

Abstract
Crystal structures have been determined for free Escherichia coli hypoxanthine phosphoribosyltransferase (HPRT) (2.9 Å resolution) and for the enzyme in complex with the reaction products, inosine 5`-monophosphate (IMP) and guanosine 5`-monophosphate (GMP) (2.8 Å resolution). Of the known 6-oxopurine phosphoribosyltransferase (PRTase) structures, E. coli HPRT is most similar in structure to that of Tritrichomonas foetus HGXPRT, with a rmsd for 150 Cα atoms of 1.0 Å. Comparison of the free and product bound structures shows that the side chain of Phe156 and the polypeptide backbone in this vicinity move to bind IMP or GMP. A nonproline cis peptide bond, also found in some other 6-oxopurine PRTases, is observed between Leu46 and Arg47 in both the free and complexed structures. For catalysis to occur, the 6-oxopurine PRTases have a requirement for divalent metal ion, usually Mg2+ in vivo. In the free structure, a Mg2+ is coordinated to the side chains of Glu103 and Asp104. This interaction may be important for stabilization of the enzyme before catalysis. E. coli HPRT is unique among the known 6-oxopurine PRTases in that it exhibits a marked preference for hypoxanthine as substrate over both xanthine and guanine. The structures suggest that its substrate specificity is due to the modes of binding of the bases. In E. coli HPRT, the carbonyl oxygen of Asp163 would likely form a hydrogen bond with the 2-exocyclic nitrogen of guanine (in the HPRT-guanine-PRib-PP-Mg2+ complex). However, hypoxanthine does not have a 2-exocyclic atom and the HPRT-IMP structure suggests that hypoxanthine is likely to occupy a different position in the purine-binding pocket.
Keywords: Crystal structure, phosphoribosyltransferase, purine salvage, Escherichia coli, enzymology
Purine nucleoside monophosphates are either synthesized de novo from simple precursors or through a salvage pathway. Most organisms, including bacteria, possess both the de novo and salvage pathways. However, some microorganisms such as the protozoan parasites do not possess the de novo pathway and therefore, depend entirely on the transport of preformed purine bases from their host cells for production of their 6-oxopurine nucleoside monophosphates (Ullman 1995). The key enzymes in the salvage pathway are the 6-oxopurine phosphoribosyltransferases (H/G/XPRTases), which catalyze the synthesis of the purine nucleoside monophosphates inosine 5`-monophosphate (IMP), guanosine 5`-monophosphate (GMP), or xanthosine 5`-monophosphate (XMP) from 5-phospho-α-D-ribosyl-1-pyrophosphate (PRib-PP) and hypoxanthine, guanine, or xanthine. A divalent cation, usually magnesium, is essential for catalysis (Musick 1981).
In contrast with most eukaryotes, which have only one 6-oxopurine PRTase, enteric bacteria such as Escherichia coli possesses two, HPRT (E.C. 2.4.2.8) and XGPRT (E.C. 2.4.2.22), the former preferentially salvaging hypoxanthine and the latter guanine and xanthine (Gots and Benson 1973; Neuhard and Nygaard 1987). Although the two enzymes share a common function, their amino acid sequence identity, 23%, is surprisingly low. In comparison, human HGPRT and E. coli HPRT are more similar, with a sequence identity of 33%. In addition to this low sequence identity, there is also considerable variation in the length of each of the polypeptides, with E. coli HPRT comprising 182 amino acids, E. coli XGPRT 152 amino acids, and human HGPRT 213 amino acids.
To date, the crystal structures of 6-oxopurine PRTases from seven different sources have been determined. These include human HGPRT (Eads et al. 1994; Balendiran et al. 1999; Shi et al. 1999b), Tritrichomonas foetus HGXPRT (Somoza et al. 1996), Toxoplasma gondii HGXPRT (Schumacher et al. 1996; Héroux 1999), E. coli XGPRT (Vos et al. 1997, 1998), Plasmodium falciparum HGXPRT (Shi et al. 1999a), Trypanosoma cruzi HGPRT (Focia et al. 1998a,b), and Giardia lamblia GPRT (Shi et al. 2000). Apart from E. coli XGPRT and T. gondii HGXPRT, all other structures have been determined only in the presence of substrates or substrate analogs, transition state analogs, or products. Thus, although there is a large volume of structural data available on the PRTases from several sources and in the presence of many different ligands, there is a relative lack of data showing the conformation of these enzymes in their unliganded state.
The 6-oxopurine PRTases have a highly conserved "core" domain with an α/β fold consisting of a four- or five-stranded parallel β-sheet flanked by three or four α-helices. They also possess a "hood domain" located above the core and consisting of amino acid residues from both the amino and carboxyl termini of the polypeptide. Unlike the core domain, the structure of the hood is poorly conserved across the 6-oxopurine PRTases. For example, it can be a very small unit, such as in E. coli XGPRT, which has only 29 residues arranged in two short parallel β-stands and an arm of 12 residues that extends into a different subunit of the tetramer (Vos et al. 1997). At the other extreme, the hood of Giardia lamblia GPRT is ∼97 residues and comprises four β-strands and three α-helices (Shi et al. 2000).
The active site in the 6-oxopurine PRTases is situated at the interface of the hood and core domains. The phosphate group of the purine nucleotide product binds to a cluster of residues in a loop that protrudes from the core domain. In all of the 6-oxopurine PRTase structures, the phosphate-binding loop consists of a five-residue peptide segment arranged such that the main chain nitrogen atoms form hydrogen bonds with the phosphate oxygen atoms from the nucleotide. In general, the purine bases use amino acid residues from both the core and hood domains for binding. In particular, a conserved lysine residue forms a hydrogen bond to the 6-oxo oxygen and a conserved aspartate forms a hydrogen bond to the N7 atom of the purine base. The interaction with the lysine residue allows the 6-oxopurine PRTases to discriminate between the 6-oxopurines and the 6-aminopurine, adenine. The aspartate has been proposed to function as a general base by facilitating deprotonation of the N7 atom during formation of the nucleotide product (Xu and Grubmeyer 1998).
A feature of the active site in the 6-oxopurine PRTases is a highly flexible section of polypeptide called the mobile loop, which has been proposed to move during catalysis to sequester the active site from solvent (Scapin et al. 1994). In general, the loop is disordered in crystal structures of the complexes of the 6-oxopurine PRTases with nucleoside monophosphates (Eads et al. 1994; Somoza et al. 1996; Vos et al. 1997). In the presence of the substrate analog HPP or 9-deazaguanine, PRib-PP and Mg2+ (Focia et al. 1998a; Balendiran et al. 1999; Héroux et al. 2000) or the transition state analog inhibitors immucillinHP or immucillinGP plus pyrophosphate and Mg2+, the loop has a well-defined structure positioned directly over the active site (Shi et al. 1999a,b; Shi et al. 2000).
One of the two E. coli 6-oxopurine PRTases, XGPRT has been studied using X-ray crystallography and kinetic analysis (Vos et al. 1997, 1998), but to date there are no published data for the second E. coli PRTase, HPRT except for activity in crude preparations (Deo et al. 1985). In this paper, we report the cloning, expression, purification, and kinetic data for E. coli HPRT, the 2.9 Å resolution crystal structure of the free enzyme and the 2.8 Å resolution crystal structures of the E. coli HPRT-IMP and E. coli HPRT-GMP complexes. Comparison of the structures of the free and complexed enzymes provides further insights into the extent of structural flexibility possible in the 6-oxopurine PRTases. Analysis of the structures of the complexes provides a basis for understanding the appreciable differences in kcat and Km values for hypoxanthine, guanine, and xanthine between the two E. coli 6-oxopurine PRTase enzymes.
