type enzyme (Table 3). Comparison of phages and of purified enzyme activities for clones Fer503-1 and Fer503-3 confirms that between three and four copies of enzymes are displayed per phage. Interestingly, the effect of ferritin binding on the activity on penicillin G of the purified enzymes from clones Fer503-1 and Fer503-3 was significantly higher than the effect measured on phage (Tables 1 and 3). As a control, 0.5 mM BSA had no effect on Fer503-1 and Fer503-3 activity (data not shown).
Values of Kd determined from activation curves (Fig. 3 ▶) showed that the affinity of purified enzymes for ferritin was slightly lower than the affinities determined on phage (Tables 2 and 3), indicating that a cooperative binding of several β-lactamases displayed on the same phage to ferritin occurs. However, affinities for ferritin of both purified enzymes remained high, between 10 and 20 nM (Kd). To confirm these values, dissociation constants of the purified enzymes Fer503-1 and Fer503-3 for ferritin were measured by a second method in which free enzyme concentrations in solution were determined by ELISA, as a function of ferritin concentration. A low concentration of purified enzymes was incubated in the presence of various concentrations of ferritin, and concentrations of uncomplexed enzymes were titrated by ELISA on microplates coated with ferritin. The experiment was carried out in conditions where the enzyme amount that got immobilized on the plate was <2.5% of the total amount incubated, to avoid disturbing the equilibrium in solution. The conditions were also set so that the ELISA response was linear as a function of free enzyme concentration. Enzyme amounts immobilized on plates were determined by directly measuring the enzyme activity on nitrocefin. Dissociation constants extracted from the titration curves in Figure 5 ▶ were nearly identical to those determined by the first method (Table 3).
Fig. 5.

Titration of purified Fer503-1 and Fer503-3 enzymes as a function of ferritin concentration by ELISA. First, 5 nM Fer503-1 or Fer503-3 was incubated in Eppendorfs with various concentrations of ferritin in solution. After the equilibrium was reached, the concentration of free β-lactamase in each sample was determined on ferritin-coated 96-well microplates by measuring on nitrocefin the β-lactamase activity that can be immobilized on the plates.
Ferritin assay
To test whether enzymes selected to recognize other proteins could be used in enzyme-linked immunosorbant assays, as substitute to antibodies conjugated to an enzyme, an assay of horse spleen ferritin was carried out using the purified Fer503-3 enzyme. For the assay, 96-well microplates were coated with a polyclonal antibody directed against horse spleen ferritin and incubated with various amounts of purified horse spleen ferritin. Bound ferritin was detected with 50 ng of purified enzyme per well. As shown in Figure 6 ▶, ferritin could be assayed in a concentration range between 0.1 nM and 0.5 μM.
Fig. 6.

Horse spleen ferritin assay. Increasing amounts of purified horse spleen ferritin were incubated on 96-well microplates coated with an anti-horse spleen ferritin polyclonal antibody. Bound ferritin was detected with 50 ng of purified Fer503-3 enzyme per well by measuring on 1 mM nitrocefin the β-lactamase activity immobilized.
Discussion
In a previous work, we isolated β-lactamase variants forming tight complexes with monoclonal antibodies. All β-lactamase clones selected were issued from L3B or L4 libraries and had a full insert only in loop C. Normally, clones from L4 should also have contained an insert in loop A, but, instead, they contained only point mutations in that region. Therefore, it is probable that the antibody-binding site is centered on loop C in most selected clones. Although no information is presently available on the structure of these complexes, the general picture that has emerged from the determination of several structures of monoclonal antibodies complexes indicates that the extended loops of the β-lactamases could fit into the relatively broad cavities of the antibodies, which are naturally designed to bind tightly protein antigens. This kind of fit could explain the medium-to-strong inhibitory effect on activity observed in many cases. Indeed, many clones had their activity strongly affected by antibody binding. The importance of the activity modulation was in general dependent on the activity of the clones; clones with high activities were more readily subject to enzyme regulation than clones that had a low activity. Although loop C does not contain residues directly involved in catalysis or substrate binding, it may represent an appropriate binding point through which complexation affects the correct positioning of some residues important for catalysis in a way that evokes an allosteric regulation (Legendre et al. 1999).
The mode of interaction of the mutants selected here may vary with the target protein. The activity of clones selected on streptavidin, ferritin, and β-galactosidase was only poorly or not at all modulated by antigen binding. At least in the cases of streptavidin and ferritin, the weakness of the effect could not be related to a weak affinity for their target.
