

Abstract
Dihydrofolate reductase (DHFR) catalyzes the nicotinamide adenine dinucleotide phosphate (NADPH)-dependent reduction of 7,8-dihydrofolate (H2F) to 5,6,7,8-tetrahydrofolate (H4F). Because of the absence of any ionizable group in the vicinity of N5 of dihydrofolate it has been proposed that N5 could be protonated directly by a water molecule at the active site in the ternary complex of the Escherichia coli enzyme with cofactor and substrate. However, in the X-ray structures representing the Michaelis complex of the E. coli enzyme, a water molecule has never been observed in a position that could allow protonation of N5. In fact, the side chain of Met 20 blocks access to N5. Energy minimization reported here revealed that water could be placed in hydrogen bonding distance of N5 with only minor conformational changes. The r.m.s. deviation between the conformation of the M20 loop observed in the crystal structures of the ternary complexes and the conformation adopted after energy minimization was only 0.79 Å. We performed molecular dynamics simulations to determine the accessibility by water of the active site of the Michaelis complex of DHFR. Water could access N5 relatively freely after an equilibration time of approximately 300 psec during which the side chain of Met 20 blocked water access. Protonation of N5 did not increase the accessibility by water. Surprisingly the number of near-attack conformations, in which the distance between the pro-R hydrogen of NADPH and C6 of dihydrofolate was less than 3.5 Å and the angle between C4 and the pro-R hydrogen of NADPH and C6 of dihydrofolate was greater than 120 degrees, did not increase after protonation. However, when the hydride was transferred from NADPH to C6 of dihydrofolate before protonation, the side chain of Met 20 moved away from N5 after approximately 100 psec thereby providing water access. The average time during which water was found in hydrogen bonding distance to N5 was significantly increased. These results suggest that hydride transfer might occur early to midway through the reaction followed by protonation. Such a mechanism is supported by the very close contact between C4 of NADP+ and C6 of folate observed in the crystal structures of the ternary enzyme complexes, when the M20 loop is in its closed conformation.
Keywords: Enzyme catalysis, active site, alternative mechanisms, molecular dynamics
Dihydrofolate reductase (DHFR) is a ubiquitous enzyme that catalyzes the reduction of 7,8-dihydrofolate (H2F) to 5,6,7,8-tetrahydrofolate (H4F) using NADPH as a cofactor (Scheme 1 ▶). The reaction occurs by hydride transfer of the pro-R hydrogen of NADPH to C6 of H2F and protonation of N5 (Charlton et al. 1979). H4F is an active form of the vitamin folic acid and a precursor of cofactors required for the production of purines, pyrimidines, and several amino acids (Blakley 1984). Therefore, DHFR is an essential enzyme in these biosynthetic pathways and as such a target for antitumor and antimicrobial drugs such as methotrexate and trimethoprim, respectively.
Scheme 1.

All structurally determined, chromosomally encoded DHFRs are single domain proteins containing an eight-stranded mixed β-sheet, which is flanked by two α-helices on either side (Fig. 1 ▶) (Sawaya and Kraut 1997; Feeney 2000). DHFRs are organized into two subdomains, the adenosine-binding domain, which binds the adenosine portion of NADPH, and the loop domain, which is dominated by three loops, namely the M20 loop, the βF–βG loop, and the βG–βH loop. The space between these subdomains forms the active site. Access to the active site in the bacterial enzymes relies on the conformation of the largely conserved M20 loop, which is situated between the first β-sheet and the first α-helix and consists of amino acids 14 to 24. From crystallographic studies this loop is known to adopt three distinct conformations, namely open, closed, and occluded (Bystroff and Kraut 1991; Sawaya and Kraut 1997). Movement of the M20 loop between these three conformations is thought to play a critical role during the catalytic cycle of DHFR (Li et al. 1992; Falzone et al. 1994; Miller and Benkovic 1998; Radkiewicz and Brooks 2000).
Fig. 1.

Representations of structures of the ternary complexes of E. coli DHFR with NADPH and folate with the M20 loop in the open (A) and closed (B) conformation (Sawaya and Kraut 1997). Met 20 is indicated, which in the open complex points away from the active site, but blocks access to N5 of the substrate in the closed form. Residues Asp 27 and Trp 22 together with substrate provide a binding site for water.
The reaction catalyzed by Escherichia coli DHFR goes through five intermediates at physiological concentrations of substrate and cofactor (Fierke et al. 1987). Rapid hydride transfer occurs in the Michaelis complex (DHFR • H2F • NADPH) (Sawaya and Kraut 1997) to yield the product complex (E • H4F • NADP+). Subsequent loss of the cofactor produces the binary E • H4F complex. NADPH then binds to this binary complex before the product is released in the rate-determining step of the catalytic cycle before binding of the substrate to reestablish the Michaelis complex.
The analysis of the data obtained from a large number of X-ray structures analogous to the five intermediate states in the catalytic cycle revealed that the M20 loop is predominantly in the closed conformation (Fig. 1B ▶) (Sawaya and Kraut 1997). In the closed conformation, the central part of the loop forms part of a short antiparallel sheet and type III′ hairpin, which seals the active site and mediates contacts with the nicotinamide part of the cofactor.
In contrast to the closed conformation, the occluded conformation, in which the central region of the M20 loop forms a 310-helix, inhibits cofactor binding by occluding the nicotinamide-binding pocket. The change from the closed to the occluded conformation has been suggested to occur by way of the intermediate open conformation (Fig. 1A ▶) (Sawaya and Kraut 1997).
Despite the wealth of structural information and the elucidation of the complete kinetic scheme for the reaction catalyzed by the E. coli enzyme (Fierke et al. 1987), many of the key chemical aspects remain unresolved. The transfer of a hydride ion from NADPH to C6 of H2F in the enzyme's active site requires proton transfer to N5. The source of this proton is unclear as in all forms of DHFR there is only one ionizable residue in the active site, either an Asp in bacterial DHFRs or a Glu in vertebrate DHFRs. Examination of crystal structures revealed that this residue is always hydrogen bonded to the amino group on C2 and N2 of H2F >5 Å away from N5 (Fig. 2 ▶). Recent evidence from NMR studies of DHFR from Lactobacillus casei indicated that the active site aspartate acted as a proton acceptor (Casarotto et al. 1999). Crystallography (Lee et al. 1996), Raman spectroscopy (Chen et al. 1994), and computational analyses (Cannon et al. 1997; Cummins and Gready 2001) support a keto-enol tautomerization of the pterin substrate. The enol form is stabilized as a consequence of the expulsion of most of the water molecules on substrate binding leading to a significantly reduced dielectric environment and an increased pKa of N5, thereby making protonation feasible (Miller and Benkovic 1998).