Results and Discussion
Cloning, expression, and purification of E. coli HPRT
The cDNA coding for E. coli HPRT was cloned into the expression vector pT7-7 as described in the Materials and Methods section. The sequence of the cloned cDNA is identical to that previously determined as part of the E. coli genome project (Fujita et al. 1994). Expression of this enzyme in Sφ606 (ara, Δpro-gpt-lac, thi, hpt, F−) cells resulted in a specific activity of 98 μmol min−1 mL−1 of cell lysate using hypoxanthine as substrate, confirming that the cDNA encoded a 6-oxopurine PRTase. The enzyme was purified to homogeneity as assessed by SDS-PAGE and mass spectrometry. The specific activity toward the three naturally occurring purine bases and the corresponding data on the specificity of E. coli XGPRT are given in Table 1. The data show that E. coli HPRT exhibits a high specific activity toward hypoxanthine, whereas E. coli XGPRT favors xanthine and guanine. This difference in base specificity is discussed later in terms of kinetic constants and structure.
Table 1.
Comparison of kinetic constants and purine base specificities for E. coli HPRT and XGPRTa
| Substrate | Specific activity (μmol • min−1 • mg−1) | Km (app) (μM) | kcatb (s−1) | kcat/Km (μM−1 • s−1) |
| E. coli HPRT | ||||
| Hypoxanthine | 177 | 12.5 ± 2.4 | 59.0 ± 3.5 | 4.9 ± 1.2 |
| Guanine | 30 | 294 ± 20 | 10.2 ± 0.7 | 0.03 ± 0.005 |
| Xanthine | 0.02 | 25 ± 6.1 | 0.008 ± 0.0001 | 0.0003 ± 0.00003 |
| PRib-PPc | 177 | 192 ± 7.0 | 50.0 ± 2 | 0.26 ± 0.04 |
| E. coli XGPRTd | ||||
| Hypoxanthine | 23 | 90.8 ± 11.3 | 13.7 ± 1.1 | 0.2 ± 0.02 |
| Guanine | 95 | 4.3 ± 0.3 | 28.0 ± 0.4 | 6.5 ± 0.5 |
| Xanthine | 114 | 30.5 ± 2.6 | 37.5 ± 0.7 | 1.2 ± 0.1 |
| PRib-PPe | 95 | 139 ± 16 | 28.0 ± 2 | 0.2 ± 0.25 |
a The kcat and Km(app) values were measured in 0.1 M Tris-HCl, 0.11 M MgCl2 at pH 8.5 and at 25°C.
b kcat calculated in terms of the concentration of subunits with four active sites per molecule of enzyme.
c Hypoxanthine as the 6-oxopurine base substrate.
e Guanine as the 6-oxopurine base substrate.
Kinetic constants for E. coli HPRT: Comparison with E. coli XGPRT
E. coli HPRT exhibits a marked preference for hypoxanthine as the purine substrate. The kcat/Km for hypoxanthine is 160-fold greater than that for guanine and 16,000-fold greater than that for xanthine (Table 1). In comparison, E. coli XGPRT prefers xanthine or guanine as its purine substrates (Table 1). The kcat value for E. coli HPRT for hypoxanthine is the highest reported in the literature, being at least ninefold higher than for human HGPRT (Keough et al. 1999). The Ki values for IMP and GMP for E. coli HPRT are 247 ± 55 μM and 526 ± 68 μM, respectively, showing that the nucleoside monophosphates bind more weakly to E. coli HPRT than to human HGPRT, which has Kd values for IMP and GMP of 61 μM and 7.1 μM, respectively (Xu et al. 1997).
Unlike mammals and most parasites, E. coli cells express two distinct 6-oxopurine PRTases, with very different specificities for hypoxanthine, guanine, and xanthine. Salvage enzymes allow a more energy efficient synthesis of purine nucleoside monophosphates compared with the de novo pathway. The kinetic analysis suggests that E. coli HPRT is mainly responsible for the synthesis of IMP and that XGPRT primarily salvages guanine and xanthine. Considering the relatively small size of its genome, it is somewhat surprising that E. coli cells have not evolved so as to produce only one 6-oxopurine PRTase, which is able to use all three bases efficiently. In this context, it is instructive to compare the efficiency of the E. coli enzymes (Table 1) to that of T. foetus HGXPRT, which can use all three 6-oxopurine bases. T. foetus HGXPRT has kcat/Km values for guanine, hypoxanthine, and xanthine of 3.0, 1.0, and 0.8 μM−1s−1 (Munagala and Wang 1998). No example has yet been found of a single 6-oxopurine PRTase that can match the combined efficiency of the two E. coli enzymes.
Subunit mass and quaternary structure of E. coli HPRT
The subunit molecular mass of purified preparations of E. coli HPRT was determined by electrospray ionization mass spectrometry. The major component had a molecular mass of 20,620 ± 3 daltons, which is in excellent agreement with the molecular mass of 20,617 daltons calculated from the complete 182 amino acid sequence. A minor component (∼3% of the total) had a mass of 20,487 ± 3 daltons, indicating that, in some molecules, the amino-terminal methionine had been cleaved. Analytical ultracentrifugation showed the molecular mass of E. coli HPRT to be 79 kD, confirming that, in solution, E. coli HPRT exists as a tetramer. There was no evidence for the presence of either monomeric or dimeric forms of E. coli HPRT under these conditions. Of the 6-oxopurine PRTase structures currently in the literature, four are tetramers (human HGPRT, T. gondii HGXPRT, E. coli XGPRT, and P. falciparum HGXPRT) and three are dimers (G. lamblia GPRT, T. foetus HGXPRT, and T. cruzi HGPRT).
Structure determination
Crystals of free E. coli HPRT and the HPRT-IMP and HPRT-GMP complexes were obtained in the presence of MgCl2, sodium citrate, and DTT (see Materials and Methods). The three-unit cells are isomorphous, except that the c axis is 2 Å longer in crystals of the free enzyme. Because the quality of the X-ray data was best for the E. coli HPRT-IMP complex, this structure was determined first using molecular replacement. The crystallographic asymmetric unit consists of two subunits, referred to as A and B, which are packed in a similar dimeric arrangement to that observed in other 6-oxopurine PRTase structures. Two other subunits located across a twofold crystallographic axis of symmetry make up the tetramer. Because the three crystals were essentially identical, the structures of the E.coli HPRT-GMP complex and the free enzyme required only refinement from the coordinates of the E. coli HPRT-IMP complex. The final Rfree, Rfactor, and model geometry for the three structures are presented in Table 2. All of these values are within the normal range for protein structures at 2.8 or 2.9 Å resolution. Inspection of the 2Fo–Fc electron density maps for both complexes and the free enzyme showed the polypeptide chain could be unambiguously traced for residues 5–72 and 83–181 in both subunits of the asymmetric unit. Beyond these residues, the electron density could not be clearly assigned. Residues 73–82 correspond to the mobile loop that closes over the active site during catalysis. Electron density for this loop has also not been observed in most other crystal structures of 6-oxopurine PRTase nucleoside monophosphate complexes. In both complexes there was sufficient electron density to assign coordinates to all of the nucleoside monophosphate in subunit A, but in subunit B there was insufficient density to unambiguously locate the purine base. The structure in subunit B may give insights into the reaction by which the nucleoside monophosphate is released from the active site—the purine base may dissociate first, followed by the rest of the molecule.
Table 2.
