

Abstract
Here we present a comparison between protein fragments produced by limited proteolysis and those identified by computational cutting based on the building block folding model. The principles upon which the two methods are based are different. Limited proteolysis of natively folded proteins occurs at flexible sites and never at the level of chain segments of regular secondary structure such as α-helices. Therefore, the targets for limited proteolysis are locally unfolded regions. In contrast, the computational cutting algorithm considers the compactness of the fragments, their nonpolar buried surface area, and their isolatedness, that is, the surface area which was buried prior to the cutting and becomes exposed subsequently. Despite the different criteria, there is an overall correspondence between sites or regions of limited proteolysis with those identified by computational cutting. The computational cutting method has been applied to several model proteins for which detailed limited proteolysis data are available, namely apomyoglobin, cytochrome c, ribonuclease A, α-lactalbumin, and thermolysin. As expected, more cuts are obtained computationally than experimentally and the agreement is better when a number of proteolytic enzymes are used. For example, cytochrome c is cleaved by thermolysin at 56–57, 45–46, and at 80–81, and by proteinase K at 48–49 and 50–51. Incubation of the noncovalent and native-like complex of cytochrome c fragments 1–56 and 57–104 with proteinase K yielded the gapped protein species 1–48/57–104 and finally 1–40/57–104. Computational cutting of cytochrome c reproduced the major experimental observations, with cuts at 47, 64–65 or 65–66 and 80–81 and an unstable 32–47 region not assigned to any building block. The next step, not addressed in this work, is to probe the ability of the generated fragments to fold independently. Since both the computational algorithm and limited proteolysis attempt to dissect the protein folding problem, the general agreement between the two procedures is gratifying. This consistency allows us to propose the use of limited proteolysis to produce protein fragments that can adopt an independent folding and, therefore, to study folding intermediates. The results of the present study appear to validate the building block folding model and are in line with the proposal that protein folding is a hierarchical process, where parts constituting local minima of energy fold first, with their subsequent association and mutual stabilization to finally yield the global fold.
Keywords: Folding intermediates, limited proteolysis, protein anatomy, protein flexibility, protein folding, protein fragments
How the amino acid sequence in the polypeptide chain directs and specifies the three-dimensional protein fold is still an open problem (Matthews 1995). A number of experimental and computational methods have been developed to address the protein folding problem. There is a growing body of evidence indicating that while folding can proceed along different routes, some paths are more populated than others (Dill and Chan 1997; Pande et al. 1998). This has been largely shown through studies of intermediate states (Kim and Baldwin 1990; Privalov 1996). However, a major difficulty in experimental studies of intermediate states is the transient nature of these states, necessitating fast techniques (Evans and Radford 1994; Eaton et al. 1997; Roder and Colon 1997; Chamberlain and Marqusee 2000).
One approach to simplify the protein folding problem is to produce protein fragments and determine whether they can form independent folding entities (Wetlaufer 1973, 1981; Peng and Kim 1994; Wu et al. 1994). Protein fragments can be produced experimentally or computationally. On the experimental side, the dissection strategy involves limited proteolytic or chemical cleavage of a protein, or alternatively synthesis of long enough peptides (Fontana et al. 1997a, 1999; Peng and Wu 2000). These are coupled with studies of the stabilities and conformational properties of the fragments and their comparisons with the corresponding native protein states. If the fragments constitute protein domains or subdomains with high enough population times, a correspondence between these and the native protein can be observed (Peng and Wu 2000). On the computational side, a reasonable strategy is to iteratively dissect the native protein fold, identifying the conformationally fluctuating building blocks. Through hierarchical assembly of these blocks, the independently folding hydrophobic units are obtained (Crippen 1978; Rose 1979; Wodak and Janin 1981; Zehfus and Rose 1986; Zehfus 1993; Panchenko et al. 1996, 1997; Tsai and Nussinov 1997; Tsai et al. 1998, 1999a, b, 2000).
Here our goal was to carry out an extensive comparison of limited proteolysis data (Fontana et al. 1997a,b; Polverino de Laureto et al. 1995, 1997, 1999, 2001) with the results of the computational cutting based on the building block folding model (Tsai et al. 2000). Overall, our results show that similar regions are cleaved by both approaches. We give an outline of the experimental strategy as well of the computational approach. We provide a case-by-case comparison of the experimental and computational results, and we conclude by speculating on the implications for protein folding schemes (Karplus and Weaver 1994).
The limited proteolysis approach
The basic premise of limited proteolysis (Neurath 1980) is that the protease can bind to the protein substrate at sites whose conformations are complementary to its active site (Fontana et al. 1993, 1997a,b, 1999). Since a rigid protein structure has a specific conformation, the chances of conformational complementarity are relatively small and thus native globular proteins are usually quite resistant to proteolysis. On the other hand, the flexible regions of the native protein imply existence in a range of local conformational isomers. Among these, there are likely to be some conformers fitting the enzyme's active site. Proteases such as thermolysin, subtilisin, proteinase K, and pepsin display broad substrate binding, suggesting their capability to accommodate a broader range of conformations of the polypeptide substrate at their active sites. Limited proteolysis does not occur at the numerous sites scattered across the protein surface, but is restricted to a few specific locations. These sites have shown a good correlation with larger crystallographic B-factors, uncertain electron density, and larger dispersion values of backbone angles. This correlation was first demonstrated with thermolysin (Fontana et al. 1986) and subsequently with other proteins (Fontana et al. 1993, 1997b). These regions have also shown higher flexibility in molecular dynamics simulations, for example, in apomyoglobin (see Fontana et al. 1997b for references). As expected, the sites of limited proteolysis in proteins are sufficiently exposed to be able to bind to the protease's active site. However, exposure is a required characteristic of the cleavage site, but not sufficient to explain the specific proteolysis. Indeed, flexibility of the chain segment suffering proteolytic attack is the key parameter dictating limited proteolysis (Fontana et al. 1986). In several studies it has been shown that there is a clear-cut correlation between sites or regions of enhanced segmental mobility and sites of limited proteolysis (Fontana et al. 1986, 1993, 1997a,b, 1999; Polverino de Laureto et al. 1995, 1999).
A modeling study by Hubbard (Hubbard et al. 1994; Hubbard 1998) showed that the site of limited proteolysis requires a conformational change of a stretch of up to 12 residues (Schechter and Berger 1967). That it is a region, rather than a specific site, is also illustrated by the finding that, if several proteases are used, one observes that cleavage takes place over a stretch of peptide bonds. Inspection of the cleavage sites on the globular protein substrate reveals that they never occur at the level of α-helices, but largely at loops (Fontana et al. 1993, 1999). Had proteolysis taken place at a helical segment, the helix is likely to have been destroyed by end-effects and loss of the cooperative hydrogen bonds that stabilize it. Furthermore, the newly created charged ends, if buried, might conceivably destabilize the protein core. Hence, limited proteolysis occurs preferentially at those loops which display inherent conformational flexibility, whereas the protein core remains quite rigid and thus resistant to proteolysis (Fontana et al. 1986, 1993, 1997a).
The limited proteolysis approach has also been used to probe the nonnative or molten globule (MG) (Ptitsyn 1995; Arai and Kuwajima 2000) state of proteins when exposed to a variety of solvent conditions (Fontana et al. 1997a). The acid-induced partly folded state (A-state) or the apo-state of α-lactalbumin at neutral pH (Polverino de Laureto et al. 1995, 1999, 2001), as well as the apo form of myoglobin (Fontana et al. 1997b) have been subjected to limited proteolysis, obtaining results consistent with those reached by NMR measurements (Arai and Kuwajima 2000). Moreover, the conformational state of proteins dissolved in aqueous trifluoroethanol (TFE) has been analyzed by using thermolysin as a proteolytic probe. Proteolysis in aqueous TFE yields specific and large protein fragments, suggesting that even under such conditions proteins retain partly rigid structures (Fontana et al. 1997a). Overall, the limited proteolysis data of several proteins in their MG state indicated a correlation between their native and MG states (Polverino de Laureto et al. 1995, 1998, 1999, 2001), in agreement with results obtained by other investigators using other physicochemical techniques, mostly NMR and hydrogen exchange measurements (Arai and Kuwajima 2000). Therefore, it appears that limited proteolysis can be used as a reliable probe of structure and dynamics of both native and partly folded proteins (Mihalyi 1978; Price and Johnson 1990; Fontana et al. 1993, 1999). Studies of the conformations of the protein fragments (Wu et al. 1994; Peng and Wu 2000) and of their complexes (Taniuchi et al. 1986; Spolaore et al. 2001) may yield information on intermediate states of proteins and their folding pathways.