For clones selected on streptavidin, β-lactamase binding is very probably mediated mostly through the HPQ sequence contained in one of the engineered loops (Table 1) because biotin has been shown to compete with HPQ-containing peptides for streptavidin binding (Kay et al. 1993; Giebel et al. 1995) and crystallographic studies have shown that the HPQ sequence fits into the biotin-binding site of the protein (Katz 1995; Katz and Cass 1997). Therefore, in streptavidin-selected β-lactamases, the enzyme is probably only a platform on which the HPQ epitope is displayed. It is, at first sight, surprising that streptavidin binding has a low effect on the activity of clones derived from L1B, as loop A lies at the entrance of the active site. This may be related to the fact that these clones have a low activity (≤1% of wild type). Streptavidin binding to HPQ in the insert does not increase the perturbation of the active site. The weak effect on more active clones derived from L3B is in contrast to the results of antibody binding. Binding to the constrained conformation of the HPQ(N/G) sequences in loop C probably does not require a conformational change and consequently has only a very weak effect.
The way β-lactamase variants selected on ferritin interact with their target protein is more difficult to deduce from the available data. The L4 library from which clone Fer503 is issued was constructed by recombination of <5 × 104 active clones from the related L1B and L1C libraries with ∼5 × 107 active clones from the L3B library (Legendre et al. 1999). Consequently, each sequence in loop A was probably associated with at least a few hundred sequences in loop C. As Fer503 was selected as a unique clone, it is very likely that the specific insert in loop C at least contributes to ferritin binding. In a search for proteins interacting with human ferritin using a ligand blot assay, the only partner that was identified is H-kininogen, a multifunctional protein that inhibits cystein protease (Torti and Torti 1998). Ceruloplasmin has also been proposed to interact with ferritin and exchange iron with it (Juan and Aust 1998). Two synthetic decapeptides derived from the ceruloplasmin sequence were shown to inhibit ceruloplasmin–ferritin interaction but at relatively high concentration (50% inhibition at 0.2 mM). Inspection of the sequences of the two extended loops did not show any similarity with the peptides derived from H-kininogen or ceruloplasmin. Affinity maturation of clone Fer503 provided clones with several mutations located on the surface of the enzyme but mostly quite far from the two insertion sites. One of the mutations, Thr114Met, is apparently sufficient to boost the affinity to the highest level reached by the clones analyzed. This residue is located ∼25 Å away from the essential serine and on the same side of the active site as the loop A insertion site, whereas loop C lies on the opposite side (Fig. 1 ▶). However, the three mutated sites are on the same plane and, therefore, should all be available for interaction with another protein. Taken altogether, these observations indicate that, at least in clones whose affinity has been matured, the region of β-lactamase interacting with ferritin encompasses a broader region than one or two loops. As an alternative explanation, Thr114Met could trigger the dimerization of Fer503. The increase in affinity would, in this case, be the result of a high avidity of the matured β-lactamases for their target. However, dimerization of β-lactamase on phage would be quite unexpected because the enzyme is in a fusion with the pIII protein and linked to the viral protein by a short linker.
The presence of extensions loops A and C on both sides of the active site of Fer503 clones does not perturb the activity. We have shown that ferritin binding has a substrate-dependent effect on activity. A similar substrate-dependent response to complexation was already observed for a β-lactamase clone isolated on a monoclonal antibody (Legendre et al. 1999). Because the Fer503 clones are nearly as active as the wild-type enzyme and the TEM β-lactamase turnover is close to the diffusion limit on penicillin substrates (Christensen et al. 1990), only a mild activation can be induced with them. The modest inhibitory effect on activity on nitrocefin is more surprising if loop C is involved in ferritin binding because antibody binding to this site was shown, in most cases, to significantly inhibit the enzyme activity. Structural information will be needed to understand this behavior.
Only low-affinity β-lactamase clones were selected on β-galactosidase. The relevance of the presence of a KS motif in some of the inserts is not clear at this stage. In their work on landscape phages in which random octapeptides were displayed at high density in fusion with the major coat protein pVIII, Petrenko and Smith (2000) isolated phages binding to the E. coli β-galactosidase. They bind outside of the main groove of the protein including the catalytic pocket, as the competitive inhibitor IPTG does not prevent binding and their affinity for the enzyme was lost upon phage denaturation, indicating that the phage environment was essential for peptide binding. Two conserved motifs emerged from sequence comparisons of the selected clones. None of the sequences of the loops in the Gal601 to Gal606 clones correspond to the consensus sequences described for the landscape phages. Again these data indicate that β-galactosidase binding involves more than the specific sequences displayed as recognition elements.
