

Abstract
Hydrogen/deuterium (H/D) exchange in combination with electrospray ionization mass spectrometry and near-ultraviolet (UV) circular dichroism (CD) was used to study the conformational properties and thermal unfolding of Escherichia coli thioredoxin and its Cys32-alkylated derivatives in 1% acetic acid (pH 2.7). Thermal unfolding of oxidized (Oxi) and reduced (Red) -thioredoxin (TRX) and Cys-32-ethylglutathionyl (GS-ethyl-TRX) and Cys-32-ethylcysteinyl (Cys-ethyl-TRX), which are derivatives of Red-TRX, follow apparent EX1 kinetics as charge-state envelopes, H/D mass spectral exchange profiles, and near-UV CD appear to support a two-state folding/unfolding model. Minor mass peaks in the H/D exchange profiles and nonsuperimposable MS- and CD-derived melting curves, however, suggest the participation of unfolding intermediates leading to the conclusion that the two-state model is an oversimplification of the process. The relative stabilities as measured by melting temperatures by both CD and mass spectral charge states are, Oxi-TRX, GS-ethyl-TRX, Cys-ethyl-TRX, and Red-TRX. The introduction of the Cys-32-ethylglutathionyl group provides extra stabilization that results from additional hydrogen bonding interactions between the ethylglutathionyl group and the protein. Near-UV CD data show that the local environment near the active site is perturbed to almost an identical degree regardless of whether alkylation at Cys-32 is by the ethylglutathionyl group, or the smaller, nonhydrogen-bonding ethylcysteinyl group. Mass spectral data, however, indicate a tighter structure for GS-ethyl-TRX.
Keywords: Thioredoxin, heat denaturation, modified protein, hydrogen deuterium exchange, electrospray ionization mass spectrometry
Amide hydrogen isotope exchange has played an important role in answering questions about protein structures and folding/unfolding mechanisms (Miranker et al. 1996; Woodward et al. 1982). There are essentially two competing processes during which hydrogen exchange takes place (Smith et al. 1997). One involves internal or local fluctuations from the native state, and the other, subglobal or global unfolding. In either case, hydrogen bonds are broken and the amide hydrogens are exposed to solvent so that exchange can occur. The inception of electrospray ionization mass spectrometry (ESI-MS) (Fenn et al. 1989; Smith et al. 1990) at the beginning of the 1990s, has made MS an increasingly popular method for monitoring hydrogen isotope exchange. Hydrogen isotope exchange and MS now have played an important role in the analysis of conformations in TRX and some of its alkylation products (Kim et al. 2001a,b).
TRX is a small, 108 amino-acid residue protein (MW = 11,673.40 Da) with a redox-active site sequence -Cys-Gly-Pro-Cys- (Holmgren 1985; Holmgren and Björnstedt 1995). Oxi-TRX was reduced and modified at Cys-32 by the episulfonium ion derived from S-(2-chloroethyl)glutathione (Fig. 1 ▶) and S-(2-chloroethyl)cysteine (Humphreys et al. 1990; Erve et al. 1995). Vicinal dihaloethanes are manufactured in large quantities and because of their volatility, they become major environmental problems. These chemicals potentially have significant toxicological effects that are in part mediated by alkylation of glutathione, which in turn alkylates thioredoxin, other proteins, and nucleic acids via the episulfonium ion (Ozawa and Guengerich 1983; Humphreys et al. 1990). It can be hypothesized that the active site of proteins containing the "thioredoxin fold", -Cys-X-X-Cys-, i.e., thioredoxin, thioredoxin reductase, and protein disulfide isomerase, are in vivo targets for alkylation by glutathione conjugates.
Fig. 1.

Simulated structure of GS-ethyl-TRX derived from the crystal structure of Oxi-TRX (Kim et al. 2001b). The glutathionyl group is represented in the ball and stick format (sulfur in yellow, nitrogen in blue, carbon in grey, oxygen in red). The TRX protein is represented as ribbons (α-helices in red, β-sheets in yellow, and turns and loops in blue and grey). The dotted lines indicate two salt bridges and three hydrogen bonds induced by the ethylglutathionyl group.
When the conformational dynamics of Oxi-TRX were probed by H/D exchange during thermal unfolding by ESI-MS, two distinct populations of molecules differing in the numbers of incorporated deuteriums were observed after short incubation periods (Maier et al. 1999). The mass spectral peaks represent the population of folded and unfolded states; time-dependent variations of these peaks were used to estimate the unfolding rate constant.
Charge-state distributions in ESI mass spectra can be used to monitor the transition of a protein in solution from the native to the denatured state (Maier et al. 1999). Such experiments rely on the empirical observation that ESI mass spectra of unfolded proteins in solution indicate a higher charge state than the identical protein in the folded state (Katta and Chait 1991; Loo et al. 1991; Mirza et al. 1993; Konermann et al. 1997). The bimodal charge-state distributions that support a two-state unfolding mechanism were analyzed to calculate the melting temperature (Tm) of TRXs. In this paper, we report on the use of amide H/D exchange and ESI-MS to show how the modification of TRX affects thermal unfolding and conformations of the proteins.