Computational dissection: The building block folding model
The building blocks are contiguous sequence fragments of variable sizes, and their stability derives from local interactions. Some building blocks are highly stable, whereas others may be only marginally so. In some cases the conformations of the building blocks that we observe in the native state are those that the building blocks would manifest as peptide fragments in solution, but this does not always occur. Nevertheless, the conformation of the building block within the native protein fold is the one that the building block likely attains early in the folding process.
According to the building block folding model, protein folding is a hierarchical event (Tsai et al. 1998, 1999a,b, 2000; Kumar et al. 1999, 2000; Ma et al. 1999, 2000). In the first step, building blocks form. Even if the native building block conformation is marginally stable and would exist in a relatively low population, the native conformer is more highly populated than all other conformers. Otherwise, it would not constitute a local minima in the building blocks fragment map (Tsai et al. 2000). In the next hierarchical step, the building blocks associate via combinatorial assembly (Tsai et al. 1998, 1999a). The only difference between the binding of building blocks and the binding of larger stable units, such as domains or subunits, or the binding of different molecules in a complex, is the shorter population time of the building block conformer. In this binding event, among the range of conformations present, the ones that bind are the most complementary. Hence, folding is largely a process of selection of building block conformations. With binding of the native conformers, the population would shift in their favor, further driving the folding reaction. Through their binding, they mutually stabilize each other, leading to the formation of stable hydrophobic folding units. In the next step, again through selection, the most complementary hydrophobic folding units within the population bind to form the domains. In each of the binding events, the changing conditions lead to shifts in the populations. Experimentally, complementing fragments provide a system for studying protein folding (Taniuchi et al. 1986; Fisher and Taniuchi 1992; Yang et al. 1998; Spolaore et al. 2001), consistent with the idea that intermolecular binding resembles intramolecular folding events (Tsai et al. 1998, 1999a). Figure 1 ▶ gives a flow chart of the cutting procedure, and its legend outlines the major steps.
Fig. 1.
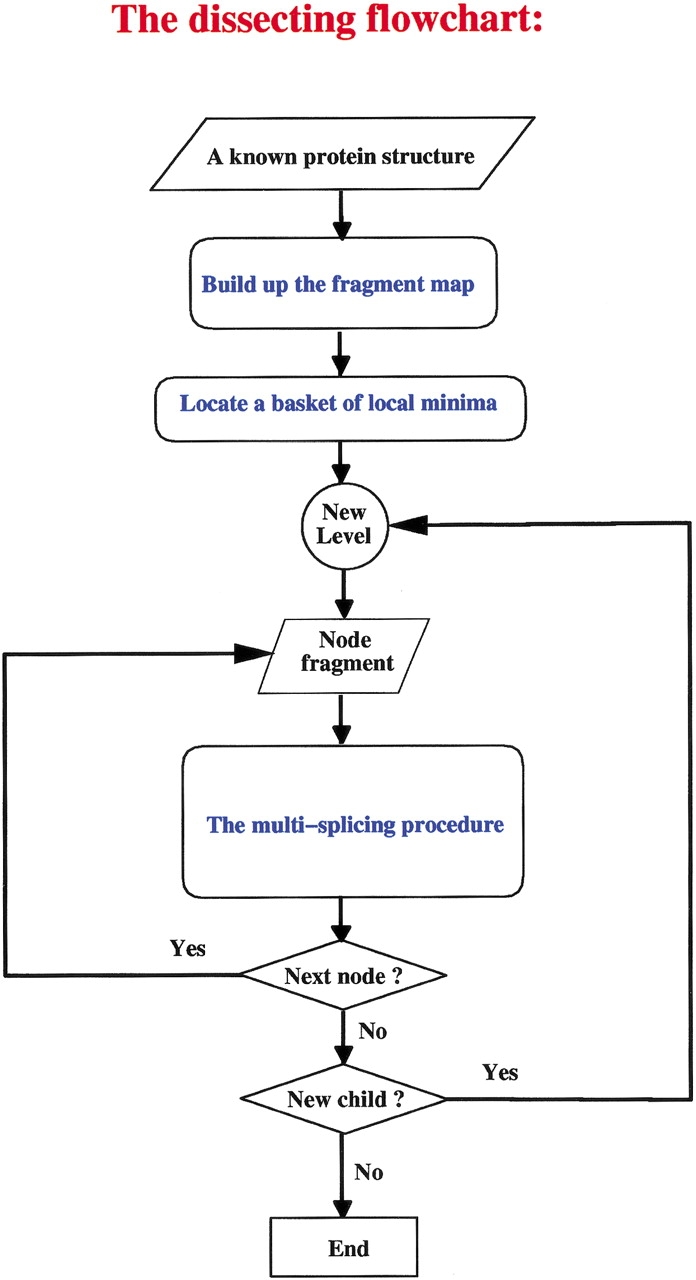
Flow chart of the algorithm. The algorithm initiates with the coordinates of the native structure, and a moving window of successively increasing length slides across the sequence. The stability of each fragment in each window is calculated using a chain length-independent scoring function. A basket of building blocks is collected by locating all local minima on the fragment map. A local minimum is the highest value in a defined local region. If a candidate building block is the highest-scoring one, the basket of building blocks is registered. The chain is iteratively cut, from the top down, allowing multiple dissections at each iterative level. This results in a hierarchy of contiguous fragments, and each node in the descending anatomy tree is a building block segment. The native structure is the root node of the tree, and the locations of the building blocks correspond to the end nodes of the top-down tree. To be able to carry out the dissection, the most critical component is a fragment size-independent scoring function, which measures the relative stability of all candidate building blocks. The empirical scoring function is based on the compactness of the building block conformation, degree of isolation, and hydrophobicity. The dissection procedure is applied progressively to the most stable fragments. An attractive feature of the anatomy tree is that, at the completion of the dissecting procedure, it yields a likely folding pathway. Moreover, examination of the minima among the fragments yields the alternate routes. Further details are given in Tsai et al. (2000).
To be able to interpret the computational data and their potential correspondence with the experimental cleavages of proteins, it is important to note some technical points about the computational cuttings and the way they are depicted in the figures. First, in the cutting procedure, a seven-residue overlap is allowed. Second, a region whose stability is not high enough to constitute a valid building block and whose addition to a neighboring building block does not increase the stability of that block is defined as unassigned. In the pictorial representation, such a building block has a light gray color. The minimum size of a building block is defined as 15 residues (Tsai et al. 2000).
For all of the protein cases described below, the tables list all building blocks minima. Here we provide only the table and the figures relating to the first protein case herewith examined (apomyoglobin, Table 1, Figure 2a–c ▶). The other tables are given in the Supplemental Material in the journal electronic site. These building blocks minima correspond to the horizontal (blue and red) lines given in the fragment maps. The building blocks relating to the major folding pathway of the protein are drawn in red, and those depicted in blue take part in alternate folding pathways. The x-axis represents the position of the building block and the y-axis its size. In general, which pathway is actually the major one is the outcome of external conditions. Hence, it is conceivable that the pathway we depict as the major one is not the most populated under a different set of physical conditions. Therefore, it is important to inspect all building block fragment minima, rather than only the major cuts.
Table 1.
