

Abstract
The activity of the Alzheimer's amyloid β-peptide is a sensitive function of the peptide's sequence. Increased fibril elongation rate of the E22Q Dutch mutant of the Alzheimer's amyloid β-peptide relative to that of the wild-type peptide has been observed. The increased activity has been attributed to a larger propensity for the formation of β structure in the monomeric E22Q mutant peptide in solution relative to the WT peptide. That hypothesis is tested using four nanosecond timescale simulations of the WT and Dutch mutant forms of the Aβ(10–35)-peptide in aqueous solution. The simulation results indicate that the propensity for formation of β-structure is no greater in the E22Q mutant peptide than in the WT peptide. A significant measure of "flickering" of helical structure in the central hydrophobic cluster region of both the WT and mutant peptides is observed. The simulation results argue against the hypothesis that the Dutch mutation leads to a higher probability of formation of β-structure in the monomeric peptide in aqueous solution. We propose that the greater stability of the solvated WT peptide relative to the E22Q mutant peptide leads to decreased fibril elongation rate in the former. Stability difference is due to the differing charge state of the two peptides. The other proposal leads to the prediction that the fibril elongation rates for the WT and the mutant E22Q should be similar under acid conditions.
Keywords: Aβ-peptide, amyloid, molecular dynamics simulation, amyloidosis, aggregation, E22Q Dutch mutant peptide, conformational analysis, hydration
A growing number of experimental studies of the structure of solvated Alzheimer's β-amyloid (Aβ) peptide and fibrils (Lee et al. 1995; Esler et al. 1996; Zhang et al. 1998, 2000) and the activity of the Aβ-peptide in the process of fibril growth (Lomakin et al. 1996; Walsh et al. 1997, 1999; Esler et al. 2000a) are beginning to define the outlines of a mechanism of amyloid fibrillogenesis (Teplow 1998; Lansbury 1999) and fibril elongation (Lomakin et al. 1997; Esler et al. 2000a,b; Massi-and-Straub 2000a). An important goal of research on Aβ-peptide aggregation is to understand the role of sequence in the peptide's activity (Wisniewski et al. 1991; Fraser et al. 1992; Davis and Van Nostrand 1996; Watson et al. 1999; Esler et al. 2000a). Two particular naturally occurring mutant forms of the wild-type Aβ-peptide, the E22Q Dutch mutant and the E22K Italian mutant, have been the focus of recent structural and activity studies (Melchor et al. 2000; Miravalle et al. 2000).
It is well established that the E22Q peptide shows enhanced activity (as measured by the rate of deposition or fibril elongation) relative to the WT peptide for both the 1–42 Aβ-peptide and the Aβ(10–35)-peptide congener. For the Aβ(10–35)-peptide congener, the E22Q mutant form of the peptide was found to deposit at a rate 215% faster than the WT peptide (Esler et al. 2000a). In a study of Austen and coworkers (Sian et al. 2000), it was found that Aβ(1–40)E22Q-peptide formed oligomers and fibrils more rapidly than the WT peptide. Using CD spectroscopy, they observed that the rate of change from mainly random coil to β-sheet was more than one order of magnitude higher in the E22Q mutant than in the WT. It was also determined that the rates of conversion from random coil to β-sheet in the WT and E22Q mutant peptides, derived from CD measurements, were an order of magnitude lower than the rate of formation of low-molecular-mass oligomers. It was suggested that the Aβ-peptide aggregates in an irregular structure and then undergoes a slower conformational transition into larger aggregates of β-sheets.
It has been proposed that the E22Q peptide has a propensity for the formation of β-structure in solution. A study by Selkoe and coworkers explored the binding of heparin to solutions of WT and E22Q mutant Aβ-peptide. Heparin binds to fibrillar, but not to nonfibrillar, Aβ-peptide. It was found that the E22Q mutant peptide assumed conformations to which heparin would bind more readily than did the WT peptide and that the affinity of heparin binding to the E22Q mutant peptide was similar to the affinity for binding of heparin to preformed β-fibrils. The results led to the conclusion that the water-aggregated E22Q mutant peptide adopted structures similar to those found in certain β-fibrils (Watson et al. 1997).
Through CD and FTIR measurements, Miravalle et al. (2000) found that whereas the WT and the E22K mutant peptide were largely in the random-coil conformation in solution, the E22Q peptide assumed a β-sheet conformation. The study explored the time dependence of peptide aggregation by CD and showed that for their sample preparations, the WT, E22K, and E22Q peptide converted to β-structure over a period of hours. Whereas CD spectra of the WT and E22K mutant peptide samples indicated that at the earliest times the peptide was in a random-coil conformation, the E22Q peptide sample showed clear signs of β-structure. However, the results in the case of the E22Q mutant peptide could be due to the presence of peptide aggregates from the earliest stages of the CD measurements.
This brief survey of recent experimental results raises two fundamental questions. In the monomeric Aβ-peptide, is there local "flickering" of conformations consistent with the larger scale formation of β-structure? Does the E22Q mutation lead to a greater propensity for the formation of β-structure in the monomeric peptide? In this computational study, we explore the hypothesis that the E22Q mutation leads to a monomeric peptide that has a higher propensity for conformational fluctuation to an "activated" β-form of the peptide. Because we are simulating only isolated monomers we cannot rule out the possibility that interactions drive the chains to adopt β-strand conformations or substantially increase β-flickering. Such interaction-driven conformational changes have been probed in prion assembly (Harrison et al. 2001; Dima and Thirumalai 2002). However, just as the propensities of amino acids to adopt specific conformations are useful in predicting secondary structures, we believe that the present simulations are instructive in ruling out a plausible hypothesis.