Fig. 2.

Hydrogen-bonding network within the active site of DHFR derived from the crystal structure of the ternary complex with NADP+ and folate. Water molecules in sites A and B are indicated (Bystroff and Kraut 1991).
This mechanism is based on the observation of conserved water molecules in X-ray structures of various forms of DHFR in a range of complexes. However, although a water molecule hydrogen bonded to Asp 27, Trp 22, and the oxygen on C4 (site A) is found in a high percentage of structures, a water molecule bonded to N5 (site B) has never been seen in a structure representing the Michaelis complex of the E. coli enzyme (Fig. 2 ▶) (Sawaya and Kraut 1997 and references therein). A water molecule in hydrogen bonding distance to N5 of the pterin ring has been observed in the open conformation of E. coli DHFR and in the complex of chicken DHFR with biopterin and NADP+, where the M20 loop is permanently in the closed conformation (McTigue et al. 1992). This water molecule is ideally located to form a bridge from the enolic proton to the lone pair on N5. Similarly, a long-lived water molecule was detected by nuclear magnetic resonance (NMR) in site B of the complex of human DHFR with methotrexate and NADPH (Meiering and Wagner 1995). Finally, a water molecule can be modeled into the active site of the Michaelis complex of the E. coli enzyme with only slight repositioning of Met 20 (vide infra).
Rather than a proton transfer by way of a water molecule, a direct proton transfer from the enol form of O4 to the lone pair of N5 could be envisaged (Morrison and Stone 1988; Uchimaru et al. 1989). However, the geometry between O4 and N5 is not favorable for proton transfer. The O4–H–N5 bond angle is ∼100° and the N5–H distance 2.5 Å rather than the optimal average hydrogen bond angle of 161° and acceptor donor distance of 1.9 Å (Olovsson and Joensson 1976; Baker and Hubbard 1984). In a related system, a QM/MM approach showed that for the triosephosphate isomerase catalyzed reaction direct intramolecular proton transfer in the enediolate is energetically unfavorable (Cui and Karplus 2001).
The importance of residues Asp 27 and Trp 22 in DHFR have been shown by site-directed mutagenesis. Replacing Asp 27 with Asn (Villafranca et al. 1983) or Ser (Howell et al. 1986) led to a decrease of the catalytic efficiency by approximately four and three orders of magnitude, respectively. In a related study, Phe or His replaced Trp 22, which led to an 18- and 73-fold reduction in steady-state catalytic activity (Warren et al. 1991).
Here we present the results from energy minimizations and molecular dynamics (MD) simulations to investigate the ability of solvent to access the active site of E. coli DHFR. The structures used were from crystallographic studies or closely modeled after crystallographic data for the Michaelis analog complex. Our results suggest that after the initial 300 psec of MD time water can gain access to N5 in the Michaelis complex of DHFR in a manner consistent with a mechanism involving either an early or late protonation step. N5 of H2F could be protonated while the M20 loop is in the open conformation and water access is facilitated, followed by a return of the loop to the closed conformation and hydride transfer from NADPH to the protonated imine of H2F. Alternatively, hydride transfer could occur early to midway on the reaction coordinate followed by a late protonation step. MD simulations suggest that after hydride transfer, water access to N5 is increased leading to easy protonation of N5.
Results and Discussion
Water access to the pterin ring of H2F
One of the central questions concerning the catalytic mechanism of DHFR is how the enzyme accomplishes protonation of N5 of the pterin ring of H2F. In the absence of any ionizable groups other than Asp 27 and because the direct transfer of the enolic proton is less favorable (vide supra), this proton must be delivered by way of a water molecule (Fig. 2 ▶) (Sawaya and Kraut 1997).
MD simulations can be used to determine the accessibility of the active site of DHFR by water molecules. Because no crystal structures for the Michaelis complex of DHFR exist, initial coordinates for DHFR were taken from ternary complexes of DHFR with folate and NADP+, which following the argument presented by Sawaya and Kraut models the Michaelis complex. This ternary complex crystallized in different space groups with the M20 loop in the open and the closed form, respectively (Sawaya and Kraut 1997). NADPH and H2F were placed into the active site by superposition onto folate and NADP+. After the removal of all crystallographic water molecules, the structure was placed in a water box.
Initially, MD simulations were performed for 210 psec on the Michaelis complex with the M20 loop in either the open or the closed conformation. When the M20 loop is in the open conformation water molecules can reach the active site relatively freely (Table 1). Twenty-six water molecules found their way into the site A of the Michaelis complex of DHFR and the site was occupied for 93% of the simulation. Only five water molecules were found in the active site when the loop was closed but the total percentage occupancy of the site (89%) was similar to that when the loop was open indicating that the water molecules were more tightly bound when the loop was closed. In the open loop conformation, 10 water molecules at some point in the simulation occupied site B, which allows the formation of a hydrogen bond to N5 of the pterin ring (Fig. 2 ▶). However, in the closed form, in which hydride transfer is believed to occur, all five water molecules were found hydrogen bonded to Asp 27 and Trp 22, whereas site B remained empty.
Table 1.
Results of two 210-psec MD simulations of the ternary complex DHFR•H2F•NADPH with the M20 loop in the open and closed conformation, respectively
| Complex | DHFR • H2F • NADPH | DHFR • H2 F• NADPH |
| M20 loop conformation | Open | Closed |
| Number of waters in site Aa | 26 | 5 |
| Percentage occupancy site Ab | 93 | 89 |
| Number of waters in site Bc | 10 | 0 |
| Percentage occupancy site Bd | 26 | 0 |
a Number of water molecules able to make a hydrogen bond to either the side chain NH of W22 or the carboxylate of D27 in at least one frame of the simulation.
b Percentage of frames that contained at least one hydrogen bond from a water molecule to either the NH of W22 or the carboxylate of D27.
c Number of water molecules able to make a hydrogen bond to N5 of H2F in at least one frame of the simulation.
d Percentage of frames that contained at least one hydrogen bond from a water molecule to N5 of H2F.