Computational cutting of horse heart myoglobina
| No. | Range | Size | Z | I | H | Score |
| 1 | 1–153 | 153 | 1.575 | 0.000 | 0.795 | 4.341 |
| 2 | 25–65 | 41 | 1.422 | 0.143 | 0.699 | 3.408 |
| 3 | 22–69 | 48 | 1.430 | 0.153 | 0.700 | 3.080 |
| 4 | 14–119 | 106 | 1.637 | 0.145 | 0.733 | 0.858 |
| 5 | 72–151 | 80 | 1.614 | 0.188 | 0.716 | 0.739 |
| 6 | 2–82 | 81 | 1.630 | 0.167 | 0.705 | 0.554 |
| 7 | 21–111 | 91 | 1.666 | 0.168 | 0.708 | 0.062 |
| 8 | 86–153 | 68 | 1.596 | 0.220 | 0.692 | 0.023 |
| 9 | 108–136 | 29 | 1.488 | 0.273 | 0.616 | −0.931 |
| 10 | 2–99 | 98 | 1.728 | 0.166 | 0.700 | −0.934 |
| 11 | 101–143 | 43 | 1.527 | 0.339 | 0.630 | −2.229 |
| 12 | 71–97 | 27 | 1.569 | 0.300 | 0.561 | −3.544 |
| 13 | 71–92 | 22 | 1.547 | 0.311 | 0.550 | −3.650 |
| 14 | 92–128 | 37 | 1.647 | 0.311 | 0.589 | −3.771 |
| 15 | 91–110 | 20 | 1.580 | 0.353 | 0.556 | −4.196 |
| 16 | 2–24 | 23 | 1.556 | 0.355 | 0.547 | −4.378 |
a The computed building blocks of horse heart myoglobin (PDB 1wla) correspond to all (blue and red) fragments depicted in Figure 2A ▶, and they are ranked (first column) by their stability. The second column gives their starting and end position in the 153-residue chain of apoMb. The third column gives their size. The three components contributing to their stability, i.e., the compactness (Z), the isolatedness (I) and the nonpolar surface area they bury (H) are listed next, with the total stability score given in the last column. See text and Tsai et al. (2000) for further details, and compare the data with those shown in Figure 2A–C ▶.
Fig. 2.


The results of the computational cutting of horse heart myoglobin (PDB 1wla). (A) The fragment map, where the x-axis is the location and the y-axis the size. All local minima are depicted in blue, and the building blocks taking part in the major folding pathway are in red. (B) The anatomy tree illustrating the major folding pathway. Each node corresponds to a fragment. The start and end positions of the building blocks are noted. These building blocks correspond to those drawn in red in part A. The stability of the building block is given next to the sequence position. The letters in parentheses denote the hydrophobic folding unit to which the algorithm assigns the building block. (C) A pictorial representation of the fold, building blocks, and hydrophobic folding units. The top row gives the iterative cuttings. Each building block is drawn in a different color. The bottom row depicts the results of the combinatorial assembly of the building blocks into hydrophobic folding units. All building blocks are specified in Table 1. To compare with the experimental dissection: Fontana et al. (1997) cut it in the 89–96 region (major cuts) and around position 30 (minor cut). These are the cumulative results by several proteolytic enzymes. NMR data indicate flexibility/unfolding of the 82–102 region (Eliezer and Wright 1996) and around 30 (Lecomte et al. 1996). The building blocks are specified in Table 1.
The anatomy tree showing the major folding pathway and the stability scores are noted in part B of the Figures. The larger the stability score value, the more stable is the building block. The stability is calculated using fragment-length independent parameters, such as nonpolar buried surface area, compactness, and isolatedness (i.e., the surface area that was buried prior to the cutting and became exposed subsequently). The larger the nonpolar buried surface area and the compactness and the smaller the isolatedness value, the more stable is the building block. Each block is given in a different color, and they correspond to the red fragments in the fragment map; their connecting red lines relate to the anatomy tree. The bottom row gives the outcome of the combinatorial assembly of the building blocks into hydrophobic folding units.
Below we compare the experimental and the computational cutting. We stress that limited proteolysis reflects the local unfolding and hence relates to the unfolding of the protein. In contrast, the computational cutting reflects protein folding pathways.
Results
Apomyoglobin
Limited proteolytic cleavage has been carried out on horse apomyoglobin (apoMb; myoglobin without the heme), a small 153-residue monomeric protein (Fontana et al. 1997b). A variety of spectroscopic studies have indicated that in solution at neutral pH, the structure of apoMb retains features of the structure of the holoprotein, although CD measurements have indicated that the helical content of apoMb is lower than that of holoMb (Hirst and Brooks 1994; Lin et al. 1994). Limited proteolysis of apoMb by subtilisin, thermolysin, chymotrypsin, and trypsin occurs at chain segment 89–96. In the crystal structure, this region encompasses the F-helix (residues 82–97), suggesting that this helix is disordered in apoMb. Indeed, NMR data have shown that at neutral pH a recombinant isotopically labeled (15N, 13C) sperm-whale apoMb has a conformation similar to that of the holoprotein, with the exception of the 82–102 region encompassing F-helix (Eliezer and Wright 1996; Eliezer et al. 1998; Cavagnero et al. 2001). Here, resonances were missing, the outcome of conformational fluctuations. A minor cleavage site has been observed near residue 31 (Fontana et al. 1997b), in line with NMR data showing chain fraying around that site (Lecomte et al. 1996). Detailed analysis of the proteolytic cleavages of apoMb (Fontana et al. 1997b) reveals that the thermolysin digest contained fragments 1–88 and 89–153, as well as fragment 1–32 as a minor component; the subtilisin digest contained fragments 90–153, 92–153, and 94–153; the trypsin digest contained fragments 1–96, 97–153, and 1–31.
The results of computational cutting of native myoglobin (PDB 1wla; Bernstein et al. 1977) are presented in Figure 2 ▶ and in Table 1. Figure 2A ▶ depicts the fragment map, Figure 2B ▶ the anatomy tree and Figure 2C ▶ shows the cuttings pictorially on the protein 3D fold, illustrating the hierarchical cutting stages (top row) and the combinatorially assembled hydrophobic folding units (bottom row). There are two cuts at the second step, around positions 22–24 and 69–72. The resulting 72–151 fragment is further split into fragments 71–97, 91–110, and 108–136. This is the major folding pathway predicted by the algorithm. Comparison of these results of computational cutting with the limited proteolysis data and the NMR structure (Eliezer and Wright 1996; Eliezer et al. 1998; Cavagnero et al. 2001) shows a nice agreement. The 91–110 building block is very unstable (a score of −4.20). The 71–97 (or 71–92) fragment also shows a marked instability score (−3.54 or −3.65, respectively). The cuts at around 91 and 108 are observed a number of times in the listing of building blocks minima in the table (for example, building block No. 6 in the table spans residues 2–82; No. 8 spans residues 86–153; No. 10 stretches between 2–99; No. 11 starts at residue 101; No. 14 initiates at residue 92). Remembering the allowed seven-residue overlap in the cutting, these results indicate that the region starting at residue 84 is repeatedly cut by the algorithm. Furthermore, while we do not see a cut near residue 31, we observe cuts around residue 24 (fragment 2–24), thus within the seven-residue overlap allowed by the computational procedure (see above). Considering that proteases do not cleave in the middle of helices and that a conformational destabilization of 10–12 residues appears to be required for proteolysis (Hubbard et al. 1994), we conclude that there is a consistency between experiments and computations. Indeed, comparison of the computationally derived fragments with those generated by proteolysis (Fontana et al. 1997b) yields the following picture. The major 1–88 and minor 1–32 thermolysin fragments are quite similar to fragment 2–82 (with a score of 0.88, see Table 1) and fragment 2–24 (with a significantly lower score, −4.38). The 90–153, 92–153, and 94–153 subtilisin fragments are quite similar to fragment 86–153 (with a stability of 0.02). The 1–96, 97–153, and 1–31 (minor) trypsin fragments are rather similar to 2–99 (with a stability of −0.93), 86–153 (0.02), and 2–24 (−4.38), respectively.
Cytochrome c
Cytochrome c (cyt c) has been subjected to limited proteolysis by thermolysin in 50% aqueous (v/v) TFE at neutral pH (Fontana et al. 1995). A major cut has been observed at peptide bond 56–57, generating the two fragments 1–56 and 57–104. Additional but minor cleavages have been observed at peptide bonds 45–46 and 80–81. Wang et al. (1998) probed the heat-induced unfolding of cyt c using proteinase K as proteolytic probe, and they observed initial cuts at peptide bonds 48–49 and 50–51. Spolaore et al. (2001) used limited proteolysis by proteinase K at neutral pH on the noncovalent and native-like complex of fragments 1–56 and 57–104. In the complex, the heme was covalently bound to fragment 1–56. Proteolysis of nicked cyt c yields a gapped protein complex given by fragments 1–48 and 57–104. Further digestion leads to fragments 1–40 and 57–104 in a still folded complex.