Data collection and refinement statistics
| HPRT | HPRT-IMP | HPRT-GMP | |
| Data collection | |||
| Temperature (K) | 100 | 100 | 100 |
| Unit cell length (Å) | a = b = 83.9, c = 169.4 | a = b = 84.1, c = 167.3 | a = b = 84.1, c = 167.3 |
| Unit cell angle (°) | α = β = 90 γ = 120 | α = β = 90 γ = 120 | α = β = 90 γ = 120 |
| Space group | P3121 | P3121 | P3121 |
| Resolution range (Å) | 50.0–2.9 | 50.0–2.8 | 50.0–2.8 |
| Unique reflections [I > 0σ(I)] | 14,653 | 17,473 | 15,697 |
| Total observations [I > 0σ(I)] | 60.922 | 58,776 | 45,289 |
| Completeness (%) | 92.0 (56.0) | 99.7 (99.8) | 88.9 (89.8) |
| Rsym (%)a | 0.103 (0.293) | 0.071 (0.276) | 0.072 (0.294) |
| ≤I>/≤σ(I)> | 7.2 (2.8) | 8.7 (4.5) | 9.1 (4.1) |
| Tetramers per asymmetric unit | 1/2 | 1/2 | 1/2 |
| Solvent content (%) | 70.6 | 70.3 | 70.3 |
| Refinement | |||
| Resolution range (Å) | 50.0–2.9 | 50.0–2.8 | 50.0–2.8 |
| Rfactor [F > 0σ(F)]b | 0.214 | 0.201 | 0.199 |
| Rfree [F > 0σ(F)]b | 0.238 | 0.243 | 0.244 |
| Average B-factor (Å2) | 30.8 | 41.7 | 37.5 |
| RMS deviations from ideal | |||
| Bond length (Å) | 0.007 | 0.006 | 0.007 |
| Bond angle (°) | 1.37 | 1.37 | 1.38 |
| Ramachandran plot statistics | |||
| Residues in most favored regions (%) | 88.9 | 87.2 | 86.5 |
| Total number | 264 | 262 | 258 |
| Residues in disallowed regions (%) | 0.0 | 0.3 | 0.3 |
| Total number | 0 | 1 | 1 |
Values in parentheses are for the outer shell of data; 3.0–2.9 Å for the free enzyme and 2.90–2.80 Å for the HPRT-IMP and HPRT-GMP complexes.
a Rsym = ∑|I = ≤I>|/∑≤I>.
b Rfactor = ∑∥Fobs| − |Fcalc∥/∑|Fobs|, where the Rfactor is calculated based on the reflections used in the refinement (90% of the total data) and Rfree (Brünger 1992a) is calculated using the remaining 10% of the data.
Structure of E. coli HPRT subunits
E. coli HPRT has a similar overall structure to other known 6-oxopurine PRTases, in that it comprises a core and hood domain. Subunit A of the E.coli HPRT-GMP complex is displayed in Figure 1 ▶. The secondary structure of the core domain is essentially identical to that found in other 6-oxopurine PRTases. However, E. coli HPRT has a small hood relative to most other 6-oxopurine PRTases, consisting of only 37 residues, 13 from the amino terminus and 24 from the carboxyl terminus. The predominant secondary structure in the hood is a small three-stranded antiparallel β-sheet (S1, 8–13; S8, 156–158; and S9, 174–178). As has been shown with other 6-oxopurine PRTases, the nucleoside monophosphate is bound in a cleft between the hood and core domains. Although Mg2+ was present in the crystallization conditions, no Mg2+ ions could be placed in the active site in the structures of the complexes, although one Mg2+ ion was observed in the free structure (see active site structure below).
Fig. 1.
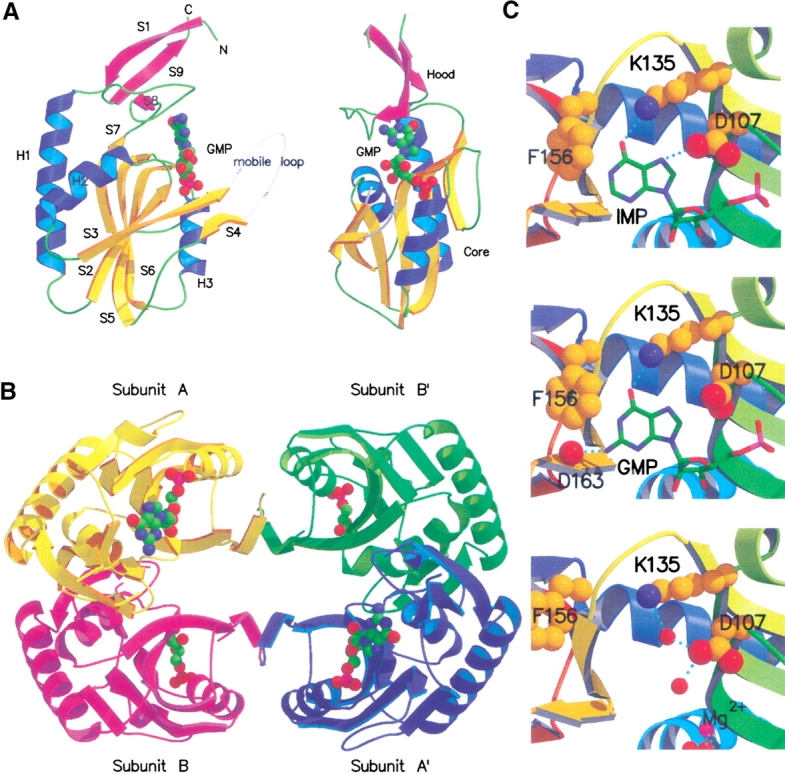
(A) Two orthogonal views of the structure of subunit A from the E. coli HPRT-GMP complex. The β-strands, shown as direction arrows are yellow in the core domain and pink in the hood domain. The mobile loop, which includes residues 73–82, is not observed in the crystal structure. To complete the structure, a hypothetical mobile loop has been modeled in and depicted as white coil. The GMP molecule is drawn as solid spheres and the atoms colored green for carbon, blue for nitrogen, red for oxygen, and pink for phosphorous. (B) The structure of the E. coli HPRT-GMP tetramer viewed down the crystallographic twofold axes. (C) The active site of subunit A of E. coli HPRT. (Top) The IMP complex. (Middle) The GMP complex. (Bottom) Free enzyme showing bound water molecules (red) and Mg2+ (pink) as solid spheres.
Tetrameric structure of E.coli HPRT
Comparison of the three E. coli HPRT structures shows that the overall quaternary structure is conserved between the native enzyme and the product complexes. In all three structures, two subunits in the asymmetric unit and two subunits located across a crystallographic twofold axis of symmetry (Fig. 1 ▶) form the tetramer. The total buried surface area between subunits A and B for the three structures is on average 2410 Å2. The intersubunit contacts at this interface include a cluster of six salt bridges involving Arg47, Asp55, Arg58, Glu65, Asp91, Asp92, and Arg169. A hydrogen bond is also observed between the side chains of Ser63 and Arg47. In addition, there are ∼28 van der Waals contacts between atoms from the side chains of Leu46, Phe50, Met51, Ala54, Cys57, and Val66. E. coli HPRT has only two cysteine residues. Cys57 is completely buried at this dimer interface and Cys130, located in strand S6 (the fifth strand in the core β-sheet) is also buried. Thus, neither cysteine residue appears capable of forming inter- or intramolecular or intersubunit disulfide bonds.
The interface between subunits A and B` or B and A` is significantly smaller than that for the A and B interface and buries on average, for the three structures, 1235 Å2 of the accessible surface area. This interface is stabilized by hydrogen bonds and salt bridges between residues Ala82, Asp83, Lys85, Asp89, Asp93, and Lys114 from subunit A and Asp83, Lys85, Asp89, Leu90, Asp93, and Lys114 from subunit B`. Van der Waals contacts, involving the side chains of Ala82, Leu90, Leu94, and Leu121 from both subunits also help to stabilize the A/B` interface. Subunits A and A` or B and B` do not form any interactions in the E. coli HPRT-IMP or GMP structures.
T. foetus HGXPRT crystallizes as a dimer, yet E. coli HPRT, which has a very similar overall structure, crystallizes as a tetramer. A comparison of the five-amino-acid residues that form the hydrogen bonds and salt bridges to stabilize the tetramer interface in E. coli HPRT with the equivalent residues in T. foetus HGXPRT suggests reasons for this difference. Of these five residues, only Asp89 is conserved between the two enzymes. On the other hand, Asp83 is changed to Asn in T. foetus HGXPRT, Lys85 to Thr, Asp93 to Asn, and Lys114 to Gln. Thus, four of the five charged residues forming the tetramer interface in E. coli HPRT are altered to uncharged residues in T. foetus HGXPRT. The loss of the bonding energy from the ion pair formed between Lys85 and Asp93 would seem to be of particular importance. The PRTases from T. gondii and T. cruzi also form dimers. In T. gondii, the equivalent five residues are Gln, Thr, Asp, Ile, and Glu, whereas in T. cruzi, they are Gly, Val, Asp, Ser, and Tyr. Again, in both enzymes, there is a significant reduction in the electrostatic binding potential for these amino acid residues when compared with E. coli HPRT. On the other hand, in human HGPRT, which does form tetramers, two of the three aspartate residues (Asp83 and Asp89 in E. coli HPRT) and Lys85 are conserved. We are currently preparing mutants of E. coli HPRT to further test our hypothesis that these five residues are critical for tetramer formation and whether it will be possible to make an active E. coli HPRT dimer by appropriate mutation.