Inspection of the crystal structure used as the initial coordinates for the closed structure revealed that a water molecule is bound at site A, whereas there was no water in site B (Fig. 1B ▶). A water molecule in site B would be the prerequisite for protonation of N5 of H2F. Analysis of the X-ray structures revealed that the side chain of Met 20 projects into the active site toward N5 and the oxygen on C4 of H2F with the terminal methyl group located in site B whenever the M20 loop is in its closed conformation (Figs. 1B and 3A ▶ ▶). However, when the M20 loop is open, the side chain of Met 20 no longer projects into the active site and water molecules are seen in sites A and B (Fig. 1A ▶) (Sawaya and Kraut 1997).
Fig. 3.

The conformation of the M20 loop in the closed conformation including water in site A as observed in the X-ray structure of the DHFR • folate • NADP+ complex (Sawaya and Kraut 1997). A water molecule was added to site B and overlapping with the terminal methyl group of Met 20. (A) In the energy minimized structure the water molecules moved significantly from their original positions in sites A and B; (B) when the coordinates of water in site B were fixed, water in site B remained there with only a slight rearrangement of the M20 loop. Water molecules are indicated as gray spheres.
Alternative conformation of the Michaelis complex
The absence of a water molecule in site B in structures of Michaelis complex analogs may result from the small structural differences between these analogs and the true reactive complex (DHFR • H2F • NADPH). To assess the possibility that a water molecule might occupy site B in the Michaelis complex the coordinates of the water molecule in site B in the crystal structure with the M20 loop open were added to the initial coordinates of the X-ray structure with the M20 loop closed. Folate and NADP+ were replaced by H2F and NADPH as before. All crystallographic water molecules with the exception of those in sites A and B were removed and the structure was minimized. Both water molecules moved from their initial positions. A direct hydrogen bond formed between the ring NH of Trp 22 and the carboxylate of Asp 27 rather than the water-mediated contact between these two residues. Water in site B was displaced significantly and moved below the plane of the pterin ring of H2F (Fig. 3A ▶). The M20 loop remained in the closed conformation seen in all structures of Michaelis complex analogs.
In a further minimization the coordinates of the water molecule in site B were fixed. The M20 loop remained in its closed conformation (Fig. 3B ▶) with only minor changes to the backbone dihedral angles of the amino acids in the loop (Fig. 4 ▶). The side chain of Met 20 rotated so that the terminal methyl group no longer protruded into the active site (Fig. 3B ▶). This allowed water to remain in site B in hydrogen bonding distance to N5 of H2F. The r.m.s. deviation between the closed M20 loop conformation observed in the crystal structure corresponding to the Michaelis analog complex of DHFR and the conformation adopted by residues 14–24 after the above minimization was only 0.79 Å for the backbone atoms. These results indicated that only minor changes in the conformation of the M20 loop were required to allow a water molecule to occupy site B in the active complex of DHFR (Fig. 4 ▶). It is noteworthy in this context that the solution of the crystal structure of chicken liver DHFR complexed with NADP+ and biopterin revealed an ordered water molecule hydrogen bonded to the oxygen on C4 and N5 of the biopterin ring (McTigue et al. 1992). For all vertebrate DHFR structures solved to date, the M20 loop is in the closed conformation. The r.m.s. deviation between the backbone atoms of the M20 loop in the chicken and the E. coli enzyme is only 1.51 Å indicating that water can be accommodated in site B of the enzyme without major repositioning of the residues of the enzyme's active site.
Fig. 4.
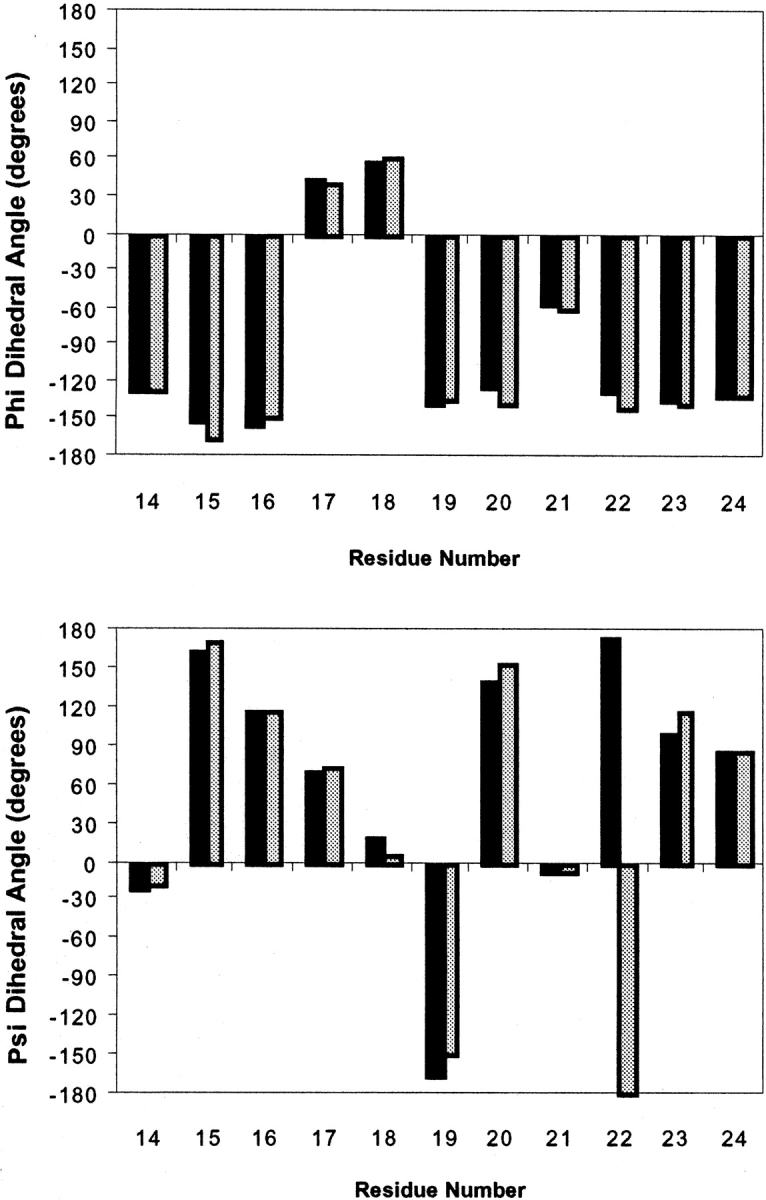
Backbone dihedral angles Ψ (bottom) and φ (top) of the residues of the closed conformation of the M20 loop (black) and of the rearranged M20 loop after minimization with a water molecule fixed in site B (gray) as depicted in Fig. 3A,B ▶, respectively.