Results
Equilibrium thermal denaturation and H/D exchange of TRXs
Thermal denaturation studies yielded information on structural changes when Red-TRX was alkylated at Cys-32 by ethylglutathionyl and ethylcysteinyl moieties. For this purpose, the charge-state distributions in ESI mass spectra were examined. Aqueous acetic acid was chosen as the solvent system as Hiraoki et al. found no signs of denaturation of TRX by proton NMR spectroscopy in this medium at pH 2.5 and 25°C (Hiraoki et al. 1988). At 1% acetic acid, a uniform charge-state distribution pattern was observed for the unmodified as well as the modified TRXs. Low pH and high temperatures showed a synergistic denaturing effect through a decrease in the melting temperature, Tm of the proteins, thereby allowing direct monitoring of thermal unfolding of TRX by mass spectrometric methods. It was demonstrated previously (Maier et al. 1999) that charge-state envelopes reflecting the folded and unfolded states in electrospray mass spectra can be used to estimate the melting temperature of Oxi-TRX. The charge-state envelopes for the folded (F) and unfolded (U) forms of Oxi-TRX are clearly different at three different temperatures (Fig. 2 ▶).
Fig. 2.

Electrospray ionization mass spectra of Oxi-TRX in 1% acetic acid at different temperatures: (A) 85°C, (B) 65°C, and (C) 25°C. Ion peaks at m/z 1668, 1460, and 1298 representing charge states 7+, 8+, and 9+, respectively, were attributed to the folded form, F. Ion peaks at m/z 1168, 1062, 973, 899, 835, and 779 representing charge states 10+ to 15+ were attributed to the unfolded form, U.
The changes in charge-state distributions as a function of temperature are evident in the denaturation transition curves (Fig. 3 ▶). F is the summation of the peak intensities of 7+ to 9+ charges. This may be an oversimplification, as many other factors besides concentration are known to play a significant role in determining ESI-MS response such as ion transmission efficiency, which depends on the charge state and an ion desorption efficiency, which depends on the protein three-dimensional geometry in solution. Nevertheless, one can use charge-state envelopes to provide a fairly accurate description of the overall conformational aspects of proteins. U is represented by the summation of 10+ to 15+ charge-state peak intensities. F/(F+U) provides an indication of the relative compactness of the protein. As the temperature is increased toward conditions that favor unfolding, the folded conformation initially changes very little. There may be increases in flexibility and localized conformational alterations, but the average structure is not changed. The protein unfolds completely within a narrow range of temperature, of about 10°C. The abruptness of the unfolding transition is indicative of a cooperative transition for thermal unfolding. Compared to the other TRX protein forms, F is highest for Oxi-TRX at room temperature. The Red-TRX has the least compact structure. The difference in F/(F+U) between Red- and Oxi-TRX is >0.2. GS-ethyl-TRX has an intermediate compactness, while Cys-ethyl-TRX and Red-TRX showed almost the same F/(F+U). The results suggest that the ethylglutathionyl group in GS-ethyl-TRX functions to make the structure of Red-TRX more compact. Thermal unfolding of the four TRXs (Oxi-, Red-, GS-ethyl-, and Cys-ethyl-TRX) in 1% acetic acid as estimated by the ESI charge-state envelopes (Fig. 3 ▶), shows that Oxi-TRX is the most stable (Tm = 67°C) while the other TRXs are substantially less stable. Red-TRX has the lowest Tm value (53°C). The difference in Tm between Oxi- and Red-TRX is about 15°C, which is in close agreement with previously reported data (∼10°C) from NMR and CD studies (Hiraoki et al. 1988). The alkylated product, GS-ethyl-TRX (Tm = 56°C) is slightly more stable than Red-TRX, while Cys-ethyl-TRX (Tm = 54°C) is about the same.
Fig. 3.

Heat denaturation curves of TRXs in 1% acetic acid deducted from the temperature-dependent, charge-state distributions obtained by electrospray ionization mass spectrometry. Tms for Oxi-, GS-ethyl-, Cys-ethyl-, and Red-TRX are 67, 56, 54, and 53°C, respectively.
Thermal denaturation of TRXs monitored by circular dichroism
CD spectra are used widely for detecting and measuring the various conformational components of proteins in solution. Bimodal CD indicates the presence of an equilibrium in solution between U and F structures in solution. Near-UV CD was used as a complementary probe to the MS-based approach, which allowed a comparison of changes in the environment of aromatic side chains of the TRXs with alteration of the charge-state distribution upon thermal denaturation. The temperature-dependent unfolding transition of Oxi-TRX was observed in the near-UV CD, which showed denaturation of the proteins as the temperature was raised (Fig. 4 ▶). The unfolding transitions based on the CD signal at 280 nm (Fig. 5 ▶) yielded melting temperatures that followed the same relative order observed by charge-state analysis (Fig. 3 ▶), but the values were lower by 3–5°C for Oxi-TRX and the alkylated TRXs, and 10°C for Red-TRX (Tm = 43°C). More importantly, the overall temperature dependence was significantly different. The CD spectra showed a gradual decrease in molar ellipticity with increasing temperatures outside of the melting region, while F/(F+U) values (Fig. 3 ▶) remained constant at temperatures above and below the melting region. At room temperature, Red-TRX showed intermediate CD signal intensities between Oxi-TRX and modified TRXs (Fig. 5 ▶), while GS-ethyl-TRX showed an intermediate F/(F+U) value in the charge-state spectrum (Fig. 3 ▶).
Fig. 4.

Near ultraviolet circular dichroism spectra of thermal denaturation of oxidized Escherichia coli thioredoxin (34 μM in 1% acetic acid).
Fig. 5.