Crystal packing
In the three E. coli HPRT structures, the solvent content is close to 70%, which is at the high end of the normal values (30%–70%) observed for most protein crystals (Matthews 1968). Analysis of the crystal packing explains the high solvent content. Neighboring molecules that are related by the 31 screw axis make contact with four small patches on a central tetramer. These contacts are at diametrically opposite corners of the central tetramer and are the only crystal packing contacts observed. There are 15 direct crystal contacts between tetramers and 7 water molecules that form bridges between adjacent tetramers. Two of the direct crystal contacts are hydrogen bonds, whereas the remainder are van der Waals interactions. Amino acid residues that form direct contacts include Glu17, Ser137, Phe150, Ser151, Ile152, Pro153, and Glu155 from subunit A and Met12, Pro14, Ser137, Ser151, Ile152, Pro153, and Lys176 from subunit B. None of these residues is predicted to be used in catalysis or involved in substrate specificity.
Comparison of the structure of E. coli HPRT with other PRTases
Despite the overall similarity in the core structure between E. coli XGPRT, E. coli HPRT, and human HGPRT, there is little amino acid sequence identity between these three enzymes. Between E. coli HPRT and E. coli XGPRT, the identity is only 22% and between E. coli HPRT and human HGPRT, only 33%. For E. coli HPRT, the highest amino acid sequence identity known is with T. foetus HGXPRT, although this is only 36%. The T. foetus enzyme uses all three purine bases with similar efficiency whereas E. coli HPRT uses only hypoxanthine efficiently. Because there are many conserved residues in the sequence, this difference in the base specificity must be due to subtle changes in the active site structures.
To analyze the differences in overall structure between the 6-oxopurine PRTases, the Cα atoms from subunit A of the E. coli HPRT-GMP complex were superimposed onto the Cα atoms of the structures of the T. foetus HGXPRT-GMP complex, E. coli XGPRT-GMP complex, and human HGPRT-GMP complex. A structure-based sequence alignment of these four enzymes is presented in Figure 2 ▶. The structure of E. coli HPRT is most similar to that of T. foetus HGXPRT with a root mean square standard deviation (rmsd) for 150 Cα atoms of 1.0 Å. The rmsd values for E. coli HPRT with E. coli XGPRT and human HGPRT are 1.7 Å for 105 Cα atoms and 1.2 Å for 135 atoms, respectively. In all four, the structure of the core is highly conserved, whereas there are significant differences in the structures of the hood. The most similar hood domains are E. coli HPRT and T. foetus HGXPRT, which have a rmsd of 1.5 Å for 32 pairs of Cα atoms (residues 7–13 and 155–179). E.coli XGPRT has a very small hood comprising 7 amino-terminal residues and 20 carboxy-terminal residues. The carboxy-terminal residues in E. coli XGPRT adopt an extended conformation that makes interactions with a neighboring subunit and probably stabilizes the tetramer (Vos et al. 1997). The very last residue in the sequence, Arg152, forms part of the active site of a symmetry-related subunit. This does not occur in E. coli HPRT.
Fig. 2.

Structure-based sequence alignment of E. coli HPRT, E. coli XGPRT, human HGPRT, and T. foetus HGXPRT. The structures were aligned using the least squares fitting algorithm in the program O (Jones et al. 1991). The secondary structure for E. coli HPRT is shown above the enzyme sequences and was calculated using the program DSSP (Kabsch and Sander 1983). Helices and strands start from the letter identifier (i.e., S1 for the first strand) and end at the arrow point. (cp) Position of the nonproline cis peptide bond; (pbl) the residues in the phosphate-binding loop; (ml) the residues in the mobile loop.
Active site structure in the enzyme complexes
The nucleoside monophosphates are located in a crevice on the surface between the hood and core domains (Fig. 1A ▶). In both E. coli HPRT complexes, the purine base in subunit B could not be unambiguously placed in the electron density and is not included in the final set of coordinates. The better density observed in subunit A correlated with lower overall B factors for this subunit. Figure 3A ▶ shows the electron density for the active site of subunit A in the E. coli HPRT-GMP complex. A comparison of the modes of binding of IMP and GMP with E. coli HPRT is presented in Figure 1C ▶ and Table 3, which lists interactions between the nucleoside monophosphates and E. coli HPRT. The phosphate oxygen atoms in the nucleoside monophosphates form an extensive network of hydrogen bonds with the main chain amide nitrogen atoms of Asp107, Gly109, Asn110, Thr111, and Leu112 and the side chain of Ser108. There are no direct interactions between the ribose ring in the GMP complex and the enzyme. However, in the IMP complex there is one van der Waals contact between the C3` atom and Cγ2 of Ile105. Thus, there appears to be little difference in the binding of the phosphate and ribose rings between the two complexes with the exception of Thr111, which adopts slightly different orientations. The major difference lies in the mode of binding of the purine bases. In both cases, a hydrogen bond is formed between the O6 atom of the purine and the amino group of Lys135. This is the highly conserved lysine that distinguishes between 6-oxopurines and 6-aminopurines (Eads et al. 1994). In the GMP complex, there is a hydrogen bond between the N2 atom in the purine base of the nucleoside monophosphate and the carbonyl oxygen of Asp163. In IMP, the N2 atom is not present. As a result, two differences are observed: first, the purine base of GMP moves 0.3 Å relative to that in IMP and second, the side chain of Asp107 is less constrained in the GMP complex than in the IMP complex, allowing it to rotate away from the purine base. In all, the distance between the N7 atom in the purine base and OD1 in Asp107 expands from 3.2 Å in the IMP complex to 3.7 Å in the GMP complex. The interaction between the Asp107 and N7 has been suggested to be important for catalysis as this residue could act as a general base to cause deprotonation of N7 (Eads et al. 1994; Xu and Grubmeyer 1998). If this closer interaction between these two atoms in IMP compared to GMP is also shown when hypoxanthine and guanine are bound in the ternary complex (Enzyme-PRib-PP-Base), it is a possible reason as to why the kcat value for hypoxanthine is fivefold faster than for guanine. Similarly, this could also explain the slow kcat for xanthine (Table 1). The 2-exocyclic oxygen in xanthine would be repelled by the carbonyl oxygen of Asp163, forcing it to adopt a different mode of binding (Km 25 μM) to the enzyme where a close approach of the N7 atom of the purine and the side chain of Asp107 is not possible.
Fig. 3.


(A) 2Fo–Fc electron density contoured at 1σ above the mean for the active site of subunit A of the E. coli HPRT-GMP complex. Two water molecules are shown as spheres. (B) Left image: 2Fo–Fc electron density contoured at 2σ above the mean for the Leu46–Arg47 dipeptide in subunit A of the free enzyme structure. In this model, the dipeptide is in the cis configuration. Right image: The same electron density for a model where the dipeptide is modeled in the trans conformation. Fo–Fc electron density −3.5σ below the mean is shown as dashed lines. This negative density is centered around the incorrectly placed backbone nitrogen atom.
Table 3.