The results of the computational cutting of cyt c (PDB 1giw) are given in Figure 3 ▶ and Supplemental Material Table 1. The table lists all building block fragments. There is a cut at position 47, with the region between residues 32 and 47 being an unassigned segment, implying that its stability is very low. This region includes the experimental minor 45–46 cut (Fontana et al. 1995) and the chain segment digested by proteinase K in the complex 1–56/57–104 (Spolaore et al. 2001). A cut is observed at residue 64 or 66. An additional cut at residue 85 is close (and within the seven-residue overlap) of the minor experimental cut at 80–81. The 64 or 66 cut of the algorithm is at a distance of eight residues from the 56–57 experimental cut. Hence, while our major cutting at residue 64 is eight residues removed from the major experimental 56 site, considering that the cutting is carried out in TFE and that a single protease is used rather than a battery of proteases, as well as the 12-residue region suggested to be distorted for proteolysis to occur (Hubbard et al. 1994), the results obtained by the experimental and computational cutting appear to be consistent. Proteolytic fragments include 1–56, 57–104, 1–45, 57–80, and 81–104 (Fontana et al. 1995). In Table 1 of the Supplemental Material we see 10–64 (with a stability of −1.16), 55–95 (1.04), 10–48 (very unstable, −3.58), 47–80 (0.12), and 81–104 (−3.61). The major computational cut observed at residue 47, with a low stability region preceding it, is in agreement with the Wang et al. (1998) cleavages at peptide bonds 48–49 and 50–51. Consistent with the results of Spolaore et al. (2001), as Figure 3 ▶ and Table 1 in the Supplemental Material show, cyt c consists of a single hydrophobic folding unit containing the entire sequence, with the 32–47 region being unstable and hence not assigned to any building block.
Fig. 3.

The fragment map obtained by computational cutting of cytochrome c (PDB 1giw). For further details see the legend for Figure 2 ▶. All of the building blocks are specified in Table 1 of the Supplemental Material. To compare with the experimental dissection: Fontana et al. (1995) cut cyt c by thermolysin in TFE at the peptide bond 56–57 (major cut) and at 45–46 and 80–81 (minor cuts). Wang et al. (1998) observed that region 48–51 of cyt c is cleaved by proteinase K.
Ribonuclease A
In an early classical study of limited proteolysis of proteins, Richards (1958) cleaved bovine pancreatic ribonuclease A (RNase) by subtilisin to yield the noncovalent, functionally active complex 1–20/21–124 (nicked RNase). Thermolysin has been used to probe both the thermal unfolding process of RNase (Arnold et al. 1996) as well as its conformational state when dissolved in aqueous TFE (Polverino de Laureto et al. 1997, 1998). CD measurements have shown that in aqueous TFE, RNase has a larger content of α-helix and reduced tertiary structure (Gast et al. 1999). Thermolysin cleaves the 124-residue chain of RNase, both in water upon moderate heating and in aqueous TFE, at peptide bond 34–35, with a slower cleavage at 45–46. In the absence of TFE, native RNase is resistant to cleavage by thermolysin. It was proposed that both TFE and heat induce a relaxed state of RNase, with a highly flexible 30–46 segment, a favored substrate for proteolysis (Polverino de Laureto et al. 1997).
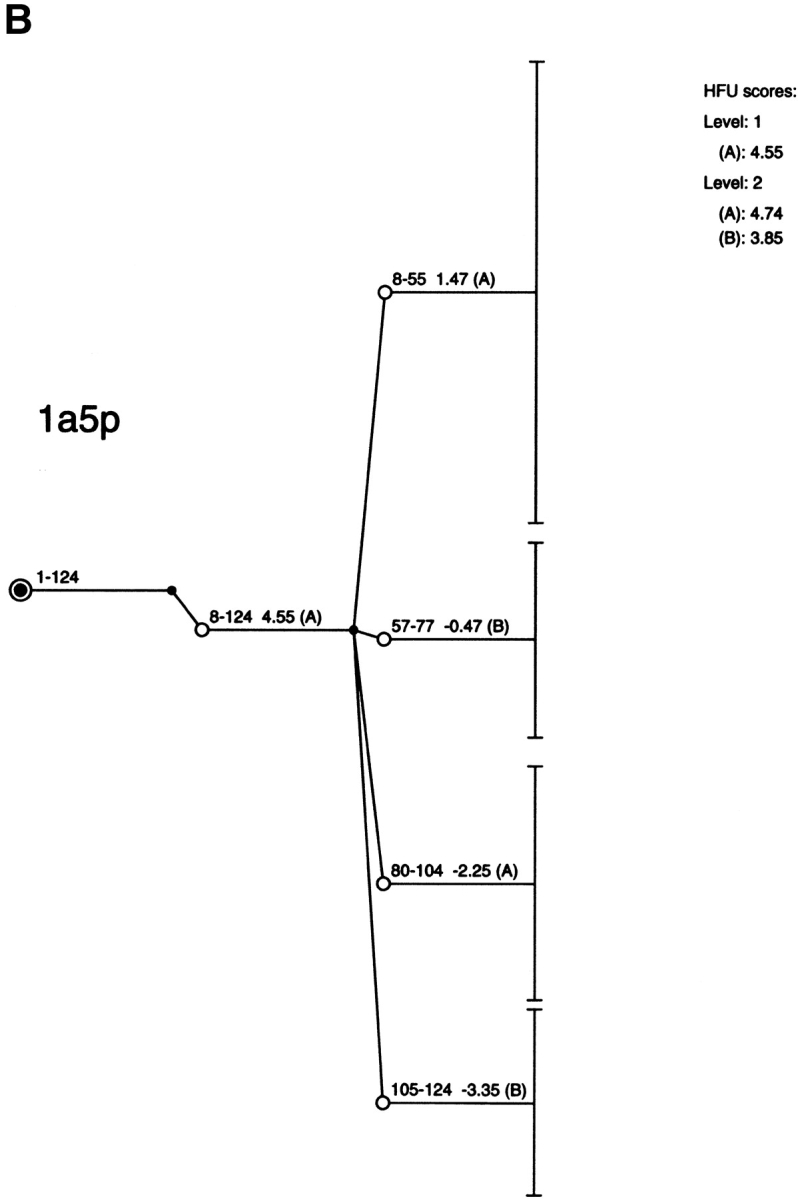
Figure 4 ▶ and Supplemental Material Table 2 present the results of the computational cutting of RNase (PDB 1a5p). No cut is observed near peptide bond 34–35. This position falls in the middle of a stable building block and, if cleaved there, it would render its component parts unstable. No major cut is observed at 45–46, whereas the major computational cut yields the 8–55 and 57–77 building blocks. The data of Table 2 also show a number of cuts around positions 43, 46, and 49. A possible reason for the disagreement with the major thermolytic 34–35 cut is that heat or TFE promote some conformational transition in this region of RNase. A cut is also observed at position 20 (building block No. 6 in Table 2, also seen in the Fig. 4 ▶ fragment map), in agreement with the limited proteolysis of RNase by subtilisin at neutral pH and ambient temperature (Richards 1958).
Fig. 4.

The fragment map obtained by computational cutting of bovine pancreatic ribonuclease A (PDB 1a5p). For further details see the legend for Figure 2 ▶. All of the building blocks are specified in Table 2 of the Supplemental Material.