Denaturation curves of Escherichia coli thioredoxins in 1% acetic acid obtained from the analyses of near ultraviolet circular dichroism spectra. Each data point is from the intensity measured at 280 nm. Tms for Oxi-, GS-ethyl-, Cys-ethyl-, and Red-TRX are 61, 53, 49, and 43°C, respectively.
Unfolding dynamics of TRXs probed by H/D exchange and ESI-MS
Conformational changes of TRXs during thermal denaturation were monitored by performing on-line H/D exchange-in experiments as a function of time and temperature. A continuous flow setup was used in which the solvent delivery line, the sample loop or reaction capillary, and the injection valve were immersed in a water bath and equilibrated at the desired temperature between 25 and 80°C (Maier et al. 1999). H/D exchange was initiated by a 50-fold dilution of a protein stock solution in 1% acetic acid into ice-cold 1% AcOD/D2O. The protein solution was injected into the sample loop, equilibrated at the desired temperature, and transferred to the ESI source. The reported incubation periods represent the time the protein spent in the sample loop exposed to H/D exchange conditions. The maximum H/D exchange reaction time depends on the volume of the sample loop and the flow rate. At a flow rate of 6 μL/min, the installed sample loop (33 μL) allowed a maximum reaction time of ∼6 min.
The thermal unfolding of small proteins often can be described by a two-state transition model (equation 1) in which F is represented as a relatively narrow distribution of structured conformations stabilized by hydrogen bonding, and U is represented as a heterogeneous distribution of less-compact conformations with no significant hydrogen bonding. Experimental rate constants k1 and k−1 for unfolding and refolding, respectively, and k2 for intrinsic isotope exchange of amide hydrogens in unstructured protein, are important in two rate-limiting processes for protein unfolding (Englander and Kallenbach 1984). When k−1>>k2, the unfolding/folding process occurs many times before the amide hydrogens in a particular unfolding region become completely deuterated. This limiting case is referred to as EX2 kinetics, where kex = k2(k1/k−1). When k2>>k−1, exchange occurs by an EX1 mechanism and kex = k1. When H/D exchange occurs by the EX2 mechanism, the mass spectrum shows a single mass peak that slowly shifts with increasing exchange-in time to higher masses. In contrast, two distinct mass peaks develop after a short exchange-in time if exchange occurs via the EX1 mechanism (Smith et al. 1997; Maier et al. 1999).
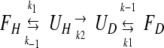 |
(1) |
Previously, it was shown that thermal unfolding of Oxi-TRX can be measured by online H/D exchange-in experiments as deuterium incorporation progresses from EX2- to EX1-type kinetics with a fairly clear demarkation somewhere in the range of 61–69°C (Maier et al. 1999). The H/D exchange evolution patterns between Oxi-TRXs and modified TRXs reflect the relative conformational stability between the different forms. First-order rate constants for unfolding can be determined when the EX1 mechanism applies.
In the charge-state distribution (Fig. 2 ▶), charge states +7, +8, and +9 were ascribed to the compact conformational state of TRX. The eightfold charged ion peak was used to make quantitative comparisons of the TRX ion peaks because it has the highest signal intensity. Peak evolution patterns of 8+ charged ion peaks at 50°C were significantly different for Oxi-TRXs and modified TRXs (Fig. 6 ▶). After 10 sec incubation, the 8+ charged ion peak of Oxi-TRX had m/z 1467.5, which indicated an incorporation of roughly 60 deuteriums (35% of the exchangeable hydrogens). These quickly incorporated deuteriums are believed to be in the side chains and terminal sites (71 sites); at pH 2–3, the exchange rates of side-chain hydrogens are much faster than those of amide hydrogens. Oxi-TRX was found to follow EX2-type kinetics in which the mass spectrum showed a single mass peak that gradually shifted with increasing exchange-in time to higher mass (Fig. 6 ▶). Maximum deuteration as reflected in the mass peaks with dashed lines were obtained from the denatured proteins that were incubated in 1% AcOD/D2O at 80°C for 1 h. In contrast, two distinct mass peaks developed after a short exchange-in period for Red-TRX and its alkylated adducts indicating exchange approaches EX1-type kinetics (Fig. 6 ▶). The higher mass peak is very close to the peak of the corresponding fully deuterated proteins. The effect of incubation temperature at comparable times (105 sec) on the EX1/EX2 kinetic limits is clearly seen for the four TRXs (Fig. 7 ▶) with their different Tms. Intermediate peaks suggesting different levels of deuteration also are visible, which may indicate the presence of partially unfolded species. Thus, exchange cannot be described solely as correlated, but rather EX2 and EX1 exchange is observed concomitantly. This also can be deduced from the difference in the mass of the deuterated species after unfolding with that of the fully deuterated species (Fig. 6 ▶).
Fig. 6.

Evolution of the eightfold charged ion peaks of Escherichia coli thioredoxins during on-line hydrogen/deuterium (H/D) exchange-in experiments in 1% AcOD/D2O at 50°C. Time points refer to H/D exchange periods. Dashed peaks were obtained by mass spectral measurements of the fully deuterated proteins.
Fig. 7.

Temperature-dependent alteration of the hydorgen/deuterium exchange mass profiles. Comparison of the eightfold charged ion peak of Escherichia coli thioredoxins at different temperatures after an exchange-in period of 105±10 sec in 1% AcOD/D2O.