Interactions between E. coli HPRT and GMP or IMP
| GMP/IMP | Protein | Distance in GMP (Å) | Distant in IMP (Å) |
| Interactions with base | |||
| N2 | D 163 O | 2.8 | a |
| N7 | D 107 ODI | (3.7) | 3.1 |
| O6 | K 135 NZ | 3.0 | 3.1 |
| C2 | I 162 CG2 | 3.7 | (4.5) |
| C6 | F 156 CE1 | 3.8 | 3.9 |
| C8 | D 107 CD | 3.5 | 3.9 |
| C2 | F 156 CZ | (4.3) | 3.7 |
| C8 | I 105 CG2 | (4.2) | 3.8 |
| Interactions with ribose ring | |||
| C3 | I 105 CD2 | (4.1) | 3.7 |
| Interactions with phosphate | |||
| O1P | G 109 N | 2.8 | 2.7 |
| O1P | S 108 OG | 3.0 | (3.5) |
| O1P | D 107 N | 2.9 | 3.0 |
| O2P | L 112 N | 3.2 | 3.1 |
| O3P | T 111 OD1 | 3.1 | (4.4) |
| O3P | T 111 N | 2.9 | 3.0 |
| O3P | N 110 N | 3.2 | 3.0 |
| O3P | S 108 OG | 2.7 | 3.1 |
a The N2 atom is not present in IMP. Numbers in brackets are measured distances, but atoms are not within ionic or van der Waals contact.
The structures of the enzyme in complex with the nucleoside monophosphates do not fully explain the 40-fold Km difference between the two purine bases hypoxanthine and guanine (9 μM vs. 311 μM). It is possible that structures of the enzyme in complex with one of the transition state analogs, ImmucillinHP or ImmucillinGP, or a structure of the enzyme in the presence of the stable PRib-PP analog, cPRib-PP, and the purine base could help answer this question.
There is very little difference in the Km value for PRib-PP for all the 6-oxopurine PRTases. This, together with the current structural data, suggests that the binding site for this substrate is highly conserved across the species. Therefore, it can be hypothesized that the wide disparity in kcat/Km values for these enzymes can be attributed to the differences in binding of the purine bases. In all of the enzymes studied kinetically to date, PRib-PP binds before the purine in an ordered sequential mechanism (Xu et al. 1997).
Active site structure in the free enzyme
At 2.9 Å resolution, two water molecules are visible in the active site of the free enzyme. One of the water molecules forms hydrogen bonds to the side chains of both Lys135 and Asp107. The second water molecule is hydrogen bonded only to Asp107. In addition to these two water molecules, a large cluster of electron density between Asp103 and Glu104 is observed in both subunits in the asymmetric unit. This density has been assigned to a Mg2+ ion and three associated water ligands. To complete the octahedral geometry for Mg2+, a fourth water is also likely to be bound to the Mg2+, but in the present structure this could not be located. The interaction between the Mg2+ and the enzyme may be important for stabilization of the enzyme before catalysis. For catalysis to occur, the 6-oxopurine PRTases have an absolute requirement for a divalent metal, usually magnesium in vivo.
The major difference between the structures of the free and product bound enzymes occurs in the purine-binding site (Fig. 4 ▶). A superposition of subunit A from the free enzyme and subunit A from the IMP complex shows the most significant conformational change can be attributed to the main chain dihedral angles in amino acids Asp154, Glu155, Phe156, and Val157 (Table 4) and to a rotation of the χ2 dihedral angle of the side chain of Phe156. These changes result in the side chain of Phe156 moving from a position that points away from the purine-binding site to a location where it makes van der Waals contact with the purine base in either IMP or GMP.
Fig. 4.

Superposition of subunit A from the E. coli HPRT-IMP complex and subunit A from the free enzyme.
Table 4.
Selected main chain dihedral angles (φ, ϕ)° and rmsd (Å) values in E. coli HPRT
| Residue | HPRT Subunit A | HPRT Subunit B | HPRT-IMP Subunit A | HPRT-IMP Subunit B | HPRT-GMP Subunit A | HPRT-GMP Subunit B |
| Pro153 | (−72.9,169.1) | (−71.9,170.3) | (−69.5,169.5) | (−74.8,177.6) | (−67.0,167.9) | (−70.4,172.0) |
| Asp154 | (−69.7,69.7) | (−70.7,66.7) | (−75.1,77.1) | (−74.6,69.3) | (−83.7,79.1) | (69.1,70.8) |
| Glu155 | (−94.4,153.4) | (−92.9,153.7) | (−111.6,155.3) | (−90.4,152.4) | (−107.5,162.4) | (−96.4,148.0) |
| Phe156 | (−75.8,96.6) | (−76.1,97.4) | (−91.2,97.2) | (−81.5,88.9) | (−92.8,103.7) | (−75.7,97.5) |
| Val157 | (−107.7,147.3) | (−108.1,147.7) | (−87.2,154.0) | (−101.6,156.6) | (−92.4,147.2) | (−110.9,153.3) |
| Val158 | (−142.6,163.8) | (−142.5,164.4) | (−145.3,178.7) | (−150.8,169.5) | (−141.1,172.1) | (−144.6,173.0) |
| 153–158a | 0.00 | 0.02 | 0.32 | 0.11 | 0.30 | 0.10 |
a rmsd values for the Cα atoms when the segment from Pro153 to Val158 is superimposed.
A comparison of subunits A and B in the free enzyme structure shows there is little variation in dihedral angle for the residues between Pro153 and Val158 with the largest deviation being 1.5 degrees. Comparison of subunit A in the free enzyme with subunit A in the two complexes shows that changes in dihedral angle of up to 20 degrees can occur. A comparison of subunit A in the free enzyme with subunit B in the complexes shows changes in dihedral angles of up to 10 degrees. Analysis of rmsd values for Cα atoms in this region also support this idea (Table 4). Thus, these data provide further evidence that subunit B molecules represent an intermediate structure between free and fully bound enzyme. As mentioned above, comparison of the electron density in the structures support this notion, as the purine base portion of IMP or GMP is visualized only in subunit A and not in subunit B.
Although main chain dihedral angles vary over a wide range for all of the 20 amino acids, when each amino acid is analyzed individually the allowed (most favored) dihedral angles are restricted to a much narrower range (Laskowski et al. 1993). Comparison of dihedral angles of Asp154 to Val157 in the free, partially bound and fully bound complexes, shows that there is a gradual progression from relatively unfavored dihedral angles in the free enzyme through to more favored dihedral angles in the complexes. Thus, the structure of the complex (in this mobile region) represents a lower energy structure than that observed for the free enzyme.
Analysis of the χ2 angle of Phe156 in all of the structures shows similar variations as to those observed for the main chain dihedral angles. In the free enzyme and in subunit B molecules, the χ2 values range from 165 to 175 degrees, which are not energetically favorable, whereas in the fully bound complexes the χ2 values for Phe156 are 65 degrees for GMP and 86 degrees for IMP, both of which are close to the most favored angles. Thus, the side chain of Phe156 is in the same orientation in the native structure and in subunit B molecules of the complex. However, in the presence of a complete nucleoside monophosphate (subunit A) the side chain orientation changes. Thus again, the fully bound complex represents a lower energy structure than that observed for the free enzyme. Analysis of the free and complexed E. coli XGPRT structures (Vos et al. 1998) reveals that similar changes in dihedral angles are observed in that enzyme, resulting in Trp134 (the equivalent residue to Phe156) moving from a position away from the active site in the free structure to where it forms hydrophobic interactions with the purine base in the purine nucleoside monophosphate enzyme complex.