Results
Molecular dynamics simulations of the fully solvated WT β(10–35)-NH2 peptide and the E22Q mutant peptide were performed. It has been proposed that the difference in activity observed for the WT and E22Q mutant peptides can be attributed to a larger propensity for the formation of β structure in the monomeric E22Q mutant peptide in solution relative to the WT peptide (Watson et al. 1997; Miravalle et al. 2000). To test this hypothesis, we have carried out an analysis of the local flickering of peptide backbone structure and side-chain contacts that are consistent with the formation of extended α or β structure in the peptide using the URMS analysis. A comparison between the conformational energy calculated during the MD simulation for the all-atom model and for a course-grained model is also presented.
Flickering of local α structure seen in LVFFA hydrophobic cluster and C terminus of WT and E22Q peptides
The structural fluctuations of the peptide were analyzed for the presence of flickering of α-like and β-like structure using local templates involving four Cα positions. The results of the URMS analysis are shown in Figure 1 ▶. In the trajectories of both the WT and E22Q mutant peptide, there is substantial flickering of α structure in the LVFFA (17–21) hydrophobic cluster region. Some flickering of α-helical structure is also apparent in the AIIGL (30–34) region of the peptide's C terminus.
Fig. 1.
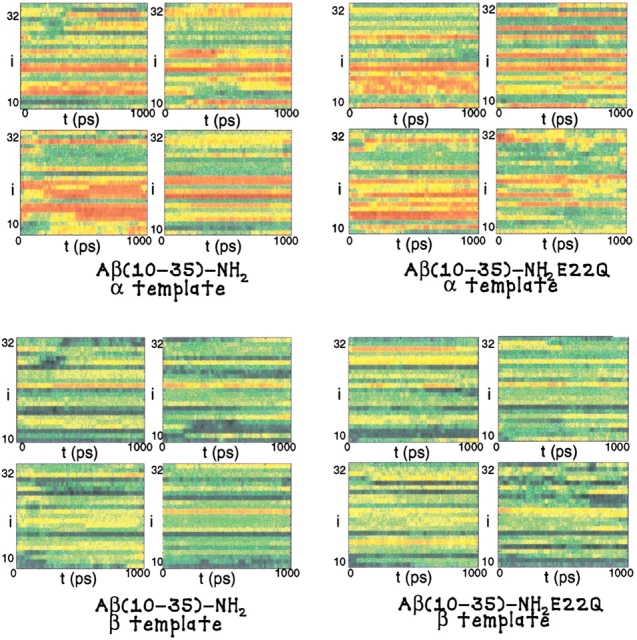
The result of the URMS analysis of the WT and E22Q mutant trajectory simulations are plotted. In the uppermost figures, the propensity for α-helical structural fluctuations is shown where red indicates strong α-like character, for the WT and E22Q mutant trajectories, respectively. In the lowermost figures, the propensity for β-sheet structure is shown where blue indicates strong β-like character, for the WT and E22Q mutant trajectories, respectively.
Barrow and colleagues (1992) have shown that the NMR-derived structure of the Aβ(1–42)-peptide, solvated in the membrane-like environment of a trifluoroethanol and water solution, consists of two short α-helices extending from 10–24, and that includes the LVFFA region, and C-terminal regions 28–42 of the peptide. Lee and coworkers have shown that for the NMR derived structure of the Aβ(10–35)-peptide in aqueous solution, there is no recognizable region of distinct α-helical structure (Zhang et al. 2000). Our analysis of the peptide's dynamics in aqueous solution indicates that one can determine the flickering of the underlying α-helical structure of the peptide. Such conformational fluctuations are apparently destabilized in aqueous solution but stabilized in the strongly hydrophobic membrane-like environments, in which the low polarity of the solvent leads to a strong enthalpic stabilization of the hydrogen bonds in regions that form α-helix.
No significant difference in β-flickering seen in WT and E22Q peptides
It can be clearly noted that in both the WT and E22Q peptide trajectories there is noticeable local formation of backbone structure consistent with the flickering β-structure. No significant flickering of extended regions of β-structure is observed in the dynamics of either the WT or E22Q mutant peptides.
The relative measure of β-flickering is quite similar in both the WT and E22Q mutant peptides. Our results indicate that on the nanosecond timescale, there is no larger propensity for the formation of local or global β-structure in the E22Q mutant peptide than in the WT peptide. Therefore, we find no support for the proposal that the E22Q mutant is prone to form significant β-structure as a monomeric peptide in solution.
Fluctuations to the β structure are essential for the aggregative activity of the Aβ-peptide. It is well known that the structure of the Aβ-peptide fibril is composed of either parallel and/or antiparallel β-strands that form an overall cross β structure in the fibril. As a result, many studies have focused on an analysis of the propensity of transition to β-sheet structure in the monomeric peptide. Our results indicate that there are only minor fluctuations of β-strand structure in the Aβ-peptide dynamics in aqueous solution. That conclusion is in agreement with the NMR-derived structure and analysis of Lee and coworkers (Lee et al. 1995; Zhang et al. 2000).
α-Flickering predominates over β-flickering
The analysis of the backbone secondary structure was performed for the coarse-grained mapping of the all-atom model trajectories, as described in Materials and Methods. The results for the torsional analysis are shown in Figure 2 ▶. Over the trajectories of the WT and E22Q mutant peptides, we computed <nH>, the average number of torsional angles adopting helical conformations, and <nS>, the average number of residues adopting strand conformations.
Fig. 2.