Our results suggested that through minor changes in the conformation of the M20 loop a water molecule that could serve as the proton donor in the reduction of H2F could be accommodated in the Michealis complex of E. coli DHFR. However, MD simulations starting from the newly defined M20 loop conformation showed that within the first 4 psec of the simulation, this water in site B was lost by the repositioning of the side chain of Met 20 to a conformation that corresponded to that observed in the structures of the Michaelis analog structures (data not shown). The side chain of Met 20 then prevented access to site B and no further water was observed in this position. Water in site A remained there throughout the course of the simulation.
These results indicated that although conformational rearrangements of the M20 loop could be made relatively easily to accommodate a water molecule in hydrogen bonding distance to N5 of H2F, this confrontation was not stable. Water could not be stabilized in site B in the closed conformation of the M20 loop. As a consequence, it is unlikely that water is trapped in site B of the complex of E. coli DHFR with cofactor and dihydrofolate.
One-nanosecond simulations with H2F and NADPH
The length of the simulation is an important question that needs careful consideration in all MD studies. There are several accounts of MD simulations of DHFR during time periods similar to the 210 psec reported here (Gorse and Greedy 1997, Verma et al. 1997; Cody et al. 2000). However, one of the most thorough studies reported in the literature uses 10 nsec simulations (Radkiewicz and Brooks 2000). All these studies highlight the question of the importance of the length of MD simulations. The absence of any proton donors in an appropriate position in the active site of DHFR (vide supra) suggests that water must act as the proton donor in the reaction (Cannon et al. 1997; Sawaya and Kraut 1997). Because during 210 psec simulations no water molecules could reach site B when the M20 loop was in its closed position, and because water molecules that were modeled into site B were unstable in that position, the length of the MD simulations was increased to ∼1 nsec.
A series of 1010 psec simulations on the Michaelis complex were performed and analyzed for hydrogen bonds as before. The r.m.s. deviation of the heavy atoms of the side chain of Met 20 from the initial minimized structure was calculated for each vrame and the trajectories were analyzed for near-attack conformations (NACs). A NAC was defined as a conformation in which the distance between the pro-R hydrogen of NADPH and the C6 of dihydrofolate was <3.5 Å and the angle between C4, the pro-R hydrogen of NADPH, and C6 of dihydrofolate was >120° (Radkiewicz and Brooks 2000).
Analysis of these longer simulations showed that water was able to access site B (Table 2). On average 10.67 water molecules were found in site A and the site was occupied for 67.33% of the simulations, whereas 20.67 water molecules were found in site B with a total occupancy of 29.67% (Table 2). Analysis of the trajectories obtained from these simulations showed that hydrogen bonds were generally only formed in site B after the first 300 psec of the simulation (Fig. 5 ▶), thus emphasizing the need for longer simulation times and explaining the absence of any water in site B during the 210 psec simulations. A plot of the r.m.s. deviation from the initial minimized structure of the heavy atoms of the side chain of Met 20 versus simulation time revealed that water molecules could only access site B when the r.m.s. approached 2Å (Fig. 5 ▶), providing an explanation why in the shorter simulations site B was inaccessible for water. Interestingly, on average ∼40% of the frames contained NACs (Table 2).
Table 2.
Averaged results of three 1010-psec MD simulations of the ternary complexes of DHFR•NADPH with H2F, and H3F+, H3F−
| Complex | DHFR • H2F • NADPH | DHFR • H3F+ • NADPH | DHFR • H3F • NADPH |
| M20 loop conformation | Closed | Closed | Closed |
| Number of waters in site Aa | 10.67 | 12.00 | 13.67 |
| Percentage occupancy site Ab | 67.33 | 81.33 | 77.67 |
| Number of waters in site Bc | 20.67 | 21.00 | 22.33 |
| Percentage occupancy site Bd | 29.67 | 30.00 | 41.33 |
| Number of NACse | 189.33 | 130.00 | na |
(na) Not applicable.
a Number of water molecules able to make a hydrogen bond to either the ring NH of W22 or the carboxylate of D27 in at least one frame of the simulation.
b Percentage of frames that contained at least one hydrogen bond from a water molecule to either the NH of W22 or the carboxylate of D27.
c Number of water molecules able to make a hydrogen bond to N5 of H2F or H3F− or the proton on N5 on H3F+ in at least one frame of the simulation.
d Percentage of frames that contained at least one hydrogen bond from a water molecule to N5 of H2F or H3F− or to the proton on N5 of H3F+.
e The number of frames which contained a near attack conformation for hydride transfer as defined in the text.
Fig. 5.