Comparison of the active sites of the two E. coli enzymes
The two E. coli 6-oxopurine PRTases adopt different binding modes for the purine bases. In most 6-oxopurine PRTases, the 6-oxo group forms a hydrogen bond with a conserved lysine residue. However, in E. coli XGPRT, although this lysine is present, the 6-oxo group is stabilized by a hydrogen bond to the backbone amide of Ile135. E. coli XGPRT has a water molecule that bridges the protein and the 2-exo group of either xanthine or guanine. This water has two contacts to the protein, one through the side chain of Glu136 and the other through Asp140. Under some circumstances, the interaction between the N7 atom of the purine and the conserved Asp (107 in E. coli HPRT) is maintained in both E. coli proteins. In the XGPRT-GMP complex the distance between the N7 atom and the carboxyl oxygen is 3.4 Å, which reduces to 2.8 Å in both XGPRT-guanine-cPRPP-Mg2+ and XGPRT-xanthine-cPRPP-Mg2+ complexes. The change in distance is due to two effects: first, there is a minor change in orientation of the purine base in the XGPRT-GMP complex compared with its position in the XGPRT-guanine-cPRPP-Mg2+ complex; second, a rotation of the side chain of Asp107 brings it closer to the N7 atom in the XGPRT-guanine-cPRPP-Mg2+ than in XGPRT-GMP. The structure of the XGPRT-XMP complex is not known, but presumably the same effect would be observed in that complex as is observed with the XGPRT complexes. In the E. coli HPRT-IMP complex, the N7 atom and side chain of Asp107 are already within hydrogen bonding distance (3.2 Å) and would, by analogy with XGRPT, presumably be in closer contact when hypoxanthine is bound together with cPRPP and Mg2+. In the E. coli HPRT-GMP complex, this distance is 3.7 Å, but, again, this distance may be reduced if guanine, cPRPP, and Mg2+, rather than GMP were bound.
The nonproline cis peptide bond
The electron density is of sufficient quality to unambiguously define a cis peptide bond between Leu46 and Arg47(Fig. 3B ▶). The cis conformation is observed in both subunits and in all three structures. Thus, in both free enzyme and in the product bound enzymes this energetically unfavorable backbone conformation is maintained. The effect of the cis peptide bond is to allow the main chain nitrogen of Arg47 to become exposed to the active site (Fig. 3 ▶). In the current structures, there are no direct contacts between this atom and any of the ligands in the active site. However, in the structures of human HGPRT-ImmucillinGP-PPi-Mg2+ (Shi et al. 1999b) and P. falciparum HGXPRT-ImucilliinHP-PPi-Mg2+ (Shi et al. 1999a), where a cis peptide bond is also observed, this nitrogen forms hydrogen bonds to the pyrophosphate oxygen atoms. The same cis peptide bond has also been noted in several other 6-oxopurine PRTase crystal structures including T. cruzi, T. foetus, and E. coli XGPRT, whereas in other structures this peptide bond has been reported in the trans conformation (Eads et al. 1994; Schumacher et al. 1996; Xu et al. 1997; Héroux et al. 1999). It has been suggested that a cis to trans isomerization may occur during catalysis with the binding of PRib-PP converting the peptide bond from trans to cis, whereas the release of pyrophosphate results in conversion of cis back to trans (Héroux et al. 1999). In E. coli HPRT, this does not appear to be the case, as in both the free and product bound structures the cis conformation is observed. These results suggest that the cis conformation is maintained throughout the catalytic cycle for this enzyme.
In E. coli HPRT, the side chain of Arg47 forms an ion pair with Glu65 from subunit B, whereas the side chain of Leu46 inserts into a hydrophobic pocket bounded by Phe68 from both subunits and Leu46 in subunit B. Comparison of the side chain orientations in the cis and trans conformations (Fig. 3 ▶) suggests that in a trans form of E. coli HPRT similar side chain interactions are possible. Thus, these structures do not reveal how this cis peptide bond is energetically stabilized. However, in the structure of a mutant form of the human HGPRT-HPP-PRibPP complex (Balendiran et al. 1999) where Lys68 (the equivalent residue to Arg47) was replaced by alanine a trans conformation was modeled, suggesting that disruption of the ionic interaction alone may be enough to revert the peptide bond back to the trans conformation.
The very high solvent content and relatively few crystal packing contacts means that these structures are likely to be highly representative of the solution structure. It has been suggested that tight packing in the human HGPRT-IMP crystals may have forced this structure into the higher energy cis configuration (Héroux et al. 1999). However, in E. coli HPRT this appears not to be the case.
Materials and methods
Materials
Guanine, xanthine, hypoxanthine, PRib-PP, IMP, and GMP were purchased from Sigma Chemical Company. DEAE-Trisacryl and Sepharose CL-4B were obtained from Amersham Pharmacia Biotech.
Amplification of the cDNA coding for E. coli HPRT
The nucleotide sequence of the gene coding for this enzyme was identified from the published genome sequence (Fujita et al. 1994). E. coli chromosomal DNA was prepared from DE3 cells (Sambrook et al. 1989) and primers were designed to amplify the cDNA based on the known nucleotide sequence. Automatic sequencing was performed at the Australian Genome Research Facility, The University of Queensland, and confirmed that the DNA sequence was identical to that previously published (Fujita et al. 1994).
Cloning, expression, and purification of E. coli HPRT
The cDNA coding for E. coli HPRT was cloned into pT7-7 as described for the cloning of human HGPRT (Free et al. 1990), P. falciparum HGXPRT (Keough et al. 1999), and E. coli XGPRT (Vos et al. 1997). The enzyme was then expressed and the cells lysed (Free et al. 1990). E. coli HPRT was purified to homogeneity by chromatography on DEAE-Trisacryl in 0.01 M phosphate at pH 7.0, 0.1 mM MgCl2, 1 mM DTT and eluted with a 0–0.3 M KCl gradient followed by chromatography on Sepharose CL-4B equilibrated in 0.05 M Tris-HCl, 0.01 M MgCl2, 1 mM DTT at pH 7.4. Fractions containing maximum specific activity were pooled and concentrated to 18 mg/mL and stored at −70 °C.
Enzyme assays and determination of protein concentrations
Protein concentrations were determined by measuring the A280 and using a molar absorption coefficient calculated by the method of Gill and Von Hippel (1989). The A1%1cm is 3.84. The activity of E. coli HPRT toward hypoxanthine, guanine, and xanthine was determined spectrophotometrically (Keough et al. 1998). The Ki values for the nucleoside monophosphates were determined in 0.1 M Tris-HCl, 12 mM MgCl2 at pH 7.4 and at 25°C.
Analytical ultracentrifugation and mass spectrometry
Before analytical ultracentrifugation, E. coli HPRT was dialyzed against 0.05 M Tris-HCl at pH 7.5, 1 mM MgCl2, 1 mM DTT. The molecular mass was determined by sedimentation equilibrium in a Beckman XL-I analytical ultracentrifuge operated at 4°C (Keough et al. 1999). Mass spectrometry was performed using a PESCIEX API 165 single quadrupole mass spectrometer equipped with an Ionspray atmospheric pressure ionization source, used in the positive mode. Before mass spectrometry the enzyme was diluted to ∼1 mg/mL and dialyzed against 0.1% acetic acid.
Crystallization
E. coli HPRT at 18 mg/mL (0.88 mM in terms of subunits) in 0.05 M Tris-HCl, 0.01 M MgCl2, 1 mM DTT at pH 7.4 was used for crystallization of the free enzyme. To prepare the enzyme–nucleoside monophosphate complexes, concentrated E. coli HPRT was incubated for 15 min with 2 mM IMP or GMP. Crystallization experiments were carried out using hanging drop vapor diffusion in VDX trays stored at 20°C; 1 μL of well solution and 1 μL of protein or complex solution were mixed together. Within 24 h, well solution consisting of 0.1 M HEPES at pH 7.5, 0.1 M sodium citrate produced diamond-shaped crystals (0.15 mm × 0.15 mm × 0.10 mm). Cryoprotection was achieved by soaking crystals for 5 min in well solution enhanced with 30% glycerol.
Data collection
X-ray data for the three structures were collected at the Advanced Photon Source, Chicago, on beam line 14D, λ = 1.000 Å using a Q1 CCD detector. Crystals were cryocooled to 100 K using an Oxford Cryosystems, Cryostream. Diffraction images were processed using the programs DENZO and SCALEPACK (Otwinowksi and Minor 1997). Results of the data collection and processing are summarized in Table 2.