α-Lactalbumin
The partly folded states of α-lactalbumin (α-LA), a 123-residue protein, have been probed using pepsin, chymotrypsin, and proteinase K (Polverino de Laureto et al. 1995, 1999, 2001). The conformational features of the A-state (acid-state) of α-LA were analyzed using pepsin as proteolytic probe at pH 2.0, while those of the TFE-state of the protein (Alexandrescu et al. 1994) were probed by thermolysin digestion (Polverino de Laureto et al. 1995). Chymotrypsin and proteinase K have been used at neutral pH to probe the apo-form of the protein obtained by EDTA-mediated removal of the calcium ion bound to the protein. Both states are considered to constitute the MG state of α-LA (Kuwajima 1996; Arai and Kuwajima 2000), a dynamic conformational state retaining most of the helices of the native structure, while the β-sheet region is largely unstructured (Alexandrescu et al. 1993; Schulman et al. 1995, 1997; Wu et al. 1995). A time-course analysis of the limited proteolytic cleavages revealed that the fast, initial cuts by all three proteases occur at the same 34–57 region, with the actual sites varying slightly with the different proteases (Polverino de Laureto et al. 1995, 1999, 2001). In the native structure, the 34–57 region encompasses the β-sheets of the protein. Subsequent cleavages took place at chain regions 31–35 and 95–105. Several of α-LA fragments have been isolated and studied. The single chain fragment 53–103, containing the calcium binding sites and crosslinked by two disulfide bridges, in the presence of calcium ions appears to possess a native-like content of α-helix. For the two-chain species 1–40/104–123 and 1–31/105–123, where the two constituting fragments are connected by two disulfide bridges, retain some secondary structure. The gapped protein species 1–34/54–,57–123, given by fragment 1–34, connected to fragment 54–123 or to 57–123 by four disulfide bridges, has an α-helix content similar to that of the native protein (Polverino de Laureto et al. 2001). Moreover, it has been shown that MG excision of the β-domain (chain region 34–57) from the α-LA does not impair the formation of the MG state of the rest of the protein in acid solution.
α-LA is a particularly good example for a comparison between experiment and computation, owing to the fact that several proteases have been employed in limited proteolysis experiments. Figure 5 ▶ and Supplemental Material Table 3 provide the results of the computational cutting of α-LA (PDB 1hfzA; see also Tsai et al. 2000). All obtained building block fragments are given in Table 3. In the first step of the cutting, the 1–123 chain remains practically intact. Only two residues are removed from its amino tail. In the second step, cuts are performed at 38–39 and 105–106 peptide bonds. This procedure yields fragments 3–38, 39–105, and 106–123. Of note, the last fragment is highly unstable, with a stability score of −9.10. The first and the last fragments associate into a single hydrophobic folding unit (marked as B in Fig. 5B ▶). The central 39–105 fragment is further split into fragments 39–55, 56–81, and 87–108, and these constitute the hydrophobic folding unit A.
Fig. 5.

The fragment map obtained by computational cutting of α-lactalbumin (PDB 1hfzA). For further details see the legend of Figure 2 ▶. All of the building blocks are specified in Table 3 of the Supplemental Material. To compare with the experimental dissection: Polverino de Laureto et al. (1999) cut α-LA by several proteolytic enzymes around 39–54, with subsequent cleavages at 31–35 and 95–105.
Overall there is an agreement between computational and experimental results. The 39–55 region observed to be initially cut by the proteases is very unstable (−4.90). The subsequent experimental cleavage at chain regions 31–35 and 95–105 correspond with main computational cutting sites at 38–39 and 105–106. Further, the two-chain folding unit (B, 3–38 and 106–123) is consistent with the two-chain species 1–40/104–123 isolated after proteolysis (Polverino de Laureto et al. 1999). The 53–103 fragment obtained by proteolysis corresponds to the association of two building blocks, 56–81 and 87–108. Further, as Table 3 in the Supplemental Material shows, there is a local minima at the 48–105 fragment (with a stability score of 2.94). The 111–120 fragment, which has been shown to be largely unstructured in solution (Kuhlman et al. 1997), corresponds to the unstable 106–123 fragment in the computational cutting. This fragment is stabilized by association with the 3–38 fragment, yielding the hydrophobic folding unit. Kuhlman et al. (1997) showed that peptides consisting of residues 72–100 of α-LA, with or without Cys73 and Cys91 replaced by Ala, are monomeric and unstructured in solution. Consistently, we observe a score of −2.95 for the 87–108 fragment and a score of −2.38 for the 70–105 fragment, as given in Figure 5B ▶ and Supplemental Material Table 3.
Thermolysin
Limited proteolysis of thermolysin has been carried out both using subtilisin and autolysis under different experimental conditions (upon heating or in the presence of 1 mM or 10 mM EDTA) (Fontana et al. 1986). Subtilisin was observed to cleave thermolysin to yield fragments 5–224(225) and 225(226)–316, which remain associated in a stable and functional complex at neutral pH (Vita et al. 1985). Thermal autolysis yielded fragments 1–221, 1–154(155), 155(156)–221, and 224–316, whereas autolysis in the presence of low EDTA concentration yielded fragments 1–129, 130–187, and 205–316 and in the presence of a higher EDTA concentration 1–196, 197–204, and 205–316 (Fassina et al. 1986). Dalzoppo et al. (1985) cut the C-terminal fragment 206–316 of thermolysin with several proteolytic enzymes. Analysis of the kinetics of the proteolytic digestions and of the isolated subfragments provided evidence that the proteases degrade fragment 206–316 in a stepwise manner proceeding from the amino terminus. The highly helical fragment 255–316 was found to be rather resistant to further proteolysis.
The computational cutting yields the two equal-sized domains 3–148 and 153–316 (PDB 2tlx) (Fig. 6 ▶, Supplemental Material Table 4), in agreement with fragments 1–154(155) and 155(156)–316 obtained by thermal autolysis of the protein. Although we do not observe a computational cut near residue 225 but do so at 233, this position is within the 10–12 residue region considered to be distorted in order to achieve proteolysis at that region (Hubbard et al. 1994). Figure 6 ▶ shows a major cut to yield 3–148 (with a high stability score, 6.68), in agreement with the identification of thermal autolysis 1–154 fragment and 233–316 (vs. 224–316), also with a high stability score (3.79). We observe the computational fragments 203–316, consistent with the EDTA autolysis (highly stable, with a score of 4.42, Table 4 in Supplemental Material), 213–316 (also very stable, with a score of 4.05), and the largely helical 265–310 building block (No. 39 in Table 4). A number of fragments initiate from around residue 205, from which the stepwise proteolysis initiates (Dalzoppo et al. 1985) (e.g., building blocks No. 11, 25, 28, or 69).
Fig. 6.

The anatomy tree obtained by computational cutting of thermolysin (PDB 2tlxA). For further details see the legend for Figure 2 ▶. All of the building blocks are specified in Supplemental Material Table 4. To compare with the experimental dissection, see the details given in the text.
Discussion
Comparison of the data of experimental limited proteolysis with those of computational cuttings indicates that while position-wise the cuttings vary, region-wise there is a satisfactory correspondence between the two methods. In carrying out such a comparison, the premises of the two methods should be considered first.
In limited proteolysis, the site of cleavage should be on the protein surface, needs to be flexible, and cannot be in the middle of α-helices (Fontana et al. 1986). In contrast, in the computational cutting cleavages can be performed in the interior of protein cores. Indeed, some building blocks are buried, mediate interactions between building blocks, and play a critical role in reaching the correct three-dimensional fold of the protein (Ma et al. 2000; Kumar et al. 2001). Furthermore, the cleavages can take place in the middle of α-helices, if a cut at this site leads to a more compact and favorable building block (Tsai et al. 2000). The agreement between the two methods is clearly better when proteolysis data are obtained by the use of several proteolytic enzymes, rather than by a single one, since in this case a chain region rather than a single peptide bond is identified. Indeed, there is a correspondence between experiment and computation in the cases of apoMb and α-LA, where several proteases have been used. Further, the correspondence becomes closer for cyt c if the proteinase K cuts (Wang et al. 1998; Spolaore et al. 2001) are added to the thermolysin cuts (Fontana et al. 1995).
The computational approach suffers from limitations. First, a drawback of the stability function which is used is that it lacks an electrostatic component (Tsai et al. 2000). It is possible that had electrostatics been taken into account, a greater correspondence would have been observed. The second limitation is the fact that the computational algorithm is based solely on the native conformation (Tsai et al. 2000). Hence, if some nonnative interactions play a role at the site of the experimental cutting, such as for example in aqueous TFE or under other solvent conditions favoring partly folded states, it may lead to an inconsistency with the computational algorithm. Indeed, the fact that several proteins in their native state are not attacked by protease indicates that the population of species fitting the active site of the proteolytic enzyme is low. Additionally, the computational cutting yields more protein fragments than the experimental limited proteolysis.