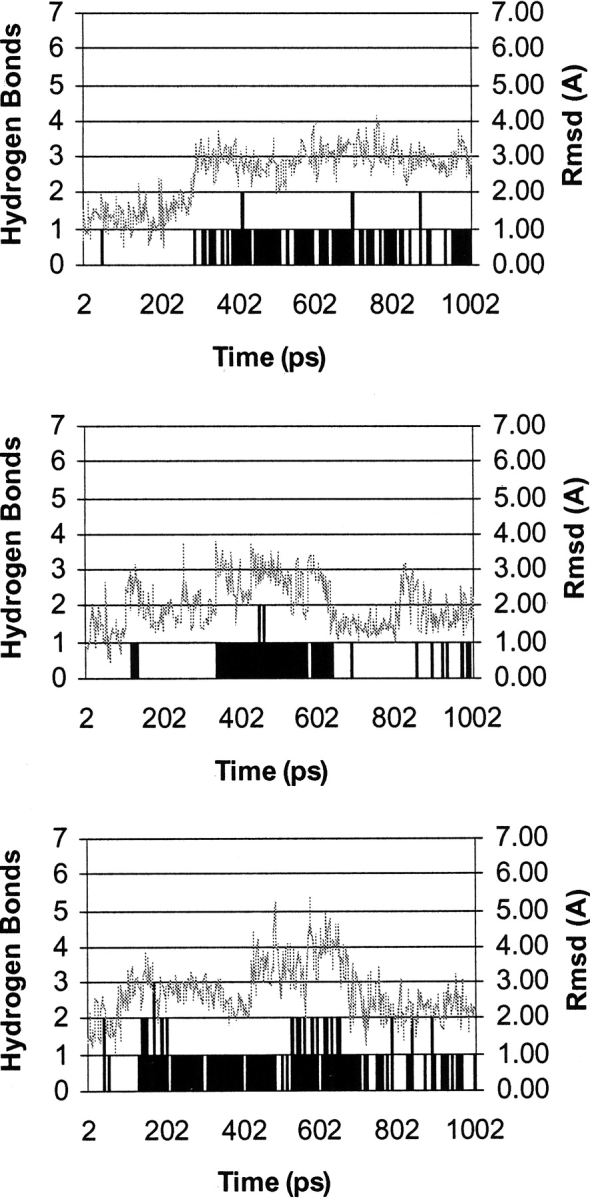
Representative results from 1010 psec MD simulations indicating the r.m.s.d. in Å of the side chain heavy atoms of Met 20 from the initial minimized structures containing H2F (top), H3F+ (middle), and H3F− (bottom). Also shown are the number of water molecules (black bars) able to make a hydrogen bond to N5 (or the proton on N5 in the case of H3F+) of the substrate during the course of the simulation.
Water access to charged pterins in the active site of DHFR
Our results showed that water has access to both sites A and B in the active site of the Michaelis complex of DHFR when the loop is open. The open conformation of the M20 loop is thought to be an intermediate between the closed and the occluded forms. Such a movement of the M20 loop may explain the obligatory keto–enol isomerization of the substrate in the ternary enzyme complex preceding hydride transfer. Similarly, rotation of the aromatic residue in position 31 of vertebrate DHFRs has been postulated to control water access to N5 of the pterin ring (McTigue et al. 1992). A possible mechanism for the reduction of H2F could be that N5 is protonated by a water molecule, when the M20 loop is in the open conformation (Fig. 6 ▶) and the active site is freely accessible by solvent molecules (vide supra) (Sawaya and Kraut 1997). Subsequently, water is displaced from the active site through the return of the M20 loop to the closed conformation. MD simulations of 1010 psec showed that water could access both sites A and B in the Michaelis complex of E. coli DHFR after protonation of N5 (DHFR • H3F+ • NADPH) when the M20 loop was in its closed form (Table 2). Accessibility was similar to that observed for the ternary reactant complex with the unprotonated substrate. An average of 12 water molecules were found in site A (occupancy 81.33%) and 21 in site B (occupancy 30%) (Table 2). In the case of H3F+ the number of water molecules that could hydrogen bond to the proton on N5 were counted. As with H2F, water molecules were only found in site B after ∼300 psec and when the r.m.s.d. of the Met 20 side chain was >2Å (Fig. 5 ▶). However, if protonation occurs in the open conformation followed by a return of the loop to the closed form and hydride transfer, water does not need to access site B in the Michaelis complex. Interestingly, the number of frames found to correspond to NACs for the protonatated substrate was ∼30% lower than for the neutral pterin ring (Table 2). The fact that an increased number of reactive conformations was not found with the protonated pterin suggested that it may not be intrinsically more reactive than H2F with respect to hydride transfer from NADPH. However, it is important to remember that conformational preferences in classical MD simulations cannot necessarily be used to draw conclusions about intrinsic reaction rates.
Fig. 6.

Different mechanisms for the reduction of dihydrofolate. Protonation of N5 of H2F can occur early to produce H3F+ followed by hydride transfer to C6. Alternatively, hydride transfer to produce H3F− can precede protonation of N5. H3F− and H3F+ can be either true intermediates of the reaction or intermediate stages along the reaction coordinate in a single transition state reaction.
Protonation in the open form, however, is not readily achieved because water access to the pterin ring is relatively free and consequently the pKa of N5 will be similar to its solution value of 2.6 (Maharaj et al. 1990). In addition, NMR experiments with apo-DHFR had revealed that the M20 loop samples two distinct environments and that interconversion between these conformations occurs with a frequency of 35 sec−1 (Falzone et al. 1994). This rate is similar to the rate of H4F release in the kinetic cycle (Fierke et al. 1987). Hydride transfer in the active site of the enzyme on the other hand occurs at the much faster rate of 950 sec−1. If the substrate bound to the active site and was protonated while the loop was open, the necessary movement of the M20 loop from the open to the closed conformation for hydride transfer would significantly slow down the chemical step.
Because of the low basicity of N5 of H2F, the difficulty to protonate N5 in the open conformation of the M20 loop and the necessity for loop movement after protonation, a mechanism by which hydride transfer preceded protonation was tested. In such a mechanism an intermediary opening of the M20 loop is not required. Hydride transfer occurs in the Michaelis complex followed by protonation of N5 at a later stage (Fig. 6 ▶). If the hydride ion is transferred from NADPH to C6 of H2F before protonation of N5 occurs, the buildup of negative charge on N5, which will increase the basicity of N5, could lead to an increase of water accessibility. The MD simulations with the DHFR complex of NADP+ and H3F− revealed that the accessibility of site B was higher after hydride transfer than for the neutral pterin ring (Table 2). During the 1010 psec MD simulations an average of 22.33 water molecules found their way into site B with a total occupancy of 41.33% of the duration of the simulation with H3F−, as compared to the 20.67 water molecules that accessed site B for 29.67% of the time in simulations with H2F (Table 2). Although the number of water molecules in site B was increased by <10%, these water molecules were clearly more tightly bound in the case of H3F−. Again movement of the side chain of Met 20 was needed before water molecules could access site B. Interestingly this opening of site B occurred already after ∼100 psec rather than 300 psec observed with H2F and H3F+ (Fig. 5 ▶).