Structure determination
The first of the three structures solved was the E. coli HPRT-IMP complex. The T. foetus HGXPRT dimer (Somoza et al. 1996) was used as the starting model for molecular replacement. Solutions for the rotation and translation functions were found using the program X-PLOR v3.851 (Brünger 1992b). The maximum peak in the rotation function was 4.0 σ above the mean. Translation searches were calculated for the space groups P3121 and P3221. The space group P3121 provided the strongest peak, 7.2 σ above the mean, and 2.0 σ above the next highest peak. Inspection of the packing of the molecules showed tetramers generated by crystallographic symmetry. Further analysis of this solution did not reveal any interpenetrating symmetry molecules. The initial Rfactor for data in the resolution range 15–4 Å was 0.498. Model building was performed using 2Fo–Fc and Fo–Fc electron density maps and the program O (Jones et al. 1991). Cycles of model building were alternated with molecular dynamics refinement, initially using X-PLOR v3.851 (Brünger 1992b) and later with program CNS (Brünger et al. 1998). Because there were two subunits per asymmetric unit and the resolution of the data moderate, noncrystallographic symmetry restraints were used until the Rfree was 0.30. Once these restraints were released, the Rfree value dropped to 0.28. 2Fo–Fc and Fo–Fc electron density was observed in both subunits for the phosphate group and ribose ring of the IMP molecule. However, only subunit A showed significant density for the base. As a result, the base in subunit B was not included in the final model. Incorporation of the nucleoside monophosphates, 98 water molecules, and fitting the cis peptide bond reduced the final Rfree to 0.243. There was insufficient electron density to model residues 1–4, 73–82, and the two carboxy-terminal residues 181–182 in both monomers. The side chains of Lys6 in both monomers and Gln166 in monomer B were modeled as alanine in the final structure.
Crystals of E.coli HPRT-GMP complex were isomorphous with crystals of the E. coli HPRT-IMP complex, thus the coordinates of the protein in the E.coli HPRT-IMP complex were used as the starting point for refinement of the GMP structure. Rfree was 0.289 after a cycle of rigid body refinement. Model building in O and simulated annealing refinement with CNS reduced Rfree to 0.260. As with the HPRT-IMP complex, 2Fo–Fc and Fo–Fc electron density was observed in both subunits for the phosphate group and ribose ring, and again only subunit A had significant density for the base. Residues and side chains not included in the HPRT-GMP structure were the same as for the HPRT-IMP structure, except that there was no side chain density for Arg82 in subunit B; therefore, this amino acid was modeled as alanine. Inclusion of 114 water molecules, individual B-factor refinement, and the cis peptide bond gave a final Rfree of 0.244.
Crystals of free E. coli HPRT had similar unit cell dimensions and the same space group as the complexes. Therefore, rigid body refinement using the protein coordinates of E.coli-HPRT-IMP was initially performed, resulting in an Rfree value of 0.270. Simulated annealing and a round of model building lowered the Rfree to 0.257. Placement of two magnesium ions (one in each monomer), 74 water molecules, the cis peptide bond, and individual B-factor refinement reduced the Rfree to a final value of 0.238. Residues and side chains not included in the free structure were the same as for the HPRT-GMP structure.
Ramachandran plots from PROCHECK (Laskowski et al. 1993) for the three structures showed all the residues, except for Leu179 of subunit A in the HPRT-GMP complex, in the most favored or allowed regions. When comparing the structures of the enzymes from E. coli, T. foetus, and human, the program lsq_improve in O (Jones et al. 1991) was used to calculate the rmsd values. The distance cutoff for inclusion of atoms in the alignment is the default value of 3.8 Å.
Accession numbers
The refined coordinates and structure factors have been deposited in the RCSB protein data bank and have the codes 1G9S and 1G9T for the IMP and GMP complexes, and 1GRV for the free enzyme, respectively.
Acknowledgments
We thank Drs. Harry Tong, Gary Navrotski, and Wilfried Schildkamp for assistance at beam line 14D, Advanced Photon Source, Argonne National Laboratory, Chicago, Illinois. We also thank Greg Dulley for some of the kinetic measurements and Thomas Sandmann and Amutha Kandasamy for assistance with crystallization. We acknowledge Professor Bryan T. Emmerson for his continued support and enthusiasm for the project. Figures were generated using MOLSCRIPT version 2.0.1 (Kraulis 1991), RASTER 3D version 2(Merrit and Murphy 1994), SETOR (Evans 1993), INSIGHT 98 (MSI Corporation), and IRIS SHOWCASE version 3.1 (Silicon Graphics Corporation). This work was supported by grants from the Australian Research Council (ARC).
The use of the BioCARS sector was supported by the Australian Synchrotron Research Program, which is funded by the Commonwealth of Australia under the Major National Research Facilities Program. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Basic Energy Sciences, Office of Energy Research, under Contract No. W-31-109-Eng-38.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
HPP, 7-hydroxy [4,3,-d] pyrazolo pyrimidine
PRib-PP, 5-phospho-α-D-ribosyl-1-pyrophosphate
cPRPP, (+)-(1S)-1-pyrophosphoryl-(2R,3R)-2,3-dihydroxy-(4S)-4-(phosphoryloxy-methyl)cyclopentane
DTT, dithiothreitol
PPi, pyrophosphate
ImmucillinHP, (1S)-1-(9-deazahypoxanthin-9-yl)-1,4-dideoxy-1,4-imino-D-ribotol 5-phosphate
ImmucillinGP, (1S)-1-(9-deazaguanin-9-yl)-1,4-dideoxy-1,4-imino-D-ribotol 5-phosphate
H/G/XPRTase, hypoxanthine/guanine/xanthine phosphoribosyltransferase. The nomenclature adopted to describe the 6-oxopurine PRTases from different sources is based on their ability to use the three naturally occurring purine bases, hypoxanthine (H), guanine (G), or xanthine (X)
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0201002.
References
- Balendiran, G. K., Molina, J.A., Xu, Y., Torres Martinez, J., Stevens, R., Focia, P.J., Eakin, A.E., Sacchettini, J.C., and Craig, S.P. III. 1999. Ternary complex structure of human HGPRTase, PRPP, Mg2+ and the inhibitor HPP reveals the involvement of the flexible loop in substrate binding. Protein Sci. 8 1023–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger, A.T. 1992a. The free R value: A novel statistical quantity for assessing the accuracy of crystal structures. Nature 355 472–474. [DOI] [PubMed] [Google Scholar]
- Brünger, A.T.1992bX-PLOR: A system for X-ray crystallography and NMR. Yale University, New Haven, CT.