The rate constants for unfolding (k1) at different temperatures in which EX1 kinetics applies were calculated from f/(f+u) = Aexp(−k1t) where f and u are the intensities of the lower and higher mass peak, respectively. The results show that Cys-ethyl-TRX and Red-TRX have similar rates of H/D exchange at 50 and 55°C, while the rate of exchange for GS-ethyl-TRX is 0.3–0.4 times as fast at these temperatures (Table 1). At temperatures above the Tms, the unfolding rates converge. Alkylation of reduced thioredoxin at Cys-32 by the ethylglutathionyl moiety provides additional stabilization that likely results from extra hydrogen bonding. Direct evidence for such hydrogen bonding was obtained by site-specific amide H/D exchange experiments, supported by energy-minimized conformational preferences by AMBER force field (Kim et al. 2001b).
Table 1.
Unfolding rate constants k1(min−1) of TRXs in 1% acetic acid solution
| T (°C) | Oxi-TRX | Red-TRX | GS-ethyl-TRX | Cys-ethyl-TRX |
| 50 | —a | 0.22 | 0.09 | 0.21 |
| 55 | —a | 1.3 | 0.48 | 1.2 |
| 60 | 0.1 | 2.5 | 2.1 | —b |
| 80 | 2.1c | — | — | — |
a EX2 kinetics.
b Only the higher mass peak from unfolded state of protein was observed.
c Measured in 2% acetic acid-d solution (Maier et al. 1999).
Kinetic H/D exchange-in
Time-dependent offline H/D exchange-in experiments at room temperature show different degrees of solvent accessibility for the various chemical forms of thioredoxin (Fig. 8 ▶). The H/D exchange-in behavior of the TRXs monitored at 25°C in 1% acetic acid (pH 2.6), showed that Oxi-TRX is most resistant to exchange with >60% of the protons remaining after 3 h. There were ∼12 more deuteriums (7%) in Red-TRX than in Oxi-TRX after 45 min, and this difference did not change significantly on incubation up to 3 h (Fig. 8A ▶). After back-exchange, the difference was only 5 deuteriums (3%) (Fig. 8B ▶), the difference in the numbers of deuteriums on peptide amide sites between the oxidized and reduced proteins. These results are comparative and consistent with those in F/(F+U) (Fig. 3 ▶), in which a significant difference in compactness was observed between Oxi- and Red-TRX when T<Tm.1H-NMR studies showed that the overall structures of Oxi- and Red-TRX are not very different, but the 14-membered disulfide loop in the oxidized form confers stability and reduces the exchange rate in this region (Hiraoki et al. 1988; Jeng and Dyson 1995).
Fig. 8.

Deuterium incorporation in Escherichia coli thioredoxins with time (A) total deuterium content and (B) after back exchange of side-chain deuteriums.
The amount of deuterium uptake by GS-ethyl-TRX and Cys-ethyl-TRX was closer to that of Oxi-TRX than to Red-TRX. This result suggests that the modifications in GS-and Cys-ethyl-TRX prevent H/D exchange of some hydrogens that would be unprotected in Red-TRX. Although nearly imperceptible (Fig. 8A ▶), there appears to have been a slight difference in deuterium uptake between Oxi-TRX, GS-ethyl-TRX, and Cys-ethyl-TRX. GS-ethyl-TRX showed slightly more resistance to exchange than Cys-ethyl-TRX as indeed expected. A tighter structure for GS-ethyl-TRX results from the presence of salt bridges between carboxylates of the γ-Glu and Gly of the glutathione and the guanidinium of Arg-73 and ɛ-amino group of Lys-90 of the protein (Fig. 1 ▶). The stabilizing effect of the salt bridges allows the induction of additional hydrogen bonding between the glutathionyl group carbonyl oxygens and the amide protons of TRX residues Ile-75 and Ala-93. Salt bridges are not possible in Cys-ethyl-TRX and similar hydrogen bonding was not detected (Kim et al. 2001b).
Discussion
The analysis of charge-state distributions showed that the relative compactness, i.e. F/(F+U), of GS-ethyl-TRX at T<Tm was constant and closer to that of Oxi-TRX than to Red-TRX, while the melting temperature for GS-ethyl-TRX was closer to those for Red-TRX and Cys-ethyl-TRX (Fig. 3 ▶). The Tm values for Oxi-TRX and the alkylated TRXs obtained by near-UV CD were similar to those obtained by charge-state analysis, but the value for Red-TRX was 10°C lower (Fig. 5 ▶). There is no ready explanation for this large difference, but it is noted that the slopes for the pre- and posttransition baselines for Red-TRX were different, indicating that the optical properties of the aromatics were not the same. The temperature-dependent unfolding profiles obtained by the two analytical approaches generally were quite different (Fig. 3 ▶ and Fig. 5 ▶). Charge-state envelopes reflect the overall conformational aspects of proteins, whereas near-UV CD provides information on localized environmental conditions. Thus, aromatic side chains of Trp and Tyr contribute to the CD absorption at 280 nm. In TRX, there are two Trps in the active-site region and two Tyrs in the second and third α helices (Fig. 1 ▶). Gradual decrease in molar ellipticity as temperatures approach Tm results from minor changes in the asymmetric environment near the side chains of Trp and Tyr and are caused by increased rotational and vibrational motions. A precipitous and complete change in the asymmetric environment of the aromatic side chains occurs upon denaturation at Tm. On the other hand, ESI-MS data showed no apparent change in the structural compactness of the protein before reaching Tm, as low energy motions were not sufficient to change the overall protein conformation and produce more charge sites.