Limited proteolysis is at sites of local unfolding, whereas the computational algorithm relates to folding. The general fair agreement between the two approaches is consistent with the hierarchical model of protein folding, which postulates that the polypeptide chain folds by parts. Several models have been proposed to describe protein folding, including (1) the framework model, (2) the nucleation and growth mechanism, (3) the diffusion-collision model, (4) the hydrophobic collapse, and (5) the hierarchical model. In the framework model (Kim and Baldwin 1982, 1990; Udgaonkar and Baldwin 1988), secondary structure formation is independent of formation of tertiary interactions and frequently occurs earlier. In the nucleation and growth (Wetlaufer 1973) or nucleation condensation mechanism (Shakhnovich et al. 1996; Fersht 1997), folding initiates by formation of a nucleus, followed by its extension. In the diffusion-collision model, secondary structure elements assemble into folds by a random diffusion and collision process and, if the assembly is favorable, they may lock into the native conformation (Karplus and Weaver 1994). In contrast to these models, the hydrophobic collapse model highlights the hydrophobic effect, the driving force of protein folding (Rackovsky and Scheraga 1977; Dill 1985, 1990). In this case, folding initiates with collapse of the molecule, consequent burial of extensive nonpolar surface area and, therefore, secondary structure formation and specific interactions follow. In the hierarchical model, protein folding initiates locally and the local folded elements assemble in a stepwise fashion to yield the final native protein fold (Baldwin and Rose 1999a,b).
The models of protein folding listed above are not necessarily exclusive of each other. The hierarchical model may include elements of hydrophobic collapse in the assembly of local folded elements. Such a hydrophobic assembly would constitute the first stage of the parts coming together, followed by the optimization of the specific (van der Waals, electrostatic, disulfide bonds, etc.) interactions. The hierarchical model may further include elements of the nucleation and growth or nucleation-condensation model. The nucleus can be a part of the polypeptide chain whose folded structure forms local minima. Such an element may subsequently act as a template for further folding of the protein. Similarly, with respect to the framework model, the formation of single secondary structure elements can be substituted by local building blocks. Thus, depending on the interpretation of these models, each may be viewed as a specific case of the more general hierarchical model (Baldwin and Rose 1999a,b).
Conclusions
Limited proteolysis has been used extensively for many years to unravel structural features and conformational transitions of proteins (Mihalyi 1978; Neurath 1980; Price and Johnson 1990; Fontana et al. 1986, 1997a). Here we have compared the results of the experimental limited proteolytic dissection and the computational cutting for several proteins. In general, there is a correspondence in the regions of the polypeptide chains being cleaved by the two methods, despite the difference in their premises. Limited proteolysis is performed at sites which are flexible and not at the level of regular secondary structure such as α-helices. Therefore, limited proteolysis occurs at regions which are locally unfolded (Fontana et al. 1986). In contrast, the building block folding model is based on protein folding, and the criteria on which the scoring function is based are compactness of the fragment, extent of the nonpolar surface area it buries, and its isolatedness, that is, the surface area which was buried prior to cutting and became exposed subsequently (Tsai et al. 2000). The overall agreement between the two methods allows us to propose that proteolytic enzymes can be used as reliable probes of protein structure, dynamics, and folding pathways. Furthermore, the fact that distinct fragments can be produced and their conformations analyzed leads us to suggest that the limited proteolysis approach can be used in studies of folding intermediates, complementing other methods in use for analyzing the transient intermediates along the folding reaction path (Chamberlain and Marqusee 2000). On the computational side, the consistency with the experimental cleavages appears to provide a validation of the building block folding model. Considering that it is increasingly becoming accepted that protein folding may initiate by folding of local fragments and proceed by their associations, fragments can be identified by computating and produced by proteolysis for further studies. This suggests a procedure of computational predictive folding; that is, initial folding of protein fragments rather than of the entire protein and subsequent fragment assembly. This approach may well simplify the prediction of the three-dimensional structure of proteins.
Acknowledgments
We thank Drs. B. Ma, S. Kumar, and, in particular, J.V. Maizel for discussions and encouragement. R.N.'s research in Israel was supported in part by the Magnet grant, Ministry of Science and Centre of Excellence in Geometric Computing and its Applications funded by the Israel Science Foundation and administered by the Israel Academy of Sciences. This work was supported by the Italian National Council of Research (Biotechnology Project) and by the Ministry of University and Research (PRIN-2000). The project was funded in whole or in part with federal funds from the National Cancer Institute, NIH, under contract number NO1-CO-12400.
The content of this publication does not necessarily reflect the view or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organization imply endorsement by the U.S. government.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
ApoMb, apomyoglobin
cyt c, cytochrome c
RNase, bovine ribonuclease A
α-LA, α-lactalbumin
MG, molten globule
A-state, partly folded state in acid solution
PDB, Protein Data Bank
TFE, trifluoroethanol
CD, circular dichroism
NMR, nuclear magnetic resonance
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.4100102.
References
- Alexandrescu, A.T., Evans, P.A., Pitkeathly, M., Baum, J., and Dobson, C.M. 1993. Structure and dynamics of the acid-denatured molten globule state of α-lactalbumin: A two state dimensional NMR study. Biochemistry 32 1707–1718. [DOI] [PubMed] [Google Scholar]
- Alexandrescu, A.T., Ng, Y.-L., and Dobson, C.M. 1994. Characterization of a TFE-induced partially folded state of α-lactalbumin. J. Mol. Biol. 235 587–599. [DOI] [PubMed] [Google Scholar]
- Arai, M. and Kuwajima, K. 2000. Role of the molten globule state in protein folding. Adv. Protein Chem. 53 209–282. [DOI] [PubMed] [Google Scholar]
- Arnold, U., Rücknagel, K.P., Schierhorn A., and Ulbrich-Hofman, R. 1996. Thermal unfolding and proteolytic susceptibility of ribonuclease. Eur. J. Biochem. 237 862–869. [DOI] [PubMed] [Google Scholar]
- Baldwin, R.L. and Rose, G.D. 1999a. Is protein folding hierarchic? I. Local structure and peptide folding. Trends Biochem. Sci. 24 26–33. [DOI] [PubMed] [Google Scholar]
- Baldwin, R.L. and Rose, G.D. 1999b. Is protein folding hierarchic? II. Folding intermediates and transition states. Trends Biochem. Sci. 24 77–84. [DOI] [PubMed] [Google Scholar]
- Bernstein, F.C., Koetzle, T.F., Williams, G.J.B., Meyer, E.F. Jr., Brice, M.D., Rodgers, J.R., Kennard, O., Shimanouchi, T., and Tasumi, M. 1977. The protein databank: A computer-based archival file for macromolecular structures. J. Mol. Biol. 112 535–542. [DOI] [PubMed] [Google Scholar]