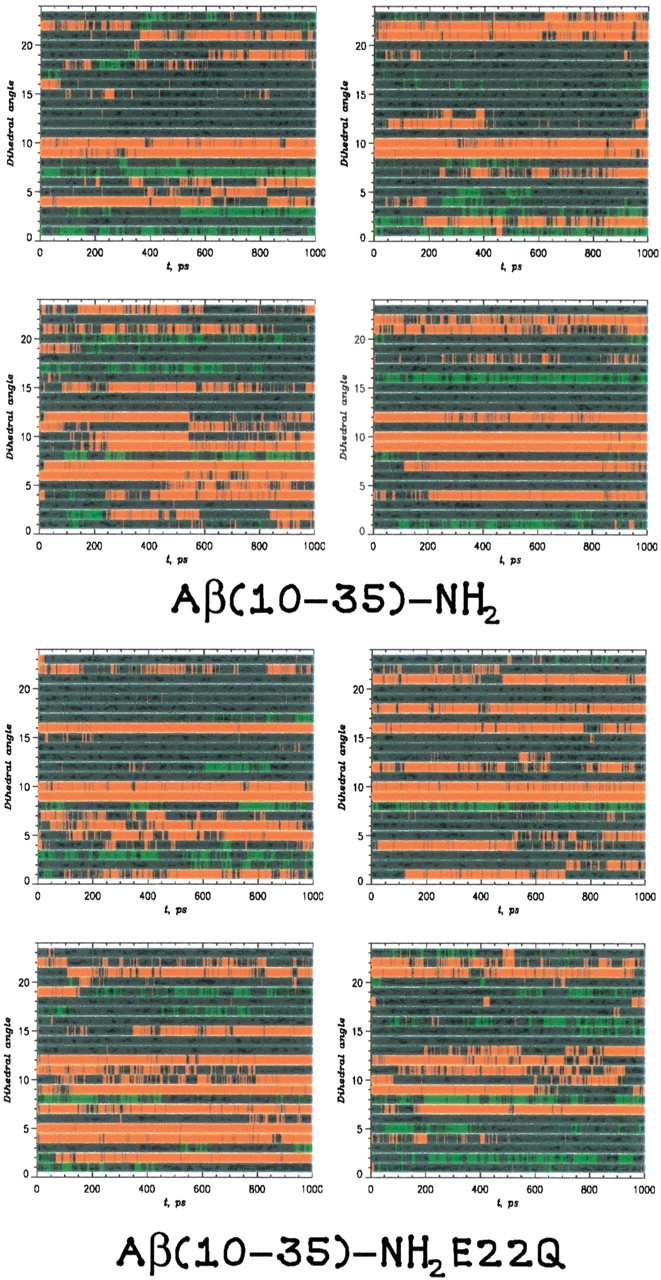
The secondary-structure formation probed through an analysis of the dihedral angle, φi, derived by mapping the all-atom model dynamics on a coarse-grained representation. The local backbone conformation was said to be helical if |φi − 60°| ≤ 30° (red) or a β-strand structure if |φi − 180°| ≤ 30° (green). All other conformations were said to be coil (blue).
For the WT peptide, the value of <nH> varied between 4 and 7, whereas the value of <nS> was ∼2. The ratio of <nH>/<nS> varied from 1.9 to 4.2 for all trajectories, indicating a significantly higher ratio of helical conformations.
For the E22Q mutant peptide, the value of <nH> varied between 4.9 and 8.2, whereas the value of <nS> varied from 0.8 to 2.5. The ratio of <nH>/<nS> had an average value of 5.2 for the E22Q mutant peptide, compared with an average of 3.3 for the WT peptide. As in the WT peptide, there is a significantly greater degree of α-helical flickering as opposed to β-strand flickering. These results are consistent with the results of the URMS analysis.
These results are consistent with the evaluation of the contact order (<s>), see Materials and Methods, which was found to be 0.26 for both WT and E22Q mutant peptides. For β-sheet proteins the contact order is typically found to be larger than 0.3. This further supports the observation that there is a lack of stable β-structure in our simulations of the WT and mutant peptides.
Stabilization of the collapsed-coil structure through plurality of side-chain contacts and hydrogen bonds
Considerable insight can be provided by a contact analysis in both analyzing all-atom model trajectories and in the parameterization of coarse-grained peptide models. Several conclusions may be drawn from our analysis.
The contact analysis led to the identification of a subset of side-chain contacts that were observed to occur with high probability Pq > 0.5 in the course of our simulations. The fact that the sets of contacts are not identical for all trajectories indicates that, as can be expected, the conformation of the peptide are not sampled ergodically over any given trajectory. However, the results do provide insight into the variety of side-chain interactions that play a role in stabilizing the peptide's structure in solution.
The results of our analysis are shown in Figure 3 ▶. A number of general features can be observed. No direct side-chain interactions are found between LVFFA (17–21) and VGS (24–26) regions. Any coupling between these regions may be due to other interactions, such as a hydrogen-bond network. In addition to hydrophobic–hydrophobic side-chain pairs (numbering 8 in the WT peptide and 8 in the E22Q mutant peptide) there are polar–polar pairs (numbering 6 in the WT and 7 in the E22Q mutant peptides). However, the majority of side-chain contacts are formed between hydrophobic–polar side-chain pairs (numbering 18 in the WT and 14 in the E22Q mutant peptides).
Fig. 3.

This matrix shows the pairs of side-chain contacts and hydrogen-bonding interactions most frequently observed in the course of our simulations of the WT and E22Q mutant peptides. The side-chain contacts are shown above the diagonal; the backbone hydrogen-bonding pairs are shown below the diagonal. All side-chain contact and hydrogen-bonding pairs that are noted occur with a probability > 50%. Data is represented for the WT peptide (black spots) and E22Q mutant peptide (red spots). The full spots are contact pairs and the open spots are hydrogen-bonding pairs.
The connectivity that is displayed shows that there is a significant number of side-chain contacts that are formed across the VGSN turn in trajectories of both the WT and E22Q mutant peptide. There is also a common side-chain contact between the V18 and A21 residues that is found repeatedly in the trajectories of the WT and E22Q mutant peptides.
Longer range contacts are also found between residues. For example, in the simulations of the WT peptide, there are side-chain contacts formed between residues of the C-terminal region and residues within the central cluster region. Overall, the average sequence separation between contacts, <|i − j|> is 7.2 in the WT peptide and 6.6 in the E22Q mutant peptide. The distribution of contacts is slightly skewed in the WT peptide toward intermediate range (7–11) contacts, whereas the E22Q mutant peptide has a higher probability of short- (3–6) and long- (12–15) range contacts.