- Brünger, A.T., Adams, P.D., Clore, G.M., Delano, W.L., Gros, P., Gross-Kunstleve, R.W., Jiang, J.-S., Kuazewski, J., Nilges, N., Pannu, N.S., Read, R.J., Rice, L.M., Simonson, T., and Warren, G.L. 1998. Crystallography and NMR System (CNS): A new software system for macromolecular structure determination. Acta Crystallogr. D 54 905–921. [DOI] [PubMed] [Google Scholar]
- Deo, S.S., Tseng, W.C., Saini, R., Coles, R.S., and Athwal, R.S. 1985. Purification and characterisation of Escherichia coli xanthine-guanine phosphoribosyltransferase produced by plasmid pSV2gpt. Biochim. Biophys. Acta. 839 233–239. [DOI] [PubMed] [Google Scholar]
- Eads, J.C., Scapin, G., Xu, Y., Grubmeyer, C., and Sacchettini, J.C. 1994. The crystal structure of human hypoxanthine-guanine phosphoribosyltransferase with bound GMP. Cell 78 325–334. [DOI] [PubMed] [Google Scholar]
- Evans, S.V. 1993. SETOR: Hardware lighted three-dimensional solid model representations of macromolecules. J. Mol. Graph. 11 134–138. [DOI] [PubMed] [Google Scholar]
- Focia, P.J., Craig, S.P. III, and Eakin, A.E. 1998a. Approaching the transition state in the crystal structure of a phosphoribosyltransferase. Biochemistry 37 17120–17127. [DOI] [PubMed] [Google Scholar]
- Focia, P.J., Craig, S.P. III, Nieves Alicea, R., Fletterick, R.J., and Eakin, A.E. 1998b. A 1.4 Å crystal structure for the hypoxanthine phosphoribosyltransferase of Trypanosoma cruzi. Biochemistry 37 15066–15075. [DOI] [PubMed] [Google Scholar]
- Free, M.L., Gordon, R.B., Keough, D.T., Beacham, I.R., Emmerson, B.T., and de Jersey J. 1990. Expression of active human hypoxanthine-guanine phosphoribosyltransferase in Escherichia coli and characterisation of the recombinant enzyme. Biochim. Biophys. Acta 1087 205–211. [DOI] [PubMed] [Google Scholar]
- Fujita, N., Mori, H., Yura, T., and Ishihama, A. 1994. Systematic sequencing of the Escherichia coli genome: analysis of the 2.4–4.1 min (110,917–193,643 bp) region. Nucleic Acid Res. 22 1637–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill, S.C. and von Hippel. P.H. 1989. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 182 319–326. [DOI] [PubMed] [Google Scholar]
- Gots, J.S. and Benson, C.E. 1974. Genetic control of bacterial purine phosphoribosyltransferase and an approach to gene enrichment. In Purine metabolism in man (eds. O. Sperling, A. de Vries, and J. Wyngaarden), pp. 33–39. Plenum Press, New York. [DOI] [PubMed]
- Héroux, A., White, E.L. Ross, L.J. Davis, R.L., and Borhani, D.W. 1999. Crystal structure of Toxoplasma gondii hypoxanthine-guanine phosphoribosyltransferase with XMP, pyrophosphate and two Mg2+ ions bound: Insights into catalytic mechanism. Biochemistry 38 14495–14506. [DOI] [PubMed] [Google Scholar]
- Héroux, A., White, E.L. Ross, L.J., Kuzin, A.P., and Borhani, D.W. 2000. Substrate deformation in a hypoxanthine-guanine phosphoribosyltransferase ternary complex: The structural basis for catalysis. Structure 8 1309–1318. [DOI] [PubMed] [Google Scholar]
- Jones, T.A, Zou, J.Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A47 110–119. [DOI] [PubMed] [Google Scholar]
- Kabsch, W. and Sander, C. 1983. Dictionary of protein secondary structure: Pattern recognition of hydrogen bond and geometrical features. Biopolymers 22 2577–2637. [DOI] [PubMed] [Google Scholar]
- Keough, D.T., Ng, A.L, Emmerson, B.T., and de Jersey, J. 1998. Expression and properties of recombinant P. falciparum hypoxanthine-guanine phosphoribosyltransferase. Adv. Exp. Med. Biol. 431 735–739. [DOI] [PubMed] [Google Scholar]
- Keough, D.T., Ng, A.L., Winzor D.J., Emmerson, B.T., and de Jersey, J. 1999. Purification and characterization of Plasmodium falciparum hypoxanthine-guanine-xanthine phosphoribosyltransferase and comparison with the human enzyme. Mol. Biochem. Parasitol. 98 29–41. [DOI] [PubMed] [Google Scholar]
- Kraulis, P.J. 1991. MOLSCRIPT—a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 24 946–950. [Google Scholar]
- Laskowski, R., Macarthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26 283–291. [Google Scholar]
- Matthews, B.W.1968. Solvent content of protein crystals. J. Mol. Biol. 33 491–497. [DOI] [PubMed] [Google Scholar]
- Merritt, E.A. and Murphy, M.E.P. 1994. Raster 3D Version 2.0: A program for photorealistic molecular graphics. Acta Crystallogr. D50 869–873. [DOI] [PubMed] [Google Scholar]
- Munagala, N.R. and Wang, C.C. 1998. Altering the purine specificity of hypoxanthine-guanine-xanthine phosphoribosyltransferase from Tritrichomonas foetus by structure-based point mutations in the enzyme protein. Biochemistry 37 16612–16619. [DOI] [PubMed] [Google Scholar]
- Musick, W.D. 1981. Structural features of the phosphoribosyltransferases and their relationship to the human deficiency disorders of purine and pyrimidine metabolism. CRC Crit. Rev. Biochem. 11 1–34. [DOI] [PubMed] [Google Scholar]
- Neuhard, J. and Nygaard, P. 1987. Purines and pyrimidines. In Escherichia coli and Salmonella typhimurium: Cellular and molecular biology, vol 1 (eds. F.C. Neihardt, J.L. Ingraham, K. Brooks Low, B. Magasanik, M. Schaechter, and H.E. Umbarger), pp. 445–473. American Society for Microbiology, Washington, D.C.
- Otwinowksi Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276 307–326. [DOI] [PubMed] [Google Scholar]
- Sambrook, J., Fritsch, E.F., and Maniatis, R. 1989. In Molecular cloning. A laboratory manual (ed. N. Ford), pp. E3–E4. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Scapin, G., Grubmeyer, C., and Sacchetini, J.C. 1994. Crystal structure of orotate phosphoribosyltransferase. Biochemistry 33 1287–1294. [DOI] [PubMed] [Google Scholar]
- Schumacher, M.A., Carter, D., Ross, D.S., Ullman, B., and Brennan, R.G. 1996. Crystal structures of Toxoplasma gondii HGXPRTase reveal the catalytic role of a long flexible loop. Nat. Struct. Biol. 3 881–887. [DOI] [PubMed] [Google Scholar]
- Shi, W., Li, C.M., Tyler, P.C., Furneaux, R.H., Cahill, S.M., Girvin, M.E., Grubmeyer, C., Schramm, V.L., and Almo, S.C. 1999a. The 2.0 Å structure of malarial purine phosphoribosyltransferase in complex with a transition-state analogue inhibitor. Biochemistry 38 9872–9880. [DOI] [PubMed] [Google Scholar]
- Shi, W., Li, C.M., Tyler, P.C., Furneaux, R.H., Grubmeyer, C., Schramm, V.L., and Almo, S.C. 1999b. The 2.0 Å structure of human hypoxanthine-guanine phosphoribosyltransferase in complex with a transition-state analog inhibitor. Nat. Struct. Biol. 6 588–593. [DOI] [PubMed] [Google Scholar]
- Shi, W., Mungala, N.R., Wang, C.C., Li, C.M., Tyler, P.C., Furneaux, R.H., Grubmeyer, C., Schramm, V.L., and Almo, S.C. 2000. Crystal structures of Giardia lamblia guanine phosphoribosyltransferase at 1.75 Å. Biochemistry 39 6781–6790. [DOI] [PubMed] [Google Scholar]
- Somoza, J.R., Chin, M.S., Focia, P.J., Wang, C.C., and Fletterick, R.J. 1996. Crystal structure of the hypoxanthine-guanine-xanthine phosphoribosyltransferase from the protozoan parasite Tritrichomonas foetus. Biochemistry 35 7032–7040. [DOI] [PubMed] [Google Scholar]
- Ullman, B. 1995. Hypoxanthine-guanine phosphoribosyltransferase as a therapeutic target in protozoal infections. Infect. Agents Dis. 4 29–40. [PubMed] [Google Scholar]
- Vos, S., de Jersey, J., and Martin, J.L. 1997. Crystal structure of Escherichia coli xanthine phosphoribosyltransferase. Biochemistry 36 4125–4134. [DOI] [PubMed] [Google Scholar]
- Vos, S., Parry, R.J., Burns, M.R., de Jersey, J., and Martin, J.L. 1998. Structures of the free and complexed forms of Escherichia coli xanthine-guanine phosphoribosyl transferase. J. Mol. Biol. 282 875–889. [DOI] [PubMed] [Google Scholar]
- Xu, Y., Eads, J., Sacchettini, J.C., and Grubmeyer, C. 1997. Kinetic mechanism of human hypoxanthine-guanine phosphoribosyltransferase: Rapid phosphoribosyl transfer chemistry. Biochemistry 36 3700–3712. [DOI] [PubMed] [Google Scholar]
- Xu, Y. and Grubmeyer, C. 1998. Catalysis in human hypoxanthine-guanine phosphoribosyltransferase: Asp 137 acts as a general acid/base. Biochemistry 37 4114–4124. [DOI] [PubMed] [Google Scholar]