The significant difference in CD signals between Oxi-, Red- and modified TRXs in the range T<Tm (Fig. 5 ▶) can be ascribed to environmental differences experienced by Trp side chains. The modified Cys-32 site is adjacent to Trp-31 and near the Trp-28 residue. The presence of the alkyl group on Cys-32 has a significant effect on the CD signal because of environmental changes around Trp-31 and Trp-28. A molecular model of GS-ethyl-TRX obtained by AMBER force-field simulation places the glutathione chain on a broad surface formed by two loop structures in the protein (Fig. 1 ▶; Kim et al. 2001b). The fact that the temperature-dependent profiles of GS-ethyl- and Cys-ethyl-TRX are nearly superimposable at temperatures above and below Tm, suggests there is a common effect on the CD response regardless of the alkylation at Cys-32. On the other hand, mass-spectral data showed that the charge-state envelopes for Cys-ethyl-TRX and Red-TRX at T<Tm are similar and indicative of a relatively large population of molecules with structural "openness" as compared to GS-ethyl-TRX, which appears to consist of a large population of compact structures. A possibility that needs to be considered is that the presence of two salt bridges removes possible charge sites from GS-ethyl-TRX that could lead to a higher F/(F+U) value. But, if this were the major reason for the difference, then the F/(F+U) values for Oxi-TRX and Red-TRX should be closer, as the relevant Arg-73 and Lys-90 residues are far removed from the active site region and there should be no difference in their availability for protonation.
The presence of two salt bridges between carboxyl groups of the ethylglutathionyl group and basic sites on the side chains of the protein's Arg-73 and Lys-90 (Fig. 1 ▶; Kim et al. 2001b) imposes a structural constraint that allows for hydrogen bonding between the carbonyl oxygens of glutathione and the amide hydrogens of Ile-75 and Ala-93 in the protein. The ethylcysteinyl group in Cys-ethyl-TRX cannot form salt brides; thus it rotates freely and induces no extra hydrogen bonds. The absence of a salt bridge in Cys-ethyl-TRX is probably responsible for the "openness" and similarity of the F/(F+U) value with that of Red-TRX when T<Tm (Fig. 3 ▶). After melting, GS-ethyl-TRX loses this additional structural bridging and its F/(F+U) value became similar to that of Red-TRX and Cys-ethyl-TRX. In contrast, Oxi-TRX with its intact disulfide retained some compactness after melting as reflected in the higher F/(F+U). The increased stability of GS-ethyl-TRX indicated by the slightly higher (∼3°C) Tm and the larger unfolding rate constant (Table 1) probably also resulted from the presence of salt bridges and extra hydrogen bonding from the ethylglutathionyl group.
All results for the thermal denaturation of TRXs point to cooperative unfolding through a two-state model (Robertson and Baldwin 1991; Chamberlain et al. 1999). Evidence from previous near-UV CD experiments also showed that thermal denaturation of Oxi-TRX is reversible (Maier et al. 1999). There is a transition from EX2- to EX1-type kinetics for thermal unfolding of proteins, which is reflected in peak broadening before Tm is reached and only when T≥Tm are two separated isotopically enriched peaks clearly visible in the mass spectra (Yi and Baker 1996; Maier et al. 1999). In this case, when H/D exchange was performed at 50°C, Oxi-TRX clearly underwent EX2-type kinetics, while two peaks evolved for Red-TRX, GS-ethyl-TRX, and Cys-ethyl-TRX (Fig. 6 ▶). These were not unexpected results, as the temperature (50°C) at which the isotope exchange was performed was well below the Tm of Oxi-TRX and equal to or slightly below the Tm values of the other TRXs. Peak broadening is evident in these profiles (Fig. 6 ▶) indicating that exchange took place within the EX2/EX1 kinetic limits (Arrington and Robertson 2000).
The unusual CD melting profile and Tm observed for Red-TRX (Fig. 5 ▶) may just be an extreme case of what occurs with the other TRXs, all of which showed lower Tms from those obtained by charge-state envelopes. Generally, when melting profiles are not superimposable, transient intermediates are suspected. The unfolding profiles obtained by H/D exchange (Fig. 6 ▶) showed small peaks between the two major mass peaks that suggest participation of unfolding intermediates. When H/D exchange was carried out at T>Tm, the intermediate peaks were not prominent, although they did not disappear entirely. The 8+ charged ion peak of Oxi-TRX incubated under H/D-exchange conditions at 82°C (Tm : 65°C), for example, showed two well-resolved mass peaks representing folded and unfolded conformers (Maier et al. 1999). Small intermediate peaks also were observed for Red- and alkylated-TRXs even when the exchange experiments were conducted at 60°C or well above the Tms (Fig. 7 ▶). The evidence suggests that unfolding of these proteins may indeed involve transient intermediates. Previous NMR H/D exchange studies of Oxi- and Red-TRX under thermal denaturing conditions also indicated that partially unfolded structures exist (Hiraoki et al. 1988; Jeng and Dyson 1995). Specifically, there is a group of slow-exchanging protons with similar rate constants in the central and protected part of the β-sheet (β2). The process of simultaneous breaking of a group of hydrogen bonds has been referred to as "regional melting" (Kosiakoff 1982).