Although the protein antigens chosen as models in this study have no interest in diagnosis, we have tested whether the kind of molecules that were isolated here could be useful as substitutes for antibodies in enzyme-linked immunosorbant assays. Using a β-lactamase selected on horse spleen ferritin, we have shown that horse spleen ferritin could be assayed by ELISA in a ferritin concentration range between 0.1 nM and 0.5 μM. Interestingly, attempts to use this enzyme to assay human spleen ferritin failed (data not shown), although horse and human spleen ferritin share a high degree of sequence and structure identity (Harrison and Arosio 1996; Hempstead et al. 1997). These data indicate that the enzymes selected on ferritin are quite specific for their target. Specificity of the enzymes selected on ferritin and β-galactosidase is also shown by the observation that the corresponding phage–enzymes are retained significantly more on streptavidin beads coated with their target antigen than on uncoated streptavidin beads, on which they behave like the wild-type phage–enzyme (data not shown).
The kind of enzyme–antibodies we have developed here may have some advantages over antibodies conjugated to an enzyme for immunodetection purposes because they bear the recognition and enzymatic functions on the same molecule and, therefore, should be easier and cheaper to produce. The yield of purified mutant enzymes in this paper was quite low as enzymes were purified from the periplasmic space of transformed bacteria (Materials and Methods). However, we have observed that mutant enzymes precipitated in E. coli as inclusion bodies and could be renatured after solubilization of the protein in guanidine hydrochloride, to yield ∼50 mg of enzyme per liter of culture (data not shown). Mutation of the main catalytic residue Ser 70 could provide catalytically inactive enzyme–antibodies that, in the case of multimeric proteins, could be used to immobilize the antigen on a support before assaying it with their active counterparts. The weak effect of antigen binding on enzyme activity makes these enzymes difficult to use in homogeneous assays. However, the substrate-dependent effect observed upon complexation of ferritin could be exploited to amplify the signal measured; the rate of hydrolysis of penicillin G and nitrocefin could be measured simultaneously in the same cuvette at two different wavelengths and the presence of ferritin determined by the ratio of activities. The method offers the advantage that the measure is less susceptible to small variations in enzyme concentration.
In conclusion, we have shown that β-lactamases able to bind three different protein antigens could be selected from β-lactamase libraries that contain insertions in loops surrounding the enzyme active site. The fact that clones could be isolated for each unrelated antigen indicates that these libraries potentially contain clones that have the capacity to bind to a large number of different targets. However, more sophisticated β-lactamase libraries, in which more than two loops of the enzyme are engineered to contain random sequences, will be probably required to reach high-affinity clones directly and to obtain enzymes featuring antibody-like binding sites. This work is currently underway. Whether the enzyme activity is modulated or not by antigen binding, these β-lactamases could prove useful in homogeneous assays or in enzyme-linked immunosorbant assays.
Materials and methods
General
Streptavidin beads and pure streptavidin were purchased from Dynal and Promega, respectively. Anti-ferritin antibodies, biotinylated and non-biotinylated horse spleen ferritin, and β-galactosidase were from Sigma. Restriction enzymes were from Roche and New England Biolabs. Penicillin G and nitrocefin were purchased from Sigma and Calbiochem, respectively.
Oligonucleotides
Oligonucleotides were all purchased from Eurogentec (Liége). Restriction sites are underlined. O1, 5`-CACAGTGCACACCCAG AAACGCTGG-3`; O2, 5`-TTCTGTATGAGGTTTTGCTAA-3`; O3, 5`-GGAATTCGGATCCCTATTACCAATGCTTAATCAGT G-3`; O4, 5`-CTAGAATTCATATGAAAAAATTATTATTCG-3`.
Libraries of β-lactamases
L1B, L3B, and L4 libraries of phage-displayed TEM-1 β-lactamases were described previously (Legendre et al. 1999). L1B and L3B contain an insertion of six random codons in substitution for sequences encoding, respectively, residues V103E104Y105 and T271 of the TEM-1 β-lactamase gene. L4 contains two insertions of six codons in substitution for V103E104Y105 or E104Y105 and T271. The Fer503L1 library was constructed on the basis of the ferritin-selected phage Fer503 (Table 1) by error-prone PCR mutagenesis. The β-lactamase gene of Fer503 was amplified with oligonucleotides O1 and O2, following the procedure of Cadwell and Joyce (1994). In this procedure, Mn2+ is introduced in the PCR reaction to diminish the Taq polymerase fidelity, which leads, in the conditions described, to a mutation frequency of 0.66% per position. After amplification, the β-lactamase gene was restricted by ApaLI and NotI and cloned in the corresponding sites of fd-tet-DOG1, as described previously (Soumillion et al. 1994). The O2 oligonucleotide hybridizes in the sequence encoding the mature g3p of fd-Fer503, slightly downstream from the NotI site. It should also be noted that oligonucleotide O1 restores the first codon of the mature β-lactamase that had been mutated previously from histidine to glutamine in the construction of the FdBla phage that displays a pseudo-wild-type TEM-1 β-lactamase (Soumillion et al. 1994). The H26Q mutation is also present in L1B, L3B, and L4 clones. The Fer503L1 library was produced by electroporating the ligation product in TG1. The size of the library is 5.3 × 106. The construction was checked by DNA sequencing, which confirmed the average mutation frequency of 1 mutation per 150 nucleotides.