Peptide energetics
The energy of each configuration was calculated for the course-grained model, as described in Materials and Methods. To compare this result with the energy of the all-atom model we considered only nonbonding side chain–side chain interactions. The distribution of the energies for all four trajectories of the WT and the Dutch-mutant peptides are shown in Figure 4 ▶. Gaussian functions were used to fit these energy distributions. To allow for a better comparison of the two models, the energy values have been normalized by the number of degrees of freedom of the respective systems. The potential energy of the all-atom model is very sensitive to conformational changes, resulting in broader distributions than those obtained for the course-grained model.
Fig. 4.
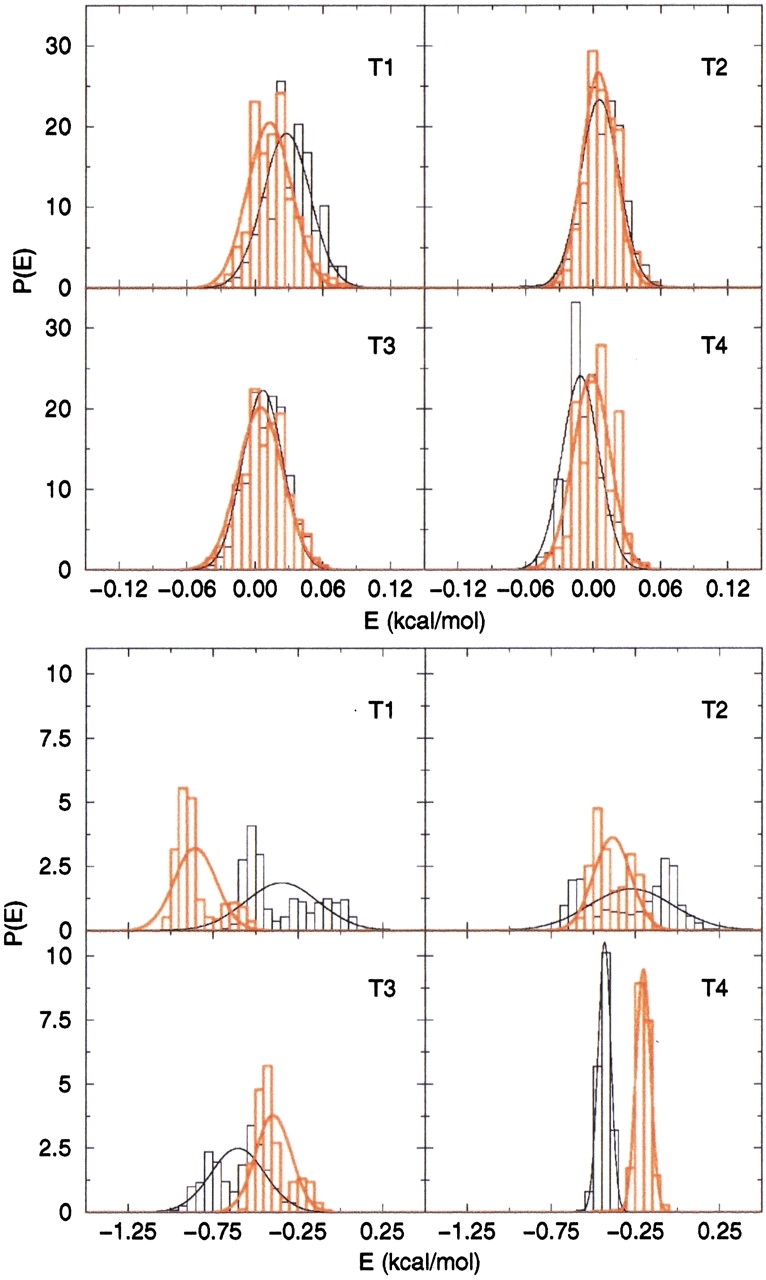
Conformational energy for the four trajectories of the WT peptide (black) and of the E22Q mutant peptide (red). The top panel shows the energies calculated from the coarse-grained model; the bottom panel shows those from the all-atom model.
The energy of the peptide calculated in the course-grained model is very similar in behavior, as a function of time, for the two peptides. The distributions are very narrow and the fluctuations in time are similar for all the trajectories, showing that this model is less sensitive to conformational changes in the peptide, which are likely "averaged over" when the peptide side chains are represented by beads placed in the relative centers of mass.
Discussion
Based on our simulations and the following simple arguments we propose that the stability of the Aβ-peptides in the monomeric state may determine the deposition rates. The relative stabilities of E22Q and WT may be estimated using naive arguments based on the effective charge on these peptides. Because the hydrophobic content in both the peptides is the same, we assume that the intrinsic stabilities are determined by electrostatic interactions. The effective charge (in units of e) on the WT and E22Q is −1 and 0, respectively. Assuming that the peptide is compact in water the radius of gyration Rg ≈ aN1/3 where a = 3.8 Å is the distance between covalently linked Cα atoms and N is the number of residues in the peptide. For the peptides considered here we find that Rg ≈ 12 Å. Based on the charge of the WT, we expect this peptide to be unstable with respect to E22Q by ∼1 kcal/mole. However, the presence of the charge enables the WT to be better solvated in water so that the hydration effect can compensate for the electrostatic destabilization. The larger stability of WT over E22Q in water makes conformational fluctuations in the former to be relatively slow. Thus, we propose that it is the ease of structural rearrangement to aggregation-prone states, rather than any intrinsic propensity to form β-structures, that is responsible for enhanced rate of amyloid formation in E22Q.
The above arguments allow us to make additional predictions. (1) Because the stability in this class of peptides is determined by a balance of hydration effects and electrostatic interactions, we predict that the fibril elongation kinetics will be affected by pH and ionic strength. It is likely that the rates of deposition in these peptides would become comparable at higher pH. (2) The deposition rates will also be altered in different solvents (for example, trifluroethanol or mixtures of solvents), which can alter the balance of interactions. (3) We predict that there should not be a significant difference in the elongation rates of WT and E22K under acidic conditions.