The effects of chemical denaturants on protein conformations has received somewhat more attention than thermal denaturation. Oxidized and reduced cytochrome c, for example, are believed to unfold through sequential intermediates under varying denaturant concentrations (Bai et al. 1995). These partially unfolded intermediates were found under equilibrium native conditions (pD 7, 30°C). Miranker and coworkers (1993) reported the participation of transient protein intermediates during refolding of hen lysozyme, which were detected by pulse-labeling H/D exchange and analysis by NMR and MS. These intermediates were detected in the nonequilibrium refolding process, which was thought to involve cooperative folding of various protein domains.
Conclusions
Site-specific amide H/D exchange experiments have revealed that there is little structural difference between Oxi- and Red-TRX except in or near the active site (Jeng and Dyson 1995; Kim et al. 2001a,b). However, global exchange in 1% acetic acid (pH 2.7) under equilibrium as well as nonequilibrium conditions showed that there are significant differences in thermal stability, unfolding kinetics, and solvent accessibility for Oxi-TRX, Red-TRX, and active-site Cys-32 alkylated TRXs. Thermal denaturation approached the EX1 kinetic limit, but MS and CD results indicated that none of the TRXs underwent unambiguous correlated unfolding. Relative to the melting temperatures at pH 7.8 (Hiraoki et al. 1988), the acidic conditions under which thermal unfolding was performed here resulted in significantly lower Tms. The acidic conditions also may have something to do with the fact that thermal unfolding did not follow simple EX1 kinetics.
Redox-active TRX is a required subunit in certain bacteriophage systems, including T7 DNA polymerase and filamentous phage assembly, where Red-TRX is functional but Oxi-TRX is not (Adler and Modrich 1983; Russel and Model 1985). The major determining factor in the bioactivity of Red-TRX in comparison to that of Oxi-TRX does not appear to be due to the presence of free thiol groups, since mutants of TRX where one or both cysteines were replaced by Ala or Ser were also partly functional (Russel and Model 1986). These observations suggest that the overall structure of the protein, and not the chemical properties of the cysteine residues, is important to the regulation of this particular biological activity. In conclusion studies by H/D exchange coupled with analysis by MS are clearly useful and complementary to other biophysical methods for characterizing structural differences between protein forms that may relate to different biological activities.
Materials and methods
Analyses of bimodal charge-state distributions
Oxi-TRX (Promega) was reduced by Tris(2-carboxyethyl)phosphine hydrochloride (TCEP.HCl, Pierce) and alkylated by the episulfonium ion derived from CEG and CEC (Erve et al. 1995). Oxi-TRX, GS-ethyl-TRX, and Cys-ethyl-TRX (0.2 μg/μL, 0.017 mM) were dissolved in 1% AcOH/H2O (pH 2.6). Red-TRX solution (0.2 μg/μL, 0.017 mM) was obtained by diluting Red-TRX stock solution (0.85 mM in 8 mM TCEP/1% AcOH/H2O) 50-fold with 1% AcOH/H2O. Protein solutions (50 μL) were introduced into a Rheodyne injection valve fitted with a 40-μL sample loop. The solvent delivery line, the sample loop, and the injection valve were immersed in a water bath and equilibrated at the desired temperature between 25 and 80°C. After the protein solutions were incubated for 2 min in the sample loop, they were transferred into the ESI source. The solvent flow (0.5 mL/h) was maintained with a syringe pump and a solvent mixture of 1% AcOH/H2O. F/(F+U) was determined as a function of temperature.
Online H/D exchange/ESI-MS experiment
Oxi-TRX, GS-ethyl-TRX, and Cys-ethyl-TRX (10 μg/μL, 0.85 mM) were dissolved in 1% AcOH/H2O (pH 2.6). Red-TRX solution (10 μg/μL, 0.85 mM) was obtained by dissolving Oxi-TRX in 8 mM TCEP/1% AcOH/H2O. TRX solutions were allowed to equilibrate for 100 min at 25°C (equilibration solution). The equilibration solutions were diluted 50-fold with 1% AcOD/D2O (labeling solution) at 0°C. Immediately, 50 μL of diluted solution was introduced into a Rheodyne injection valve fitted with a 40-μL sample loop by precooled syringe. The sample loop (peek tubing i.d. 0.01 in., o.d. 1/16 in., 65 cm) was used as a reaction capillary. The solvent delivery line, the sample loop, and the injection valve were immersed in a water bath and equilibrated at the desired temperature between 25 and 80°C. As soon as the sample was injected into the sample loop, the protein solution in the sample loop was transferred into the ESI source via a fused silica capillary (i.d. 0.075 mm, 100 cm), which was immersed in an ice-water bath to quench H/D exchange and to facilitate refolding of thermally unfolded TRX. The solvent flow (0.5 mL/h) was maintained with a syringe pump and a solvent mixture of 1% AcOD/D2O. The time necessary for the transport of the protein from the sample loop to the ESI needle was ∼60 ± 5 sec. The incubation periods reported represent the time that the protein sample spent in the sample loop.