Phage production and purification
Phages from libraries or from individual clones were amplified in TG1, in liquid LB medium containing 7.5 μg/mL tetracycline, at 23°C for 60 h. Phages were recovered from the culture supernatant by two successive polyethylene glycol precipitations and were resuspended in TBS (150 mM NaCl, 50 mM Tris at pH 7.5). Between the first and the second precipitations, the phage solutions were filtrated through a 0.45-μm filter. Stock phage concentrations were determined by measuring their absorbance at 265 nm using 8.4 × 107 M−1 cm−1 as extinction coefficient.
Selections
Biopannings were carried out on streptavidin-coated magnetic beads (selections on streptavidin) or on streptavidin-coated beads saturated with biotinylated ferritin or biotinylated β-galactosidase. Beads were washed with TBS (150 mM NaCl, 50 mM Tris at pH 7.5) and blocked by incubation with a 2% solution of nonfat dried milk. Then 1013 phages (1 mL of 1 OD265) were incubated overnight in the presence of 100 μg of beads under gentle stirring on a rotating wheel at room temperature. After that incubation period, beads were washed with TTBS (TBS containing 0.1% Tween 20), and bound phages were eluted from the beads upon resuspension of the beads in 0.025 N HCl (pH adjusted to 2.2 with glycine). After neutralization by addition of Tris buffer at pH 9.0, the eluted phages were amplified by infecting log phase TG1 cells. Eluted phages were titrated by plating the infected bacteria on LB medium containing 7.5 μg/mL tetracycline. Depending on the selection, three to six consecutive rounds of selection-amplification were carried out. The number of TTBS washes increased along the rounds, from 3 to 4 for the first round to up to 15 for the last round.
Analysis of binding properties of phage–enzymes by ELISA
The 96-well microplates were coated with 10 μg of purified streptavidin, β-galactosidase, or ferritin per well, in 200 μL of PBS (100 mM NaCl, 50 mM phosphate at pH 7.0), at 4°C for one night. Plates were then washed 3 times with PBS and blocked with 200 μL of MPBS (PBS + 2% nonfat dried milk) per well, at room temperature for 1 h. Dilutions of phages from 1013 to 107 were incubated in 200 μL of MPBS per well at 4°C overnight. Unbound phages were removed by 3 washes with TPBS (PBS + 0.1% Tween 20) and 1 wash with PBS. Bound phages were detected with an anti-M13 monoclonal antibody conjugated to horseradish peroxidase (Amersham Pharmacia Biotech). The antibody was diluted 5000 times in MPBS and incubated on the microplates at room temperature for 1 h. Excess antibody was washed away by 3 washes with TPBS and 1 wash with PBS. Peroxidase activity was measured on ABTS [2,2`-azinobis(3-ethylbenzthiazoline-6-sulfonic acid]. The substrate solution was prepared by dissolving 2 mg of ABTS in 10 mL of 0.05 M citric acid (pH 4). Just before use, 8 μL of 35% hydrogen peroxide was added to the solution, and 200 μL of the solution was dispensed per well. Readings were carried out every minute at 405 nm.
Cloning of ferritin-selected β-lactamases in pET26b expression vector
β-lactamase genes from Fer503-1 and Fer503-3 clones were amplified by PCR using primers O3 and O4. The amplified fragments were restricted by NdeI and BamHI and cloned between the NdeI and BamHI sites of the pET26b(+) expression vector (Novagen) to produce plasmids pFer503-1 and pFer503-3, respectively. Ligation products were electroporated in JM109 cells. DNA from transformants was analyzed by restriction and sequencing. Correctly constructed clones were then transferred into BL21-DE3 cells (Novagen) for expression.