Several lines of evidence support a mechanism in which hydride transfer from NADPH to C6 of H2F occurs before protonation of N5. First, the distance between C4 of NADP+ and C6 of folate in the ternary complexes of E. coli DHFR is on ∼3.32 Å when the M20 loop is in its closed conformation (Sawaya and Kraut 1997). This is significantly shorter than the distance observed in ternary complexes with the M20 loop in its open conformation (3.49 Å) (Sawaya and Kraut 1997) and only slightly larger than the computationally determined optimal C–C distance in the transitional state of 2.7 Å (Wu and Houk 1987) and the sum of the van der Waals radii for the two nuclei. When the E. coli DHFR • NADPH complex was superimposed on the complex of DHFR with folate, the distance between C6 of the substrate and C4 of NADPH was even shorter (2.9 Å) (Reyes et al. 1995). As a consequence, the attractive forces within the complex of cofactor and substrate are optimized and the reactants within this ground state complex are poised to enter the transitional state for hydride transfer.
Second, a detailed analysis by semiempirical methods of the reaction profile for the reduction of pyruvate by dogfish lactate dehydrogenase revealed a midway transitional state for hydride transfer from NADPH to the substrate carbon and a very late transitional state for the acid catalyzed protonation (Almarsson et al. 1992; Almarsson and Bruice 1993).
Third, it is evident from the X-ray structures of the complexes of E. coli DHFR with folate and NADP+ that the nicotinamide ring is bound in part by interactions between the carbonyl oxygens of Ile 14 and 94 and the γ oxygen of Thr 46 with C2, C4, C5, and C6. Although aromatic C–H bonds are generally not very strongly polarized, these interactions will nevertheless stabilize NADP+ relative to NADPH in the active site and thereby help initiated hydride transfer to H2F. Such a mechanism would be similar, albeit less pronounced, to the positioning of a positive charge in the environment of the proton donor observed in dogfish lactate dehydrogenase, an effect that was shown theoretically by use of a MAR diagram (Wilkie and Williams 1992).
Fourth, local density functional theory suggested that upon binding, DHFR induced long-range polarization effects of unprotonated H2F resulting in a reduction of the π bond and an increase of the σ bond character of the 5,6 bond of H2F (Bajorath et al. 1991).
Finally, UV difference (Maharaj et al. 1990) and NMR spectroscopy (Selinsky et al. 1990) have failed to reveal protonation of N5 of H2F over a wide pH range in binary complexes. In addition, as pointed out there is no obvious charge or polar contact near N5 that could sustain a preprotonation mechanism. Because the pKa of the protonated form of free H2F is 2.6 and the pKa of Asp 27 of DHFR is 6.5 in all ligand-bound complexes, Adams et al. (1991) have pointed out that the concentration of the protonated species must be negligible and have previously proposed the possibility of a concerted or late proton transfer within the enzyme's active site.
In conclusion, the results reported provide further insights into the catalytic mechanism of DHFR from E. coli. A slight rearrangement of the M20 loop from the conformation observed in crystal structures representing the Michaelis complex was possible, thereby allowing water access to N5 of dihydrofolate. However, this conformation was not stable and reverted to the regular closed conformation within <5 psec of MD simulation. Water could access site B in the active site of dihydrofolate after an equilibration time of ∼300 psec and after movement of the side chain of Met 20. This rearrangement of the M20 loop was found to be a prerequisite of protonation. Protonation of N5 of H2F did not increase either the accessibility of the active site by water or the number of NACs compared with neutral substrate. However, hydride transfer from NADPH to C6 of H2F before protonation increased the water accessibility of the active site, indicating the feasibility of the early hydride transfer mechanism. Semi-empirical and ab initio calculations are underway to determine the order of events during the reduction of dihydrofolate by NADPH in the active site of DHFR.
DHFR appears to catalyze the reduction of H2F by providing an architecture that complements the reaction. Binding free energy between the substrate, the cofactor, and the enzyme is used to force the reactants into close proximity where the attractive forces between cofactor and substrate are optimized. The reactants within this complex are thus poised to enter the transitional state for hydride transfer. The results reported here indicate that the creation of a negative charge in the low dielectric environment of the active site of DHFR leads to an enhancement of the water accessibility of the pterin ring and protonation.
Materials and methods
Initial coordinates for DHFR with the open conformation of the M20 loop were taken from 1RA2.pdb and coordinates for DHFR with the closed conformation of the M20 loop were taken from 1RX2.pdb (Sawaya and Kraut 1997). Both these structures are ternary complexes containing folate and NADP+ • H2F and NADPH structures were built using Insight II (Biosym Technologies Inc., San Diego, CA) and positioned in the active site by superposition onto (and subsequent deletion of) the folate and NADP+ structures. The parameters and charges for NADPH and NADP+ of Ulf Ryde (pers. comm.) were used, and RESP charges for H2F were obtained at the HF/3–21G level.
Calculations were performed using the AMBER 4.1 program with the Cornell all atom forcefield (Cornell et al. 1995). Unless stated otherwise all crystallographic waters were removed and each structure resolvated with a water box of ∼4000 TIP3P water residues (Jorgensen et al. 1983). All structures were minimized using a protocol of ∼5000 steps of the steepest descent algorithm followed by a further 95,000 steps of the conjugate gradient mechanism.
The MD simulations were run at constant volume. Initial velocities were assigned according to the Maxwell-Boltzmann distribution at 300 K, the system was coupled to a temperature bath with a time constant of 0.2 psec. An electrostatic cutoff of 12 Å was used in all calculations. All bonds to hydrogen atoms were constrained using the SHAKE algorithm (Ryckaert et al. 1977) allowing a time step of 0.002 psec and coordinates were saved every 2 psec. Trajectories were analyzed using the Moil-view program (Simmerling et al. 1995) to search for water molecules capable of making hydrogen bonds with the ring NH of W22, the carboxylate of D27, and N5 of H2F. Results are from an average of three separate calculations for 1010 psec simulations, whereas results from 210 psec simulations are from single experiments.