Summary and conclusions
A number of independent, all-atom molecular dynamics simulations of the WT and E22Q mutant Aβ(10–35)-peptides in aqueous solution were computed. The structure and fluctuations of the peptide were analyzed to address two fundamental questions related to the activity of the Aβ-peptide. In the monomeric Aβ-peptide, is there noticeable local flickering of conformations consistent with the larger scale formation of β-structure? Does the E22Q mutation lead to a greater propensity for the formation of β-structure in the monomeric peptide? From the results of our simulations, a number of conclusions can be drawn. (1) Our simulation data indicate that the central core structure of the Aβ(10–35)-peptide, characterized by an LVFFA (17–21) hydrophobic cluster and VGSN (24–27) turn region, are stable in aqueous solution in both the WT and E22Q mutant sequences as observed in the NMR studies of Lee and coworkers (Lee et al. 1995; Zhang et al. 2000). (2) The E22Q peptide is more flexible in solution, supporting an early hypothesis that the equilibrium structural fluctuations of the E22Q mutant peptide were larger than those of the WT peptide (Esler et al. 2000b). (3) The peptide adopts predominantly helical-like structures and shows much weaker β-strand propensity in all trajectories. Stable helix is typically formed near the hydrophobic cluster region LVFFA (17–21). The peptide's C terminus also displays strong helical propensity AIIGL (30–34). There is no consistent pattern of β-strand flickering in the WT or E22Q mutant peptide. These observations are consistent with a recent study by Teplow and colleagues, who argue, based on CD spectra, that the formation of cross β-fibril structures may be preceded by helix formation (Kirkidatze et al. 2002). Thus, the transition from the random coil to fiber involve the pathway RC ↔ Helix ↔ β. (4) Few direct side-chain interactions are observed between the LVFFA (17–21) and VGSN (24–27) regions in the WT peptide. However, in both the WT and E22Q mutant peptides it is observed that the LVFFA cluster and VGSN turn region interact through intermitant hydrogen bonding (Massi et al. 2001; Massi and Straub 2001b).
Our simulation results do not support the hypothesis that the Dutch E22Q mutation leads to a higher probability of formation of β-structure in the monomeric peptide in aqueous solution. A number of experimental studies have concluded that the Dutch mutant of the monomeric Aβ-peptide undergoes structural transition to a β form in aqueous solution (Miravalle et al. 2000; Sian et al. 2000). Our results suggest that it is likely that those observations result from the formation of solvated oligomeric peptide clusters where the peptide's β structure is stabilized by peptide–peptide interactions.
It is expected that the desolvation of the E22Q mutant peptide is significantly more favorable than that of the WT peptide (Massi and Straub 2001a, b). As a result, it is possible that the increased activity of the E22Q mutant peptide is due to an increased rate of association and aggregation of the monomeric peptide in solution.
The invalidity of the assumption that E22Q has enhanced tendency to form β structure, which we have established using all-atom molecular dynamics simulations, should be viewed with caution. Experimental studies show that β-hairpin can form in about 5 μsec (Munoz et al. 1997). All atom molecular dynamics simulations have shown that the time for forming β-turns in small peptides (like SYPFDV) is ∼1 nsec (Mohanty et al. 1997). We expect that β-structures in Aβ peptides form is a small region that, perhaps, includes residues 16–22. The experimental results on hairpins and molecular dynamics simulations on small peptides indicate that such secondary structures are likely to be populated on timescales ranging from a few nanoseconds to about a microsecond. Because our simulation timescales are shorter, we cannot rule out the possibility that E22Q may exhibit a significant propensity for forming β-structures on much longer timescales than 1 nanosecond.
Materials and methods
The initial conditions for our simulations of the WT β(10–35)-NH2 peptide and the E22Q mutant peptide were obtained from the NMR solution structure of Lee and coworkers (1995) derived from distance geometry calculations employing NMR-derived NOE restraints (Zhang et al. 2000). The structure of the E22Q mutant was modeled from the WT structure. Currently, there is no NMR-derived structure of the E22Q mutant peptide analogous to the structure of the WT Aβ(10–35)-peptide congener. However, NMR measurements of Hα proton chemical shifts for the WT and E22Q mutant Aβ(10–35)-peptides are consistent with a structure of the E22Q mutant that is indistinguishable from the known collapsed-coil structure of the WT peptide (Zhang 1999).
The E22Q Dutch mutant peptide congener is depicted in Figure 5 ▶. The colored regions are Tyr 10–Glu 11–Val 12–His 13–His 14–Gln 15–Lys 16 (blue), Leu 17–Val 18–Phe 19–Phe 20–Ala 21 (red), Glu 22 (green), Asp 23 (blue), Val 24–Gly 25–Ser 26–Asn 27 (yellow), Lys 28–Gly 29–Ala 30–Ile 31–Ile 32–Gly 33–Leu 34–Met 35 (blue). The dominant structural motifs in the peptide are the hydrophobic cluster LVFFA 17–21 segment (red), the turn 24–27 VGSN segment (yellow), and the glutamine residue Q22 that is positioned at the interface between the hydrophobic cluster and turn regions.
Fig. 5.

The E22Q mutant form of the congener Aβ(10–35)-NH2 peptide is depicted. From the N terminus the groups are Tyr 10–Glu 11–Val 12–His 13–His 14–Gln 15–Lys 16 (blue), Leu 17–Val 18–Phe 19–Phe 20–Ala 21 (red), Glu 22 (green), Asp 23 (blue), Val 24–Gly 25–Ser 26–Asn 27 (yellow), and Lys 28–Gly 29–Ala 30–Ile 31–Ile 32–Gly 33–Leu 34–Met 35 (blue). The figure compares the all-atom model employed in the peptide simulations with a coarse-grained representation of the peptide used in the side-chain contact analysis.