Enzyme purification
An overnight culture of the pFer503-1 or pFer503-3 construct in BL21-DE3 cells was diluted 40× in 2.4 L of LB medium containing 50 μg/mL kanamycin and grown at 30°C until the culture reached an OD600 of 0.8. Then 100 μM IPTG (isopropyl β-D-thiogalactopyranoside) was added to the medium, and the culture was grown under stirring at 30°C for 3 h. Cells were harvested by centrifugation and resuspended in 30 mL of 20% sucrose, 30 mM Tris-HCl (pH 8.0). After resuspension of the cells, 60 μL of 0.5 M EDTA (pH 8.0) was added and the solution was placed under stirring at room temperature for 10 min. Cells were centrifuged at 4°C, and the pellet was resuspended in 30 mL of 5 mM MgSO4. The suspension was put under stirring at 4°C for 10 min. Cells were then discarded by centrifugation, and the supernatant, containing the periplasmic fraction, was recovered by transferring the supernatant to a fresh tube. A tab of an inhibitor cocktail (Complete, Roche) was added to the solution at this stage. The extract was dialyzed against 10 mM Tris-HCl (pH 7.5) at 4°C overnight. The extract was then loaded at 4°C on a Q-sepharose fast flow (Pharmacia) column (h = 20 cm, d = 2.9 cm), equilibrated with the dialysis buffer. Proteins were eluted by an NaCl gradient (0–1 M) in 10 mM Tris-HCl (pH 7.5) at 0.4 mL/min, and 68 7-mL fractions were collected, of which 4 contained the active β-lactamase. These fractions were pooled for further purification. The semipurified enzyme was dialyzed against 100 mM phosphate (pH 8.0) at 4°C overnight. After dialysis, the protein was loaded on a Ni2+ (Affiland) column (h = 25.5 cm, d = 1.2 cm) equilibrated in the same phosphate buffer at 4°C. Elution was performed with an imidazole gradient (0–100 mM) in 100 mM phosphate buffer (pH 8.0) in a total volume of 120 mL. Four active fractions of 4 mL were recovered. The enzyme purity was checked by SDS-PAGE and the protein concentration was determined by the bicinconinic assay (Uptima). We obtained 7.5 mg of pure Fer503-1 and 0.78 mg of pure Fer503-3 (the lower yield of Fer503-3 results from the fact that the purification protocol was set up on Fer503-3 and then optimized on Fer503-1).
Kinetic measurements
The β-lactamase activities of phage–enzymes and of purified enzymes were determined at 20°C in 50 mM phosphate buffer (pH 7.5). Specific activities of phage-displayed enzymes and kcat from purified enzymes were extracted from complete progress curves of product formation. The change in absorbance was measured as a function of time with penicillin G at 232 nm (Δɛ = 1042 M−1 cm−1) and nitrocefin at 482 nm (Δɛ = 17,400 M−1 cm−1). Specific activities of phage-displayed enzymes are determined per mole of phage (enzyme molarities on phage are difficult to estimate because more than 1 mole of β-lactamase is displayed per mole of phage and the extent of display may vary from one mutant to another). The effect of protein binding on enzyme activity was measured after incubation of the phage–enzymes or the purified enzymes with a defined concentration of the protein for at least 10 min before adding the substrate. Some enzymes whose activity was affected by protein binding were further characterized with various protein concentrations to establish inhibition/activation curves. From these curves, maximal effects of protein binding on enzyme activity were deduced.
Kd measurements
Dissociation constants between enzymes and proteins were determined either from inhibition/activation curves or by ELISA.
Dissociation constants deduced from the inhibition/activation curves presented in Figure 3 ▶ were obtained by fitting to the experimental points the function based on the following equations:
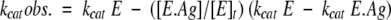 |
 |
where [E]t and [Ag]t are the total enzyme and protein (Antigen) concentrations, respectively; [E-Ag] is the enzyme–protein complex concentration; Kd is the dissociation constant of the enzyme–protein complex; kcat E and kcat E-Ag are the catalytic constants (or specific activities) of the enzyme (or phage–enzyme) and the enzyme (or phage–enzyme)–protein complex, respectively; and kcat obs. is the kcat or specific activity observed.