Acknowledgments
We thank Peter J. Knowles for computer time, Ulf Ryde for providing parameters and the charges of NADPH and NADP+, Oliver S. Smart and Lesley A. Tannahill for critical reading of the manuscript, and Daniel Flint, Oliver Smart, Richard Ward, and John Wilkie for helpful discussions. This work was supported by the University of Birmingham's School of Chemical Sciences and the BBSRC (6/B15285).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.5060102.
References
- Adams, J.A., Fierke, C.A., and Benkovic, S.J. 1991. The function of amino acid residues contacting the nicotinamide ring of NADPH in dihydrofolate reductase from Escherichia coli. Biochemistry 30 11046–11054. [DOI] [PubMed] [Google Scholar]
- Almarsson, O. and Bruice, T.C. 1993. Evaluation of the factors influencing reactivity and stereospecificity in NAD(P)H dependent dehydrogenase enzymes. J. Am. Chem. Soc. 115 2125–2138. [Google Scholar]
- Almarsson, O., Karaman, R., and Bruice, T.C. 1992. Kinetic importance of conformations of nicotinamide adenine dinucleotide in the reactions of dehydrogenase enzymes. J. Am. Chem. Soc. 114 8702–8704. [Google Scholar]
- Baker, E.N. and Hubbard, R.E. 1984. Hydrogen-bonding in globular proteins. Prog. Biophys. Mol. Biol. 44 97–179. [DOI] [PubMed] [Google Scholar]
- Bajorath, J., Kraut, J., Li, Z., Kitson, D.H., and Hagler, A.T. 1991. Theoretical studies on the dihydrofolate reductase mechanism: Electronic polarization of bound substrates. Proc. Natl. Acad. Sci. 88 6423–6426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakley, R.L. 1984. Chemistry and biochemistry of folates. In Folates and pterins. (eds. R.L. Blakley and S.J. Benkovic), pp. 191–253. Wiley, New York, NY.
- Bystroff, C. and Kraut, J. 1991. Crystal structure of unliganded Escherichia coli dihydrofolate reductase. Ligand-induced conformational changes and cooperativity in binding. Biochemistry 30 2227–2239. [DOI] [PubMed] [Google Scholar]
- Cannon, W.R., Garrison, B.J., and Benkovic, S.J. 1997. Electrostatic characterization of enzyme complexes: Evaluation of the mechanism of catalysis of dihydrofolate reductase. J. Am. Chem. Soc. 119 2386–2395. [Google Scholar]
- Casarotto, M.G., Basran, J., Badii, R., Sze, K.-H., and Roberts, G.C.K. 1999. Direct measurement of the pKa of aspartic acid 26 in Lactobacillus casei dihydrofolate reductase: Implications for the catalytic mechanism. Biochemistry 38 8038–8044. [DOI] [PubMed] [Google Scholar]
- Charlton, P.A., Young, D.W., Birdsall, B., Feeny, J., and Roberts, G.C.K. 1979. Stereochemistry of reduction of folic acid using dihydrofolate reductase. J. Chem. Soc. Chem. Commun. 20 922–924. [Google Scholar]
- Chen, Y.Q., Kraut, J., Blakley, R.L., and Callender, R. 1994. Determination by raman spectroscopy of the pka of N5 of dihydrofolate bound to dihydrofolate reductase. Mechanistic implications. Biochemistry 33 7021–7026. [DOI] [PubMed] [Google Scholar]
- Cody, V., Chan, D., Galitsky, N., Rak, D., Luft, J.R., Pangborn, W., Queener, S.F., Laughton, C.A., and Stevens, M.F.G. 2000. Structural studies on bioactive compounds. 30. Crystal structure and molecular modeling studies on the Pneumocystis carinii dihydrofolate reductase cofactor complex with TAB, a highly selective antifolate. Biochemistry 39 3556–3564. [DOI] [PubMed] [Google Scholar]
- Cornell, W.D., Cieplak, P., Bayly, C.I., Gould, I.R., Merz, K.J., Ferguson, D.M., Spellmeyer, D.C., Fox, T., Caldwell, J.W., and Kollman, P.A. 1995. A 2nd generation force-field for the simulation of proteins, nucleic-acids, and organic molecules. J. Am. Chem. Soc. 117 5179–5197. [Google Scholar]
- Cui, Q. and Karplus, M. 2001. Triosephosphate isomerase: A theoretical comparison of alternative pathways. J. Am. Chem. Soc. 123 2284–2290. [DOI] [PubMed] [Google Scholar]
- Cummins, P.L. and Gready, J.E. 2001. Energetically most likely substrate and active-site protonation sites and pathways in the catalytic mechanism of dihydrofolate reductase. J. Am. Chem. Soc. 123 3418–3428. [DOI] [PubMed] [Google Scholar]
- Falzone, C.J., Wright, P.E., and Benkovic, S.J. 1994. Dynamics of a flexible loop in dihydrofolate reductase from Escherichia coli and its implication for catalysis. Biochemistry 33 439–442. [DOI] [PubMed] [Google Scholar]
- Feeney, J. 2000. NMR studies of ligand binding to dihydrofolate reductase. Angew. Chem. Int. Ed. 39 290–312. [DOI] [PubMed] [Google Scholar]
- Fierke, C.A., Johnson, K.A., and Benkovic, S.J. 1987. Construction and evaluation of the kinetic scheme associated with dihydrofolate reductase from Escherichia coli. Biochemistry 26 4085–4092. [DOI] [PubMed] [Google Scholar]
- Gorse, A.-D. and Greedy, J.E. 1997. Molecular dynamics simulations of the docking of substituted N5-deazapterins to dihydrofolate reductase. Protein Engineering 10 23–30. [DOI] [PubMed] [Google Scholar]
- Howell, E.E., Villafranca, J.E., Warren, M.S., Oatley, S.J., and Kraut, J. 1986, Functional role of aspartic acid 27 in dihydrofolate reductase revealed by mutagenesis. Science 231 1123–1128. [DOI] [PubMed] [Google Scholar]