We chose a fragment of the Aβ-peptide because fibril formation with enhanced β-sheet content does not require the full length chain. The local propensity for β-flickering can be examined using Aβ-peptide fragments. However, the mechanism of conversion from a random coil to fiber and the resulting morphology might depend on the length of the peptide.
Simulation model and protocol
For the fully solvated WT and mutant peptides, four independent one-nanosecond trajectories were simulated. Each trajectory originated from one of a set of four initial peptide structures that were chosen from two families of conformers characterized by variations in their C-terminal regions. The initial structures resulted from the work of Lee and coworkers (1995) who used a combination of distance geometry refinement and molecular dynamics annealing/minimization procedures employing experimentally derived NOE restraints (Zhang 1999; Zhang et al. 2000; Massi et al. 2001). The core regions of the peptide, including the LVFFA and VGSN substructures, were largely similar in the four starting configurations. However, outside of that core structure there was significant disorder in the N- and C-terminal regions of the peptide due to the small number of experimentally derived restraints in those regions.
The simulation protocol, summarized below, has been described elsewhere in detail (Massi et al. 2001; Massi and Straub 2001b). For the simulations of the WT and mutant peptides, the solute was centered in a rhombic dodecahedron cell that was carved from a cubic box of 50 Å on a side and then filled with 2113 water molecules. Periodic boundary conditions were applied to avoid edge effects. The energetics of the Aβ peptide in water was simulated using the version 22 potential energy function of the CHARMM program (Mackerell et al. 1998). Nonbonded interactions were truncated at 12 Å and Ewald summation was used to evaluate the electrostatic interactions. SHAKE was employed to constrain bonds involving hydrogen atoms to their equilibrium values. A time step of integration of 2 fsec was employed in the Verlet algorithm in the CHARMM program (Brooks et al. 1983). After the equilibration period of 200 psec, a production run of 1 nsec was completed with an average temperature of 300 K. Every 200 fsec, coordinates and energetic data were collected.
The local secondary structure of the WT and E22Q mutant peptides was analyzed through comparison with structural templates and direct measurement of local torsional angle fluctuations. The formation of contacts between side chains, proposed to play an important role in stabilizing the structure of the WT peptide, was also analyzed.
Peptide substructural comparison—contact analysis
The conformational analysis of our trajectories generated for Aβ(10–35)-NH2 peptide has been performed by first mapping the all-atom structure of the peptide onto a coarse-grained model. Instead of considering the peptide in all-atom detail, we retained only the positions of Cα carbons in the backbone and replaced the side chains of amino acids with the positions of their centers of mass. This reduced the 26-mer peptide to a system of 52 interaction sites. Using these trajectories, we calculated several conformational quantities by making several assumptions.
We assumed that the contact between a pair of side chains was formed if their centers of mass were less than 5.2 Å apart. This allowed us to calculate the contact map Cij, in which Cij = 1, if the contact between residues i and j is "on," and is 0 otherwise. The total number of contacts between amino acids was, therefore, C = ∑ Cij (|i − j| ≥ 3).
The energy of a conformation Ep was calculated using the contact map and the contact potentials derived from the statistical analysis of PDB structures. In particular, the contact interactions Bij taken from the Table 3 of Kolinski et al. (KGS; Kolinski et al. 1993) were used. Thus,
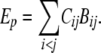 |
(1) |
To examine the balance between local and nonlocal interactions (contacts) along simulated trajectories, we have calculated the contact order
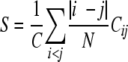 |
(2) |
where |i − j| is the distance along the sequence between residues i and j, N is the number of amino acids (26), and C is the number of amino acid contacts in a given structure.
To reduce the structural fluctuations, we applied cluster analysis based on the pattern recognition method (Klimov and Thirumalai 1998). The idea of this approach is to group together structurally similar conformations sampled along a trajectory and compute an average conformation corresponding to each cluster. Each conformation is described by the vector composed of the distances between all pairs of side chains, which are at least three residues apart. In other words, we only monitored the formation of contacts between side chains. To group conformations into a cluster, we used the conformation distance cutoff of 7 Å.
The clustering is meaningful only when 1 ≪ Ncl ≪Nconf where Ncl is the number of clusters and Nconf is the number of conformations for a given trajectory. On average, the ratio Nconf/Ncl is ∼4.4. We assume that a given pair of side chains forms a contact if the distance between them in a cluster (average structure) is less than the cutoff contact distance of 6 Å. Both cutoff distances can be varied within a certain interval without any qualitative changes in the results.
Using the representation of trajectories as a series of clusters we obtained the probabilities of occurrence for all topologically possible contacts Pq (q = 1,..,276) as time averages over each trajectory. (Index q corresponds to a unique pair of residues i,j, for which |i − j| ≥ 3. In all, there are 276 such pairs for the 26-mer peptide.) The results allowed us to identify the most probable contacts sampled by the peptide during the course of our simulations.
The secondary structure formation was further probed using the measures nH and nS, which are the numbers of dihedral angles in helical and β-strand positions, respectively. To this end, we assumed that a dihedral angle, φi, adopts a helical conformation if |φi − 60°| ≤ 30°. The dihedral angle is said to adopt β-strand structure when |φi − 180°| ≤ 30°.
Peptide substructural comparison—URMS calculations
Peptide substructural comparison was performed using the URMS method developed by Elber and coworkers (Kedem et al. 1999). The difference vector for each pair of Cα atoms along the peptide chain was computed for each stored configuration and used to define a unit difference vector. The resulting unit vectors were given a common origin so as to map the geometry of the peptide backbone onto a unit sphere. This was done for all the configurations stored from the simulation. One particular snapshot configuration was then chosen to be the template for comparison (see Fig. 6 ▶). The comparison was carried out in two steps: (1) the ith configuration was rotated to minimize the sum of the squared distances between the unit vectors of the snapshot configuration and the unit vectors of the template configuration; (2) the URMS difference was set equal to the square root of the minimum of that mean squared sum.