Dissociation constants determined by ELISA were obtained following the procedure described by Friguet et al. (1985). In the first place, we determined on 96-well microplates coated with ferritin the range of β-lactamase concentrations that gave a linear signal, directly proportional to the enzyme concentration. Different amounts of purified β-lactamase from clones Fer503-1 or Fer503-3 were incubated at room temperature for 1 h on a microtitration plate coated with 10 μg of ferritin per well and blocked with 2% nonfat dried milk in PBS (100 mM NaCl, 50 mM phosphate at pH 7.0). Wells were washed three times with TPBS (PBS + 0.1% Tween 20) and once with PBS, and the amount of enzyme bound to the plate was detected by measuring the enzyme activity on 1 mM nitrocefin. Readings were carried out every minute for ∼1 h at 490 nm. In the conditions used, the linear response domain for Fer503-1 and Fer 503-3 was between 0 and ∼7 nM of enzyme per well (the amount of enzyme bound to the plate represented <2.5% of the amount incubated per well). To determine the dissociation constants, 5 nM of Fer503-1 or Fer503-3 was incubated in Eppendorf with various concentrations of ferritin in solution at room temperature for 1 h under stirring. After the equilibrium was reached, the concentration of free β-lactamase in each sample was determined on ferritin-coated microplates as described above. Because <2.5% of the enzyme is trapped by the coated ferritin, the equilibrium in solution is not significantly modified, and the activity measured is proportional to the free enzyme concentration. By plotting the activities measured as a function of the concentration of ferritin added to the β-lactamase samples, a titration curve was obtained that obeys the following equation:
 |
where [E]0 and [Fer]0 are, respectively, the initial concentration of β-lactamase and the total concentration of ferritin in solution; Kd is the dissociation constant; A0 is the activity (ΔAbs/ΔT) measured on microplates without preincubation of the enzyme in the presence of ferritin; and A is the activity (ΔAbs/ΔT) measured by ELISA after incubation of the enzyme with various concentrations of ferritin. The Kd was obtained by fitting the function to the experimental points.
Ferritin assay
The 96-well microplates were coated with 10 μg of anti-horse spleen ferritin polyclonal antibodies in 200 μL of PBS (100 mM NaCl, 50 mM phosphate at pH 7.0) per well and blocked with MPBS (PBS + 2% nonfat dried milk). Increasing amounts of purified horse spleen ferritin were then incubated at room temperature for 1 h in 200 μL of MPBS per well. Microplates were washed 3 times with TPBS (PBS + 0.1% Tween 20) and once with PBS. Bound ferritin was detected with 50 ng of purified Fer503-3 enzyme per well, in 200 μL of MPBS at room temperature for 1 h. Wells were washed with TPBS, and PBS and β-lactamase activity was measured on 1 mM nitrocefin in PBS at 490 nm.
Acknowledgments
We thank Pascale Mathonet for several kinetic measurements and Christine Evrard for helping in the illustration of Figure 1 ▶. Purified TEM-1 β-lactamase was a gift from M. Galleni (Université de Liége). Patrice Soumillion is a research associate of the Belgian Fonds National de la Recherche Scientifique.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.023102
References
- Benito, A., Feliu, J.X., and Villaverde, A. 1996. β-Galactosidase enzymatic activity as a molecular probe to detect specific antibodies. J. Biol. Chem. 271 21251–21256. [DOI] [PubMed] [Google Scholar]
- Blum, J.H., Dove, S.L., Hochschild, A., and Mekalanos, J.J. 2000. Isolation of peptide aptamers that inhibit intracellular processes. Proc. Natl. Acad. Sci. 97 2241–2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan, C., Christianson, K., Surowy, T., and Mandecki, W. 1994. Modulation of enzyme activity by antibody binding to an alkaline phosphatase-epitope hybrid protein. Protein Eng. 7 509–514. [DOI] [PubMed] [Google Scholar]
- Brennan, C.A., Christianson, K., La Fleur, M.A., and Mandecki, W. 1995. A molecular sensor system based on genetically engineered alkaline phosphatase. Proc. Natl. Acad. Sci. 92 5783–5787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadwell, R.C. and Joyce, G.F. 1994. Mutagenic PCR. PCR Methods Applic. 3 S136–S140. [DOI] [PubMed] [Google Scholar]