Offline H/D exchange/ESI-MS experiment
Oxi-TRX, GS-ethyl-TRX, and Cys-ethyl-TRX (10 μg/μL, 0.85 mM) were dissolved in 1% AcOH/H2O (pH 2.6). Red-TRX solution (10 μg/μL, 0.85 mM) was obtained by dissolving Oxi-TRX in 8 mM TCEP/1% AcOH/H2O. TRX solutions were allowed to equilibrate for 100 min at 25°C (equilibration solution). H/D exchange was initiated by diluting the equilibration solution 50-fold with 1% AcOD/D2O at 25°C (labeling solution). At each time point, 5 μL (0.43 nmol) of labeling solution was introduced into an injection valve fitted with a 5-μL sample loop by a precooled syringe. The solvent delivery line, the sample loop, and the injection valve were immersed in an ice-water bath to minimize H/D exchange of all exchangeable hydrogens during infusion into the mass spectrometer. The solvent flow (0.3 mL/h) was maintained with a syringe pump and a solvent mixture of 1% AcOD/D2O. Back-exchanged results of side-chain deuterons were obtained by using 1% AcOH/H2O as a delivery solvent mixture. The percentage of deuterium incorporation was calculated using equation 2. Molecular weights of deuterated proteins were calculated by deconvolution with multiply charged ions from 7+ to 9+ because they showed strong peak intensities.
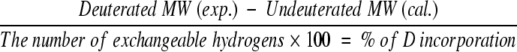 |
(2) |
Mass spectrometry
All ESI-MS data were acquired on a Perkin-Elmer Sciex API III triple quadrupole mass spectrometer (Thornhill) with pneumatically assisted electrospray source. Air was used as the nebulizer gas and nitrogen as curtain gas. The ionspray voltage was at 4700 V, and the orifice voltage was at 80 V. Mass spectral data were acquired in the range of m/z 1250 to 1800 for the online and offline H/D exchange experiments and m/z 700 to 1800 for the analyses of charge-state distributions. All mass spectra reported here were recorded in positive ion mode.
Circular dichroism
All CD spectra were recorded on a Jasco J720 spectropolarimeter equipped with a home-built, thermoelectrically controlled cell holder. Near-UV CD spectra of TRXs (34.1 μM, 1% AcOH/H2O) were measured in the near-UV range (250–360 nm) in cylindrical 1-cm path length quartz cuvettes. The temperature control of the sample cell was maintained by circulating water from a thermostatically controlled water bath through a built-in water jacket sleeve that is mounted around the CD cell. Temperature readings were measured online with a thermocouple element glued to the outside of the cuvette wall. Cuvettes were allowed a 5-min equilibration period after reaching the desired operating temperature before scanning. The temperature was manually increased from 25 to 80°C. Equilibrium thermal denaturation of TRXs was monitored at 280 nm. All spectra were smoothed with software provided with the instrument and base-line corrected for the CD signal in the absence of protein.
The mean residue molar ellipticity [θ]λ at a given wavelength was calculated according to equation 3:
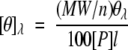 |
(3) |
where θλ is the observed ellipticity in degrees, MW is the molecular weight of TRXs, n is the number of residues (n = 108), [P] is the protein concentration in mol dm−3, and l is the path length in centimeters.
Acknowledgments
This work was supported by the NIH (NIEHS grants ES00040 and ES00210).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
AMBER, assisted model building with energy refinement
CD, circular dichroism
CEC, S-(2-chloroethyl)cystein
CEG, S-(2-chloroethyl)glutathione
CID, collision-induced dissociation
F, folded state
H/D, hydrogen/deuterium
HPLC, high-performance liquid chromatography
MS, mass spectrometry
NMR, nuclear magnetic resonance
TCEP, Tris(2-carboxyethyl)phosphine
Tm, melting temperature
TRX, Escherichia coli thioredoxin
Oxi-, oxidized-
Red-, reduced-
GS-ethyl-, Cys-32-ethylglutathionylated-
Cys-ethyl-, Cys-32-ethylcysteinylated-
U, unfolded state
UV, ultraviolet
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.3140102.
References
- Adler, S. and Modrich, P. 1983. T7-induced DNA polymerase. Requirement for thioredoxin sulfhydryl groups. J. Biol. Chem. 258 6956–6962. [PubMed] [Google Scholar]
- Arrington, C.B. and Robertson, A.D. 2000. Correlated motions in native proteins from MS analysis of NH exchange: Evidence for a manifold of unfolding reactions in ovomucoid third domain. J. Mol. Biol. 300 221–232. [DOI] [PubMed] [Google Scholar]
- Bai, Y., Sosnick, T.R., Mayne, L., and Englander, S.W. 1995. Protein folding intermediates: Native-state hydrogen exchange. Science 269 192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain, A.K., Fischer, K.F., Reardon, D., Handel, T.M., and Marqusee, S. 1999. Folding of an isolated ribonuclease H core fragment. Protein Sci. 8 2251–2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englander, S.W. and Kallenbach N.R. 1984. Hydrogen exchange and structural dynamics of proteins and nucleic acids. Quart. Rev. Biophys. 16 521–655. [DOI] [PubMed] [Google Scholar]