Fig. 6.

A schematic diagram describing the definition of the URMS order parameter used to measure the fluctuations of local peptide main-chain geometries consistent with β-structure.
In the URMS analysis performed in this work, the templates for α or β structure were composed of four Cα atoms each. When the value of the URMS is near 0, there is a strong similarity between the instantaneous and template structures. When the URMS is as large as 1 to 1.4, there is less similarity.
Acknowledgments
This work was supported by grants from National Institutes of Health (IR01 NS41356–01; D.T. and J.E.S.) and the National Science Foundation and the Petroleum Research Fund of the American Chemical Society (J.E.S.). We also acknowledge the Center for Computational Science at Boston University that provided essential computational resources. We thank Klara Kedem and Ron Elber for helpful discussion on the URMS analysis and Jonathan Lee for helpful discussions.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
Aβ, β-amyloid peptide
WT, wild type
CD, circular dichroism spectroscopy
FTIR, Fourier transform infrared spectroscopy
NMR, nuclear magnetic resonance spectroscopy
NOE, nuclear Overhauser enhancement spectroscopy
MD, molecular dynamics
URMS, unit vector root mean square difference
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.3150102.
References
- Barrow, C.J., Yasuda, A., Kenny, P.T., and Zagorksi, M.G. 1992. Solution conformations and aggregational properties of synthetic amyloid β-peptides of Alzheimer's disease—Analysis of circular-dichroism spectra. J. Mol. Biol. 225: 1075–1093. [DOI] [PubMed] [Google Scholar]
- Brooks, B.R., Bruccoleri, R., Olafson, B., States, D., Swaninathan, S., and Karplus, M. 1983. CHARMM: A program for macromolecular energy minimization and dynamics calculations. J. Comp. Chem. 4: 187–217. [Google Scholar]
- Cheng, Y.K., Sheu, W.S., and Rossky, P.J. 1999. Hydrophobic hydration of amphipatic peptides. Biophys. J. 76: 1734–1743. [DOI] [PMC free article] [PubMed]
- Davis, J. and Van Nostrand, W.E. 1996. Enhanced pathologic properties of Dutch-type mutant amyloid β-protein. Proc. Natl. Acad. Sci. USA 93: 2996–3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dima, R.I. and Thirumalai, D. 2002. Exploring protein aggregation and self-propagation using lattice models: Phase diagram and kinetics. Protein Sci. 11: 1036–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esler, W.P., Stimson, E.R., Ghilardi, J.R., Yu, Y.A., Felix, A.M., Vinters, H.V., Mantyh, P.W., Lee, J.P., and Maggio, J.E. 1996. Point substitution in the central hydrophobic cluster of a human β-amyloid congener disrupts peptide folding and abolishes plaque competence. Biochemistry 35: 13914–13921. [DOI] [PubMed] [Google Scholar]
- Esler, W.P., Felix, A.M., Stimson, E.R., Lachenmann, M.J., Ghilardi, J.R., Lu, Y., Vinters, H.V., Mantyh, P.W., Lee, J.P., and Maggio, J.E. 2000a. Activation barriers to structural transition determine deposition rates of Alzheimer's disease. J. Struct. Biol. 130: 174–183. [DOI] [PubMed] [Google Scholar]
- Esler, W.P., Stimson, E.R., Jennings, J.M., Vinters, H.V., Ghilardi, J.R., Lee, J.P., Mantyh, P.W., and Maggio, J.E. 2000b. Alzheimer's disease amyloid propagation by a template-dependent dock-lock mechanism. Biochemistry 39: 6288–6295. [DOI] [PubMed] [Google Scholar]
- Fraser, P.E., Nguyen, J.T., Inouye, H., Surewicz, W.K., Selkoe, D.J., Podlisny, M.B., and Kirschner, D.A. 1992. Fibril formation by primate, rodent, and dutch-hemorrhagic analogues of Alzheimer amyloid β-protein. Biochemistry 31: 10716–10723. [DOI] [PubMed] [Google Scholar]
- Harrison, P.M., Chan, H.S., Prusiner, S., and Cohen, F.E. 2001. Conformational propagation with prion-like characteristics in a simple model of protein folding. Protein Sci 10: 819–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedem, K., Chew, L.P., and Elber, R. 1999. Unit-vector RMS (URMS) as a tool to analyze molecular dynamics trajectories. Proteins 37: 554–564. [PubMed] [Google Scholar]
- Kirkidatze, M.D., Condron, M.M., and Teplow, D.B. 2002. Identification and characterization of a key kinetic intermediate in amyloid β-protein fibrillogenesis. J. Mol. Biol. 312: 1103–1119. [DOI] [PubMed] [Google Scholar]
- Klimov, D.K. and Thirumalai, D. 1998. Lattice models for proteins reveal multiple folding nuclei for nucleation-collapse mechanism. J. Mol. Bio. 282: 471–492. [DOI] [PubMed] [Google Scholar]
- Kolinski, A., Godzik, A., and Skolnick, J. 1993. A general method for the prediction of the 3-dimensional structure and folding pathway of globular proteins—Application to designed helical proteins. J. Chem. Phys. 98: 7420–7433. [Google Scholar]
- Lansbury Jr., P.T. 1999. Evolution of amyloid: What normal protein folding may tell us about fibrillogenesis and disease. Proc. Natl. Acad. Sci. USA 96: 3342–3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J.P., Stimson, E.R., Ghilardi, J.R., Mantyh, P.W., Lu, Y.A., Felix, A.M., Llanos, W., Behbin, A., Cummings, M., Van Criekinge, M., Timms, W., and Maggio, J.E. 1995. 1H NMR of Aβ amyloid peptide congeners in water solution. Conformational changes correlate with plaque competence. Biochemistry 34: 5191–5200. [DOI] [PubMed] [Google Scholar]