- Christensen, H., Martin, M.T., and Waley, S.G. 1990. β-Lactamases as fully efficient enzymes. Determination of all the rate constants in the acyl-enzyme mechanism. Biochem. J. 266 853–861. [PMC free article] [PubMed] [Google Scholar]
- Cohen, B.A., Colas, P., and Brent, R. 1998. An artificial cell-cycle inhibitor isolated from a combinatorial library. Proc. Natl. Acad. Sci. 95 14272–14277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colas, P., Cohen, B., Jessen, T., Grishina, I., McCoy, J., and Brent, R. 1996. Genetic selection of peptide aptamers that recognize and inhibit cyclin-dependent kinase 2. Nature 380 548–550. [DOI] [PubMed] [Google Scholar]
- Dennis, M.S. and Lazarus, R.A. 1994. Kunitz domain inhibitors of tissue factor-factor VIIa. J. Biol. Chem. 269 22129–22144. [PubMed] [Google Scholar]
- Dennis, M.S., Herzka, A., and Lazarus, R.A. 1995. Potent and selective Kunitz domain inhibitors of plasma kallikrein designed by phage display. J. Biol. Chem. 270 25411–25417. [DOI] [PubMed] [Google Scholar]
- Devlin, J.J., Panganiban, L.C., and Devlin, P.E. 1990. Random peptide libraries: A source of specific protein binding molecules. Science 249 404–406. [DOI] [PubMed] [Google Scholar]
- Fabbrizio, E., Le Cam, L., Polanowska, J., Kaczorek, M., Lamb, N., Brent, R., and Sardet, C. 1999. Inhibition of mammalian cell proliferation by genetically selected peptide aptamers that functionally antagonize E2F activity. Oncogene 18 4357–4363. [DOI] [PubMed] [Google Scholar]
- Feliu, J.X., Ramirez, E., and Villaverde, A. 1998. Distinct mechanisms of antibody-mediated enzymatic reactivation in β-galactosidase molecular sensors. FEBS Letts. 438 267–271. [DOI] [PubMed] [Google Scholar]
- Ferrer-Miralles, N., Feliu, J.X., Vandevuer, S., Müller, A., Cabrera-Crespo, J., Ortmans I., Hoffmann, F., Cazorla, D., Rinas, U., Prévost, M., and Villaverde, A. 2001. Engineering regulatable E. coli β-galactosidases as biosensors for anti-HIV antibody detection in human sera. J. Biol. Chem. 276 40087–40095. [DOI] [PubMed] [Google Scholar]
- Friguet, B., Chaffotte, A.F., Djavadi-Ohaniance, L., and Goldberg, M.E. 1985. Measurements of the true affinity constant in solution of antigen–antibody complexes by enzyme-linked immunosorbent assay. J. Immunol. Methods 77 305–319. [DOI] [PubMed] [Google Scholar]
- Giebel, L.B., Cass, R.T., Milligan, D.L., Young, D.C., Arze, R., and Johnson, C.R. 1995. Screening of cyclic peptide phage libraries identifies ligands that bind streptavidin with high affinities. Biochemistry 34 15430–15435. [DOI] [PubMed] [Google Scholar]
- Gunneriusson, E., Nord, K., Uhlen, M., and Nygren, P. 1999 Affinity maturation of a Taq DNA polymerase specific affibody by helix shuffling. Protein Eng. 12 873–878. [DOI] [PubMed]
- Hansson, M., Ringdahl, J., Robert, A., Power, U., Goetsch, L., Nguyen, T.N., Uhlen, M., Stahl, S., and Nygren, P.A. 1999. An in vitro selected binding protein (affibody) shows conformation-dependent recognition of the respiratory syncytial virus (RSV) G protein. Immunotechnology 4 237–252. [DOI] [PubMed] [Google Scholar]
- Harrison, P.M. and Arosio, P. 1996. The ferritins: Molecular properties, iron storage function and cellular regulation. Biochim. Biophys. Acta 1275 161–203. [DOI] [PubMed] [Google Scholar]
- Hempstead, P.D., Yewdall, S.J., Fernie, A.R., Lawson, D.M., Artymiuk, P.J., Rice, D.W., Ford, G.C., and Harrison, P.M. 1997. Comparison of the three-dimensional structures of recombinant human H and horse L ferritins at high resolution. J. Mol. Biol. 268 424–448. [DOI] [PubMed] [Google Scholar]
- Hoogenboom, H.R. 1997. Designing and optimizing library selection strategies for generating high-affinity antibodies. Trends Biotechnol. 15 62–70. [DOI] [PubMed] [Google Scholar]
- Hoogenboom, H.R., de Bruine, A.P., Hufton, S.E., Hoet, R.M., Arends, J.W., and Roovers, R.C. 1998. Antibody phage display technology and its applications. Immunotechnology 4 1–20. [DOI] [PubMed] [Google Scholar]
- Jacobson, R.H., Zhang, X.J., DuBose, R.F., and Matthews, B.W. 1994. Three-dimensional structure of β-galactosidase from E. coli. Nature 369 761–766. [DOI] [PubMed] [Google Scholar]