- Jorgensen, W.L., Chansrasekhar, J., Madura, J., Impley, R.W., and Klein, M.L. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79 926–935. [Google Scholar]
- Lee, H., Reyes, V.M., and Kraut, J. 1996. Crystal structures of Escherichia coli dihydrofolate reductase complexed with 5-formyltetrahydrofolate (folinic acid) in two space groups: Evidence for enolization of pteridine O4. Biochemistry 35 7012–7020. [DOI] [PubMed] [Google Scholar]
- Li, L., Falzone, C.J., Wright, P.E., and Benkovic, S.J. 1992. Functional role of a mobile loop of Escherichia coli dihydrofolate reductase in transition-state stabilization. Biochemistry 31 7826–7833. [DOI] [PubMed] [Google Scholar]
- Maharaj, G., Selinsky, B.S., Appleman, J.R., Perlman, M., London, R.E., and Blakley, R.A. 1990. Dissociation constants for dihydrofolic acid and dihydrobiopterin and implications for mechanistic models for dihydrofolate reductase. Biochemistry 29 4554–4560. [DOI] [PubMed] [Google Scholar]
- McTigue, M.A., Davies J.F.II, Kaufman, B.Y., and Kraut, J. 1992. Crystal structure of chicken liver dihydrofolate reductase complexed with Nadp+ and Biopterin. Biochemistry 31 7264–7273. [DOI] [PubMed] [Google Scholar]
- Meiering, E.M. and Wagner, G. 1995. Detection of long lived bound water molecules in complexes of human dihydrofolate reductase with methotrexate and NADPH. J. Mol. Biol. 247 294–308. [DOI] [PubMed] [Google Scholar]
- Miller, G.P. and Benkovic, S.J. 1998. Stretching exercises—flexibility in dihydrofolate reductase catalysis. Chem. Biol. 5 R105–R113. [DOI] [PubMed] [Google Scholar]
- Morrison, J.F. and Stone, S.R. 1988. Mechanism of the reaction catalyzed by dihydrofolate reductase from Escherichia coli—Ph and deuterium isotope effects with NADPH as the variable substrate. Biochemsistry 27 5499–5506. [DOI] [PubMed] [Google Scholar]
- Olovsson, I. and Joensson, P.-G. 1976. X-ray and neutron diffraction studies of hydrogen bonded systems. In Hydrogen bond, Vol. II. (eds. P. Schuster, G. Zundel, and C. Sandorfy), pp. 393–456. North-Holland, Amsterdam.
- Radkiewicz, J.L. and Brooks C.L. III. 2000. Protein dynamics in enzymatic catalysis: Exploration of dihydrofolate reductase. J. Am. Chem. Soc. 122 225–231. [Google Scholar]
- Reyes, V.M., Sawaya, M.R., Brown, K.A., and Kraut, J. 1995. Isomorphous crystal structures of Escherichia coli dihydrofolate reductase complexed with folate, 5-deazafolate, and 5,10-dideazatetrahydrofolate—Mechanistic implications. Biochemistry 34 2710–2723. [DOI] [PubMed] [Google Scholar]
- Ryckaert, J.P., Ciccotti, G., and Berendsen, H.J.C. 1977. Numerical investigation of the cartesian equations of motion of the system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 23 327–341. [Google Scholar]
- Sawaya, M.R. and Kraut, J. 1997. Loop and subdomain movements in the mechanism of Escherichia coli dihydrofolate reductase: Crystallographic evidence. Biochemistry 36 586–603. [DOI] [PubMed] [Google Scholar]
- Selinsky, B.S, Perlman, M.E., London, R.E., Unkefer, C.J., Mitchell, J., and Blakley, R.L. 1990. C13 and N15 nuclear magnetic resonance evidence of the ionisation state of substrates bound to bovine dihydrofolate reductase. Biochemistry 29 1290–1296. [DOI] [PubMed] [Google Scholar]
- Simmerling, C., Elber, R., and Zhang, J. 1995. Moil-view—A program for visualization of structure and dynamics of biomolecules and STO—A program for computing stochastic paths. In Modelling of biomolecular structure and mechanisms. (eds. A. Dullman et al.), pp. 241–265. Kluwer, Netherlands.
- Uchimaru, T., Tsuzuki, S., Tanabe, K., Benkovic, S.J., Furukawa, K., Taira, K. 1989. Computational studies on pterins and speculations on the mechanism of action of dihydrofolate reductase. Biochem. Biophys. Res. Commun. 161 64–68. [DOI] [PubMed] [Google Scholar]
- Verma, C.S., Caves, L.S.D., Hubbard, R.E., and Roberts, C.K. 1997. Domain motions in dihydrofolate reductase: A molecular dynamics study. J. Mol. Biol. 266 776–796. [DOI] [PubMed] [Google Scholar]
- Villafranca, J.E., Howell, E.E., Voet, D.H., Strobel, M.J., Ogden, R.C., Abelson, J.N., and Kraut, J. 1983. Directed mutagenesis of dihydrofolate reductase. Science 222 782–788. [DOI] [PubMed] [Google Scholar]
- Warren, S.M., Brown, K.A., Farnum, M.F., Howell, E.E., and Kraut, J. 1991. Investigation of the functional role of tryptophan 22 in Escherichia coli dihydrofolate reductase by site directed mutagenesis. Biochemistry 30 11092–11103. [DOI] [PubMed] [Google Scholar]
- Wilkie, J. and Williams, I.H. 1992. Transition state structural variation in a model for carbonyl reduction by lactate dehydrogenase: Computational validation of empirical predictions based upon Albery-More O'Ferrall-Jencks diagrams. J. Am. Chem. Soc. 114 5423–5425. [Google Scholar]
- Wu, Y.D. and Houk, K.N. 1987. Theoretical transition structures for hydride transfer to methyleniminium ion from methylamine and dihydropyridine—On the nonlinearity of hydride transfers. J. Am. Chem. Soc. 109 2226–2227. [Google Scholar]