- Lomakin, A., Chung, D.S., Benedek, G.B., Kirscher, D.A., and Teplow, D.B. 1996. On the nucleation and growth of amyloid β-protein fibrils: Detection of nuclei and quantitation of rate constants. Proc. Natl. Acad. Sci. USA 93: 1125–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomakin, A., Teplow, D.B., Kirschner, D.A., and Bendek, G.B. 1997. Kinetic theory of fibrillogenesis of amyloid β-protein. Proc. Natl. Acad. Sci. USA 94: 7942–7947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackerell Jr., A.D., Bashford, D., Bellott, M., Dunbrack Jr., R.L., Evanseck, J.D., Field, M.J., Fischer, S., Gao, J., Guo, H., Ha, S., Joseph-McCarthy, D., Kuchnir, L., Kuczera, K., Lau, F.T.K., Mattos, C., Michnick, S., Ngo, T., Nguyen, D.T., Prodhom III, B., Reiher, W.E., Roux, B., Schlenkricha, M., Smith, J.C., Stote, R., Straub, J.E., Watanabe, M., Wiokiewicz-Kuczera, J., Yin, D., and Karplus, M. 1998. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102: 3586–3616. [DOI] [PubMed] [Google Scholar]
- Massi, F. and Straub, J.E. 2001a. Energy landscape theory for Alzheimer's amyloid β-peptide fibril elongation. Proteins 42: 217–229. [DOI] [PubMed] [Google Scholar]
- ———. 2001b. Probing the origins of increased activity of the E22Q "Dutch" mutant of the Alzheimer's β-amyloid peptide. Biophys. J. 81: 697–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massi, F., Peng, J.W., Lee, J.P., and Straub, J.E. 2001. Simulation study of the structure and dynamics of the Alzheimer's amyloid peptide congener in solution. Biophys. J. 80: 31–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melchor, J.P., McVoy, L., and Van Nostrand, W.E. 2000. Charge alterations of e22 enhance the pathogenic properties of the amyloid β-protein. J. Neurochem. 74: 2209–2212. [DOI] [PubMed] [Google Scholar]
- Miravalle, L., Tokuda, T., Chiarle, R., Giaccone, G., Bugiani, O., Ragliavini, F., Frangione, B., and Ghiso, J. 2000. Substitutions at codon 22 of Alzheimer's aβ peptide induce diverse conformational changes and apoptotic effects in human cerebral endothelial cells. J. Biol. Chem. 275: 27110–27116. [DOI] [PubMed] [Google Scholar]
- Mohanty, D., Elber, R., Thirumalai, D., Beglov, D., and Roux, B. 1997. Kinetics of peptide folding: Computer simulations of SYPFDV and peptide variants in water. J. Mol. Biol. 272: 423–442. [DOI] [PubMed] [Google Scholar]
- Munoz, V., Thompson, P.A., Hofrichter, J., and Eaton, W.A. 1997. Folding dynamics and mechanism of β-hairpin formation. Nature 390: 196–199. [DOI] [PubMed] [Google Scholar]
- Sian, A.K., Frears, E.R., El-Agnaf, O.A., Patel, B.P., Manca, M.F., Siligardi, G., Hussain, R., and Austen, B.M. 2000. Oligomerization of β-amyloid of the Alzheimer's and the dutch-cerebral-haemorrhage. Biochem. J. 349: 299–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teplow, D.B. 1998. Structural and kinetic features of amyloid β-protein fibrillogenesis. Amyloid 5: 121–142. [DOI] [PubMed] [Google Scholar]
- Walsh, D.M., Lomakin, A., Benedek, G.B., Condron, M.M., and Teplow, D.B. 1997. Amyloid β-protein fibrillogenesis: Detection of a protofibrillar intermediate. J. Biol. Chem. 272: 22364–22372. [DOI] [PubMed] [Google Scholar]
- Walsh, D.M., Hartley, D.M., Kusumoto, Y., Fezoui, Y., Condron, M.M., Lomakin, A., Benedek, G.B., Selkoe, D.J., and Teplow, D.B. 1999. Amyloid β-protein fibrillogenesis: Structure and biological activity of protofibrillar intermediates. J. Biol. Chem. 274: 25945–25952. [DOI] [PubMed] [Google Scholar]
- Watson, D.J., Landers, A.D., and Selkoe, D.J. 1997. Heparin-binding properties of the amyloidogenic peptides aβ and amylin. J. Biol. Chem. 272: 31617–31624. [DOI] [PubMed] [Google Scholar]
- Watson, D.J., Selkoe, D.J., and Teplow, D.B. 1999. Effects of the amyloid precursor protein glu693→gln `dutch' mutation on the production and stability of amyloid β-protein. Biochem. J. 340: 703–709. [PMC free article] [PubMed] [Google Scholar]
- Wisniewski, T., Ghiso, J., and Frangione, B. 1991. Peptides homologous to the amyloid protein of Alzheimer's disease containing a glutamine for glutamic acid substitution have accelerated amyloid fribril formation. Biochem. Biophys. Res. Commun. 179: 1247–1254. [DOI] [PubMed] [Google Scholar]
- Zhang, S. 1999. `Studies of β amyloid congeners directed toward understanding the molecular mechanism underlying the formation of amyloid deposits in Alzheimer's disease.` Ph.D. thesis, Boston Univeristy, MA.
- Zhang, S.S., Casey, N., and Lee, J.P. 1998. Residual structure in the Alzheimer's disease peptide: Probing the origin of a central hydrophobic cluster. Fold. Des. 3: 413–422. [DOI] [PubMed] [Google Scholar]
- Zhang, S., Iwata, K., Lachenman, M.J., Peng, J.W., Li, S., Stimson, E.R., Lu, Y.A., Felix, A.M., Maggio, J.E., and Lee, J.P. 2000. The Alzheimer's peptide aβ peptide adopts a collapsed coil structure in water. J. Struct. Bio. 130: 130–141. [DOI] [PubMed] [Google Scholar]