- Juan, S.-H. and Aust, S.D. 1998. Studies on the interaction between ferritin and ceruloplasmin. Arch. Biochem. Biophys. 355 56–62. [DOI] [PubMed] [Google Scholar]
- Katz, BA. 1995. Binding to protein targets of peptidic leads discovered by phage display: Crystal structures of streptavidin-bound linear and cyclic peptide ligands containing the HPQ sequence. Biochemistry 34 15421–15429. [DOI] [PubMed] [Google Scholar]
- Katz, B.A. and Cass, R.T. 1997. In crystals of complexes of streptavidin with peptide ligands containing the HPQ sequence the pKa of the peptide histidine is less than 3.0. J. Biol. Chem. 272 13220–13228. [DOI] [PubMed] [Google Scholar]
- Kay, B.K., Adey, N.B., He, Y.S., Manfredi, J.P., Mataragnon, A.H., and Fowlkes, D.M. 1993. An M13 phage library displaying random 38-amino-acid peptides as a source of novel sequences with affinity to selected targets. Gene 128 59–65. [DOI] [PubMed] [Google Scholar]
- Koide, A., Bailey, C.W., Huang, X., and Koide, S. 1998. The fibronectin type III domain as a scaffold for novel binding proteins. J. Mol. Biol. 284 1141–1151. [DOI] [PubMed] [Google Scholar]
- Legendre, D., Soumillion, P., and Fastrez, J. 1999. Engineering a regulatable enzyme for homogeneous immunoassays. Nat. Biotechnol. 17 67–72. [DOI] [PubMed] [Google Scholar]
- Lu, Z., Murray, K.S., Van Cleave, V., LaVallie, E.R., Stahl, M.L., and McCoy, J.M. 1995. Expression of thioredoxin random peptide libraries on the Escherichia coli cell surface as functional fusions to flagellin: A system designed for exploring protein–protein interactions. Biotechnology 13 366–372. [DOI] [PubMed] [Google Scholar]
- Markland, W., Ley, A.C., and Ladner, R.C. 1996. Iterative optimization of high-affinity protease inhibitors using phage display. Biochemistry 35 8045–8067. [DOI] [PubMed] [Google Scholar]
- Nord, K., Gunneriusson, E., Ringdahl, J., Stahl, S., Uhlen, M., and Nygren, P.A. 1997. Binding proteins selected from combinatorial libraries of an α-helical bacterial receptor domain. Nat Biotechnol. 15 772–777. [DOI] [PubMed] [Google Scholar]
- Norman, T.C., Smith, D.L., Sorger, P.K., Drees, B.L., O'Rourke, S.M., Hughes, T.R., Roberts, C.J., Friend, S.H., Fields, S., and Murray, A.W. 1999. Genetic selection of peptide inhibitors of biological pathways. Science 285 591–595. [DOI] [PubMed] [Google Scholar]
- Petrenko, V.A. and Smith, G.P. 2000. Phages from landscape libraries as substitute antibodies. Protein Eng. 13 589–592. [DOI] [PubMed] [Google Scholar]
- Roberts, B.L., Markland, W., Ley, A.C., Kent, R.B., White, D.W., Guterman, S.K., and Ladner, R.C. 1992. Directed evolution of a protein: Selection of potent neutrophil elastase inhibitors displayed on M13 fusion phage. Proc. Natl. Acad. Sci. 89 2429–2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottgen, P. and Collins, J. 1995. A human pancreatic secretory trypsin inhibitor presenting a hypervariable highly constrained epitope via monovalent phagemid display. Gene 164 243–250. [DOI] [PubMed] [Google Scholar]
- Skerra, A. 2000. Engineered protein scaffolds for molecular recognition. J. Mol. Recognit. 13 167–187. [DOI] [PubMed] [Google Scholar]
- Smith, G.P., Patel, S.U., Windass, J.D., Thornton, J.M., Winter, G., and Griffiths, A.D. 1998. Small binding proteins selected from a combinatorial repertoire of knottins displayed on phage. J. Mol. Biol. 277 317–332. [DOI] [PubMed] [Google Scholar]
- Soumillion, P., Jespers, L., Bouchet, M., Marchand-Brynaert, J., Winter, G., and Fastrez, J. 1994. Selection of β-lactamase on filamentous bacteriophage by catalytic activity. J. Mol. Biol. 237 415–422. [DOI] [PubMed] [Google Scholar]
- Torti, S.V. and Torti, F.M. 1998. Human H-kininogen is a ferritin binding protein. J. Biol. Chem. 273 13630–13635. [DOI] [PubMed] [Google Scholar]
- Wang, C.I., Yang, Q., and Craik, C.S. 1995. Isolation of a high affinity inhibitor of urokinase-type plasminogen activator by phage display of ecotin. J. Biol. Chem. 270 12250–12256. [DOI] [PubMed] [Google Scholar]
- Weber, P.C., Ohlendorf, D.H., Wendoloski, J.J., and Salemme, F.R. 1989. Structural origins of high-affinity biotin binding to streptavidin. Science 243 85–88. [DOI] [PubMed] [Google Scholar]