- Cavagnero, S., Nishimura, C., Schwarzinger, S., Dyson, H.J., and Wright, P.E. 2001. Conformational and dynamic characterization of the molten globule state of an apomyoglobin mutant with an altered folding pathway. Biochemistry 40 14459–14467. [DOI] [PubMed] [Google Scholar]
- Chamberlain, A.K. and Marqusee, S. 2000. Comparison of equilibrium and kinetic approaches for determining protein folding mechanisms. Adv. Protein Chem. 53 283–328. [DOI] [PubMed] [Google Scholar]
- Crippen, G.M. 1978. The tree structural organization of proteins. J. Mol. Biol. 126 315–332. [DOI] [PubMed] [Google Scholar]
- Dalzoppo, D., Vita, C., and Fontana, A. 1985. Folding of thermolysin fragments: Identification of the minimum size of a carboxyl-terminal fragment that can fold into a stable native-like structure. J. Mol. Biol. 182 331–340. [DOI] [PubMed] [Google Scholar]
- Dill, K.A. 1985. Theory for the folding and stability of globular proteins. Biochemistry 24 1501–1509. [DOI] [PubMed] [Google Scholar]
- Dill, K.A. 1990. Dominant forces in protein folding. Biochemistry 31 7134–7155. [DOI] [PubMed] [Google Scholar]
- Dill, K.A. and Chan, H.S. 1997. From Levinthal to pathways to funnels. Nat. Struct. Biol. 4 10–19. [DOI] [PubMed] [Google Scholar]
- Eaton, W.A., Munioz, V., Thompson, P.A., Chan, C.K., and Hofrichter, J. 1997. Submillisecond kinetics of protein folding. Curr. Opin. Struct. Biol. 7 10–14. [DOI] [PubMed] [Google Scholar]
- Eliezer, D. and Wright, P.E. 1996. Is apomyoglobin a molten globule? Structural characterization by NMR. J. Mol. Biol. 263 531–538. [DOI] [PubMed] [Google Scholar]
- Eliezer, D., Yao, J., Dyson, H.J., and Wright, P.E. 1998. Structural and dynamic characterization of partially folded states of apomyoglobin and implications for protein folding. Nat. Struct. Biol. 5 148–155. [DOI] [PubMed] [Google Scholar]
- Evans, P.A. and Radford, S.E. 1994. Probing the structure of folding intermediates. Curr. Opin. Struct. Biol. 4 100–106. [Google Scholar]
- Fassina, G., Vita, C., Dalzoppo, D., Zamai, M., Zambonin, M., and Fontana, A. 1986. Autolysis of thermolysin: Isolation and characterization of a folded three-fragment complex. Eur. J. Biochem. 156 221–228. [DOI] [PubMed] [Google Scholar]
- Fersht, A.R. 1997. Nucleation mechanism in protein folding. Curr. Opin. Struct. Biol. 7 3–9. [DOI] [PubMed] [Google Scholar]
- Fisher, A. and Taniuchi, H. 1992. A study of core domains and the core domain-domain interactions of cytochrome c fragment complex. Arch. Biochem. Biophys. 96 1–16. [DOI] [PubMed] [Google Scholar]
- Fontana, A., Fassima, G., Vita, C., Dalzoppo, D., Zamai, M., and Zambonin, M. 1986. Correlation between sites of limited proteolysis and segmental mobility in thermolysin. Biochemistry 25 1847–1851. [DOI] [PubMed] [Google Scholar]
- Fontana, A., Polverino de Laureto, P., and De Filippis, V. 1993. Molecular aspects of proteolysis of globular proteins. In: Protein stability and stabilization (eds. W. Van der Tweel, A. Harder, and M. Buitelaar), pp. 101–110. Elsevier Sci. Publ., Amsterdam.
- Fontana, A., Polverino de Laureto, P., De Filippis, V., Scaramella, E., and Zambonin, M. 1997a. Probing the partly folded states of proteins by limited proteolysis. Folding Des. 2 R17–R26. [DOI] [PubMed] [Google Scholar]
- Fontana, A., Polverino de Laureto, P., De Filippis, V., Scaramella, E., and Zambonin, M. 1999. Limited proteolysis in the study of protein conformation. In: Proteolytic enzymes: Tools and targets (eds. E.E. Sterchi, W. Stocker), pp. 257–284. Springer-Verlag, Heidelberg.
- Fontana, A., Zambonin, M., De Filippis, V., Bosco, M., and Polverino de Laureto, P. 1995. Limited proteolysis of cytochrome c in trifluoroethanol. FEBS Lett. 362 266–270. [DOI] [PubMed] [Google Scholar]
- Fontana, A., Zambonin, M., Polverino de Laureto, P., De Filippis, V., Clementi, A., and Scaramella, E. 1997b. Probing the conformational state of apomyoglobin by limited proteolysis. J. Mol. Biol. 266 223–230. [DOI] [PubMed] [Google Scholar]
- Gast, K., Zirwer, D., Müller-Frohne, M., and Damaschun, G. 1999. Trifluoroethanol-induced conformational transitions of proteins: Insights gained from the differences between α-lactalbumin and ribonuclease A. Protein Sci. 8 625–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst, J.D. and Brooks, C.L. 1994. Helicity, circular dichroism and molecular dynamics of proteins. J. Mol. Biol. 243 173–178. [DOI] [PubMed] [Google Scholar]
- Hubbard, S.J. 1998. The structural aspects of limited proteolysis of native proteins. Biochim. Biophys. Acta 1382 191–206. [DOI] [PubMed] [Google Scholar]
- Hubbard, S.J., Eisenmenger, F., and Thornton, J.M. 1994. Modelling studies of the change in conformation required for cleavage of limited proteolytic sites. Protein Sci. 3 757–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karplus, M. and Weaver, D.L. 1994. Protein folding dynamics: The diffusion-collision model and experimental data. Protein Sci. 3 650–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, P.S. and Baldwin, R.L. 1982. Specific intermediates in the folding reactions of small proteins and the mechanism of protein folding. Annu. Rev. Biochem. 51 459–489. [DOI] [PubMed] [Google Scholar]
- Kim, P.S. and Baldwin, R.L. 1990. Intermediates in the folding reactions of small proteins. Annu. Rev. Biochem. 59 631–660. [DOI] [PubMed] [Google Scholar]
- Kuhlman, B., Boice, J.A., Wu, W.J., Fairman, R., and Raleigh, D.P. 1997. Calcium binding peptides from α-lactalbumin: Implications for protein folding and stability. Biochemistry 36 4607–4615 [DOI] [PubMed] [Google Scholar]
- Kumar, S., Ma, B., Tsai, C.J., Sinha, N., and Nussinov, R. 2000. Folding and binding cascades: Dynamic landscapes and population shifts. Protein Sci. 9 10–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, S., Ma, B., Tsai, C.J., Wolfson, H., and Nussinov, R. 1999. Folding funnels and conformational transitions via hinge-bending motions. Cell. Biochem. Biophys. 31 23–46. [DOI] [PubMed] [Google Scholar]
- Kumar, S., Sham, Y.Y., Tsai, C.J., and Nussinov, R. 2001. Folding and function: The N-terminal building block in adenylate kinase. Biophys. J. 80 2439–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwajima, K. 1996. The molten globule state of α-lactalbumin. FASEB J. 10 74–78. [DOI] [PubMed] [Google Scholar]
- Lecomte, J.T.J., Kao, Y.H., and Cocco, M.J. 1996. The native state of apomyoglobin described by proton NMR spectroscopy: The A-B-G-H interface of wild-type sperm whale apomyoglobin. Proteins: Struct. Funct. Genet. 25 267–285. [DOI] [PubMed] [Google Scholar]
- Lin, L., Pinker, R.J., Forde, K., Rose, G.D., and Kallenbach, N.R. 1994. Molten globular characteristics of the native state of apomyoglobin. Nat. Struct. Biol. 1 447–451. [DOI] [PubMed] [Google Scholar]
- Ma, B., Kumar, S., Tsai, C.-J., and Nussinov, R. 1999. Folding funnels and binding mechanisms. Protein Eng. 12 713–720. [DOI] [PubMed] [Google Scholar]
- Ma, B., Tsai, C.-J., and Nussinov, R. 2000. Binding and folding: In search of intramolecular chaperone-like building block fragments. Protein Eng. 13 617–627. [DOI] [PubMed] [Google Scholar]
- Matthews, C.R. 1995. Pathways of protein folding. Annu. Rev. Biochem. 62 36–42. [DOI] [PubMed] [Google Scholar]
- Mihalyi, E. 1978. Application of proteolytic enzymes to protein structure studies. CRC Press, Boca Raton, Florida.
- Neurath, H. 1980. Limited proteolysis, protein folding and physiological regulation. In: Protein folding (ed. R. Jaenicke), pp. 501–504. Elsevier/North Holland Biomedical Press, Amsterdam/New York.