- Erve, J.C.L., Barofsky, E., Barofsky, D.F., Deinzer, M.L., and Reed, D.J. 1995. Alkylation of E. coli thioredoxin by S-(2-chloroethyl)glutathione and identification of the adduct on the active site Cys-32 by mass spectrometry. Chem. Res. Toxicol. 8 934–941. [DOI] [PubMed] [Google Scholar]
- Fenn, J.B., Mann, M., Meng, C.K., Wong, S.K., and Whitehouse, C.M. 1989. ESI of large biomolecules. Science 246 64–71. [DOI] [PubMed] [Google Scholar]
- Hiraoki, T., Brown, S.B., Stevenson, K.J., and Vogel, H.J. 1988. Structural comparison between oxidized and reduced E. coli thioredoxin. Biochemistry 27 5000–5008. [DOI] [PubMed] [Google Scholar]
- Holmgren, A. 1985. Thioredoxin. Annu. Rev. Biochem. 54 237–271. [DOI] [PubMed] [Google Scholar]
- Holmgren, A. and Björnstedt, M. 1995. Thioredoxin and thioredoxin reductase. Methods Enzymol. 252 199–208. [DOI] [PubMed] [Google Scholar]
- Humphreys, W.G., Kim, D.-H., Cmarik, J.L., Shimada, T., and Guengerich, F.P. 1990. Comparison of the DNA-alkylating properties and mutagenic responses of a series of S-(2-haloethyl)-substituted cysteine and glutathione derivatives. Biochemistry 29 10342–10350. [DOI] [PubMed] [Google Scholar]
- Jeng, M.-F. and Dyson, H.J. 1995. Comparison of the hydrogen exchange behavior of reduced and oxidized E. coli thioredoxin. Biochemistry 34 611–619. [DOI] [PubMed] [Google Scholar]
- Katta, V. and Chait, B.T. 1991. Conformational changes in proteins probed by hydrogen-exchange electrospray-ionization mass spectrometry. Rapid Commun. Mass Spectrom. 5 214–217. [DOI] [PubMed] [Google Scholar]
- Kim, M.-Y., Maier, C.S., Reed, D.J., and Deinzer, M.L. 2001a. Site-specific amide hydrogen/deuterium exchange in E. coli thioredoxins measured by electrospray ionization mass spectroscopy. J. Am. Chem. Soc. 123 9860–9866. [DOI] [PubMed] [Google Scholar]
- Kim, M.-Y., Maier, C.S., Reed, D.J., Shing Ho, P., and Deinzer, M.L. 2001b. Intramolecular interactions in chemically modified E. coli thioredoxin monitored by H/D exchange and electrospray ionization mass spectrometry. Biochemistry 40 14413–14421. [DOI] [PubMed] [Google Scholar]
- Konermann, L., Collings, B.A., and Douglas, D.J. 1997. Conformational changes in proteins probed by hydrogen-exchange electrospray ionization mass spectrometry. Biochemistry 36 5554–5559. [DOI] [PubMed] [Google Scholar]
- Kossiakoff, A.A. 1982. Protein dynamics investigated by the neutron diffraction-hydrogen exchange technique. Nature 296 713–721. [DOI] [PubMed] [Google Scholar]
- Loo, J.A., Loo, R.R., Udseth, H.R., Edmonds, C.G., and Smith, R.D. 1991. Solvent-induced conformational changes of polypeptides probed by electrospray-ionization mass spectrometry. Rapid Commun. Mass Spectrom. 5 101–105. [DOI] [PubMed] [Google Scholar]
- Maier, C.S., Schimerlik, M.I., and Deinzer, M.L. 1999. Thermal denaturation of E. coli thioredoxin studied by H/D exchange and electrospray ionization mass spectrometry: Monitoring a two-state protein unfolding transition. Biochemistry 38 1136–1143. [DOI] [PubMed] [Google Scholar]
- Miranker, A., Robinson, C.V., Radford, S.E., Aplin, R.T., and Dobson, C.M. 1993. Detection of transient protein folding populations by MS. Science 262 896–900. [DOI] [PubMed] [Google Scholar]
- Miranker, A., Robinson, C.V., Radford, S.E., and Dobson, C.M. 1996. Investigation of protein folding by mass spectrometry. FACEB J. 10 93–101. [DOI] [PubMed] [Google Scholar]
- Mirza, U.A., Cohen, S.L., and Chait, B.T. 1993. Heat-induced conformational changes in proteins studied by electrospray ionization mass spectrometry. Anal. Chem. 65 1–6. [DOI] [PubMed] [Google Scholar]
- Ozawa, N. and Guengerich, F.P. 1983. Evidence for the formation of an S-[2-(N7-guanyl)ethyl]glutathione adduct in glutathione mediated binding of the carcinogen 1,2-dibromoethane to DNA. Proc. Natl. Acad. Sci. 80 5266–5270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson, A.D. and Baldwin, R.L. 1991. Hydrogen exchange in thermally denatured ribonuclease A. Biochemistry 30 9907–9914. [DOI] [PubMed] [Google Scholar]
- Russel, M. and Model, P. 1985. Thioredoxin is required for filamentous phage assembly. Proc. Natl. Acad. Sci. 82 29–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russel, M. and Model, P. 1986. The role of thioredoxin in filamentous phage assembly. J. Biol. Chem. 261 14997–15005. [PubMed] [Google Scholar]
- Smith, D.L., Deng, Y., and Zhang, Z. 1997. Probing the non-covalent structure of proteins by amide hydrogen exchange and mass spectrometry. J. Mass Spectrom. 32 135–146. [DOI] [PubMed] [Google Scholar]
- Smith, R.D., Loo, J.A., Edmonds, C.G., Barinaga, C.J., and Udseth, H.R. 1990. Electrospray ionization. Anal. Chem. 62 882–899. [DOI] [PubMed] [Google Scholar]
- Woodward, C., Simon, I., and Tüchsen, E. 1982. Hydrogen exchange and the dynamic structure of proteins. Mol. Cell. Biochem. 48 135–160. [DOI] [PubMed] [Google Scholar]
- Yi, Q. and Baker, D. 1996. Direct evidence for a two-state protein unfolding transition from hydrogen-deuterium exchange, mass spectrometry, and NMR. Protein Sci. 5 1060–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]