- Panchenko, A.R., Luthey-Schulten, Z., and Wolynes, P.G. 1996. Foldons, protein structural modules and exons. Proc. Natl. Acad. Sci. 93 2008–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panchenko, A.R., Luthey-Schulten, Z., Cole, R., and Wolynes, P.G. 1997. The foldon universe: A survey of structural similarity and self-recognition of independently folding units. J. Mol. Biol. 272 95–105. [DOI] [PubMed] [Google Scholar]
- Pande, V.S., Grosberg, A.Y., Tanaka, T., and Rokhsar, D.S. 1998. Pathways for protein folding: Is a new view needed? Curr. Opin. Struct. Biol. 8 66–79. [DOI] [PubMed] [Google Scholar]
- Peng, Z.-Y. and Kim, P.S. 1994. A protein dissection study of a molten globule. Biochemistry 33 2136–2141. [DOI] [PubMed] [Google Scholar]
- Peng, Z.-Y. and Wu, L.C. 2000. Autonomous protein folding units. Adv. Protein Chem. 53 1–47. [DOI] [PubMed] [Google Scholar]
- Polverino de Laureto, P., De Filippis, V., Di Bello, M., Zambonin, M., and Fontana, A. 1995. Probing the molten globule state of α-lactalbumin by limited proteolysis. Biochemistry 34 12596–12604. [DOI] [PubMed] [Google Scholar]
- Polverino de Laureto, P., Scaramella, E., De Filippis, V., Bruix, M., Rico, M., and Fontana, A. 1997. Limited proteolysis of ribonuclease A with thermolysin in trifluoroethanol. Protein Sci. 6 860–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polverino de Laureto, P., Scaramella, E., Frigo, M., Gefter-Wondrich, F., De Filippis, V., Zambonin, M., and Fontana, A. 1999. Limited proteolysis of bovine α-lactalbumin: Isolation and characterization of protein domains. Protein Sci. 8 2290–2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polverino de Laureto, P., Scaramella, E., Zambonin, M., De Filippis, V., and Fontana, A. 1998. Limited proteolysis of proteins by thermolysin in trifluoroethanol. In: Stability and stabilization of biocatalysts (eds. A. Ballesteros, F.J. Plou, J.L Iborra, and P.J. Halling), pp. 381–392. Elsevier Sci., Amsterdam.
- Polverino de Laureto, P., Vinante, D., Scaramella, E., Frare, E., and Fontana, A. 2001). Stepwise proteolytic removal of the β-subdomain in α-lactalbumin: The protein remains folded and can form the molten globule in acid solution. Eur. J. Biochem. 268 4324–4333. [DOI] [PubMed] [Google Scholar]
- Price, N.C. and Johnson, C.M. 1990. Proteinases as probes of conformation of soluble proteins. In: Proteolytic enzymes: A practical approach (R.J. Beynon and J.S. Bond), pp. 163–180, IRL Press, Oxford.
- Privalov, P.L. 1996. Intermediate states in protein folding. J. Mol. Biol. 258 707–725. [DOI] [PubMed] [Google Scholar]
- Ptitsyn, O.B. 1995. Molten globule and protein folding. Adv. Protein Chem. 47 83–229. [DOI] [PubMed] [Google Scholar]
- Rackovsky, S. and Scheraga, H.A. 1977. Hydrophobicity, hydrophilicity and the radial and orientational distributions of residues in native proteins. Proc. Natl. Acad. Sci. 74 5248–5251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, F.M. 1958. On the enzymatic activity of subtilisin-modified ribonuclease. Proc. Natl. Acad. Sci. 44 162–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roder, H. and Colon, W. 1997. Kinetic role of early intermediates in protein folding. Curr. Opin. Struct. Biol. 7 15–28. [DOI] [PubMed] [Google Scholar]
- Rose, G.D. 1979. Hierarchic organization of domains in proteins. J. Mol. Biol. 134 447–470. [DOI] [PubMed] [Google Scholar]
- Schechter, I. and Berger, A. 1967. On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Comm. 27 157–162. [DOI] [PubMed] [Google Scholar]
- Schulman, B.A., Kim, P.S., Dobson, C.M., and Redfield, C. 1997. A residue-specific NMR view of the non-cooperative unfolding of a molten globule. Nat. Struct. Biol. 4 630–634. [DOI] [PubMed] [Google Scholar]
- Schulman, B.A., Redfield, C., Peng, Z.-Y., Dobson, C.M., and Kim, P.S. 1995. Different subdomains are most protected from hydrogen exchange in the molten globule and native states of human α-lactalbumin. J. Mol. Biol. 253 651–657. [DOI] [PubMed] [Google Scholar]
- Shakhnovich, E., Abkevich, V., and Ptitsyn, O. 1996. Conserved residues and the mechanism of protein folding. Nature 379 96–98. [DOI] [PubMed] [Google Scholar]
- Spolaore, B., Bermejo, R., Zambonin, M., and Fontana, A. 2001. Protein interactions leading to conformational changes monitored by limited proteolysis: Apo form and fragments of horse cytochrome c. Biochemistry 40 9460–9468. [DOI] [PubMed] [Google Scholar]
- Taniuchi, H., Parr, G.R., and Juillerat, M.A. 1986. Complementation in folding and fragment exchange. Methods Enzymol. 131 185–217. [DOI] [PubMed] [Google Scholar]
- Tsai, C.-J. and Nussinov, R. 1997. Hydrophobic folding units derived from dissimilar monomer structures and their interactions. Protein Sci. 6 24–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai, C.-J., Kumar, S., Ma, B., and Nussinov, R. 1999a. Folding funnels, binding funnels and protein function. Protein Sci. 8 1181–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai, C.-J., Maizel, J.V., and Nussinov, R. 1999b. Distinguishing between sequential and non sequentially folded proteins: Implications for folding and misfolding. Protein Sci. 8 1591–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai, C.-J., Maizel, J.V., and Nussinov, R. 2000. Anatomy of protein structures: Visualizing how a 1D protein chain folds into a 3D shape. Proc. Natl. Acad. Sci. 97 12038–12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai, C.-J., Xu, D., and Nussinov, R. 1998. Protein folding via binding and vice versa. Folding Des. 3 R71–R80. [DOI] [PubMed] [Google Scholar]
- Udgaonkar, J.B. and Baldwin, R.L. 1988. NMR evidence for an early framework intermediate on the folding pathway of ribonuclease A. Nature 335 694–699. [DOI] [PubMed] [Google Scholar]
- Vita, C., Dalzoppo, D., and Fontana, A. 1985. Limited proteolysis of thermolysin by subtilisin: Isolation and characterization of a partly active enzyme derivative. Biochemistry 24 1798–1806. [DOI] [PubMed] [Google Scholar]
- Wang, L., Chen, R.X., and Kallenbach, N.R. 1998. Proteolysis as a probe of thermal unfolding of cytochrome c. Proteins: Struct. Funct. Genet. 30 435–441. [PubMed] [Google Scholar]
- Wetlaufer, D.B. 1973. Nucleation, rapid folding and globular intrachain regions in proteins. Proc. Natl. Acad. Sci. 70 697–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetlaufer, D.B. 1981. Folding of protein fragments. Adv. Protein Chem. 34 61–92. [DOI] [PubMed] [Google Scholar]
- Wodak, S.J. and Janin, J. 1981. Location of structural domains in proteins. Biochemistry 20 6544–6552. [DOI] [PubMed] [Google Scholar]
- Wu, L.C., Grandori, R., and Carey, J. 1994. Autonomous subdomains in protein folding. Protein Sci. 3 369–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, L.C., Peng, Z.Y., and Kim, P.S. 1995. Bipartite structure of the α-lactalbumin molten globule. Nat. Struct. Biol. 2 281–286. [DOI] [PubMed] [Google Scholar]
- Yang, X.M., Yu, W.F., Li, J.H., Fuchs, J., Rizo, J., and Tasayco, M.L. 1998. NMR evidence for the reassembly of an α/β domain after cleavage of an α-helix: Implications for protein design. J. Amer. Chem. Soc. 120 7985–7986. [Google Scholar]
- Zehfus, M.H. 1993. Improved calculations of compactness and a re-evaluation of continuous compact units. Proteins Struct. Func. Genet. 16 293–300. [DOI] [PubMed] [Google Scholar]
- Zehfus, M.H. and Rose, G.D. 1986. Compact units in proteins. Biochemistry 25 5759–5765. [DOI] [PubMed] [Google Scholar]