

Abstract
Reliability in docking of ligand molecules to proteins or other targets is an important challenge for molecular modeling. Applications of the docking technique include not only prediction of the binding mode of novel drugs, but also other problems like the study of protein-protein interactions. Here we present a study on the reliability of the results obtained with the popular AutoDock program. We have performed systematical studies to test the ability of AutoDock to reproduce eight different protein/ligand complexes for which the structure was known, without prior knowledge of the binding site. More specifically, we look at factors influencing the accuracy of the final structure, such as the number of torsional degrees of freedom in the ligand. We conclude that the Autodock program package is able to select the correct complexes based on the energy without prior knowledge of the binding site. We named this application blind docking, as the docking algorithm is not able to "see" the binding site but can still find it. The success of blind docking represents an important finding in the era of structural genomics.
Keywords: Binding site, drug research, complex, flexible ligand
Structure-based drug design builds on the availability of a reliable structure of a complex of a target molecule and a drug. The most important experimental source for such structures is X-ray crystallography. There is, however, a need for additional methods to predict complex structures, and computer-based molecular modeling is an obvious choice.
In the past decade, molecular docking has proven to be an important tool of computer-aided drug design. Basically, three steps are necessary for successful prediction of a target/ligand complex: (1) definition of the structure of the target molecule, (2) location of the binding site, and (3) determination of the binding mode. Ideally, the structure of the target molecule should be determined experimentally, although some applications of docking have been reported based on a modelled target (Stigler et al. 1999; Menziani et al. 2001; Hetényi et al. 2002). The second step, location of the binding site, can be taken computationally as well, and there are several approaches for finding binding pockets on a protein molecule. The simplest algorithms use shape-based fitting of the ligand to the macromolecular surface (Hendlich et al. 1997; Brady and Stouten 2000). Alternatively, an empirical method for identifying interaction sites based on known protein-ligand complexes (Verdonk et al. 2001) has been reported. The third step is the "typical" application for docking algorithms: Given the binding site on a target molecule, determine the binding mode of a ligand. A large number of programs have been developed to this end, for example, DOCK (Shoichet and Kuntz 1993), AutoDock (Morris et al. 1996, 1998), and some more recent algorithms described in Budin et al. (2001) and Pang et al. (2001).
In most published docking applications, only the third step is taken, and the binding modes of small ligands have been reproduced (Stigler et al. 1999; Sotriffer et al. 1999, 2000). In all these cases, the binding site was predetermined, and therefore, the search space was limited to that region of the protein in the docking simulations. The convincing results of such studies hint to the possibility of applying the same methodology to searching a space larger than a single binding site. In a recent report, protein-protein interactions were reproduced successfully by a combination of ab initio docking and nuclear magnetic resonance data (Morelli et al. 2001). These investigators used shape-based fitting as the main step of the searching process because of the large size of the proteins. Protein-protein interactions were modeled by docking a completely rigid oligopeptide fragment of a protein using the entire surface of the neighboring protein as a target (Neurath et al. 1996). However, for molecules with many degrees of freedom (e.g., flexible peptides), such shape-based fitting or docking of a rigid ligand to the protein would not be fruitful (Klepeis et al. 1998). We have previously scanned the entire surface of an amyloid peptide for possible binding sites of flexible β-sheet breaker peptides (Hetényi et al. 2002) and predicted a complex with features that agree very well with nuclear magnetic resonance data. There are, however, very few reports dealing with the latter kind of blind docking, in which a flexible ligand was docked to a target without prior knowledge of the binding site.
In the present study, we used the AutoDock program package (Morris et al. 1998) to test whether it is possible to find the binding sites (and binding modes) of flexible peptides on a protein without any prior knowledge of their location and conformation. A parameter set based on the AMBER force field (Cornell et al. 1995) and the possibility of using flexible as well as fixed torsions for the ligands during the docking procedure make AutoDock an appropriate tool for such a test.
Results and Discussion
To verify the method of blind docking of ligands to proteins, a set of different protein-peptide complexes was chosen from the Brookhaven Protein Databank. Peptides are ideal test molecules, because they have several possible torsional degrees of freedom, they have different functional groups, and they are composed of amino acids, the same force field parameters can be used as for the target molecule. Target proteins of up to 316 residues were selected for the investigations to keep the computational cost within reasonable limits. Properties of the investigated protein-ligand systems (with increasing number of torsions) are presented in Table 1. In addition to the different protein-peptide systems, the well-known benzamidine-trypsin complex (A) was used as a first test, because benzamidine is a small and rigid molecule that binds to a well-defined pocket on trypsin.
Table 1.
Properties of the protein-ligand systems investigated in this study
| Letter code | PDB code | Protein | No. of residues | Ligand | Refa | No. of free torsions | No. of heavy atoms | Edocked of crystal (kcal/mole) |
| A | 3ptb | Trypsin | 229 | Benzamidine | I | 0 | 9 | −7.77 |
| B | 1ak4 | cyclophilin A | 165 | Ace-AGP-Nme | II | 6 | 21 | −7.45 |
| C | 3tpi | Trypsinogen | 281 | IV | I | 7 | 16 | −8.91 |
| Cw | 3tpi | Trypsinogen | 281 | IV | I | 7 | 16 | −9.13 |
| D | 3cpa | carboxypeptidase A | 307 | GY | III | 7 | 17 | −3.86 |
| E | 8gch | γ-chymotrypsin | 237 | GAW | IV | 8 | 24 | 4.64b |
| F | 5sga | SG protease A | 181 | Ace-APY | V | 9 | 28 | −8.08 |
| G | 1ak4 | cyclophilin A | 165 | Ace-HAGP-NMe | II | 9 | 31 | −9.01 |
| H | 1lna | Thermolysin | 316 | VK | VI | 10 | 17 | −7.77 |
| Hw | 1lna | Thermolysin | 316 | VK | VI | 10 | 17 | −9.13 |
| I | 5sga | SG protease A | 181 | Ace-PAPY | V | 11 | 35 | −8.31 |
| J | 1sua | subtilisin BPN1 | 262 | ALAL | VII | 12 | 27 | −11.88 |
a References: I. Marquart et al. 1983, II. Gamble et al. 1996, III. Rees and Lipscomb 1983, IV. Harel et al. 1991, V. James et al. 1980, VI. Holland et al. 1995, and VII. Almog et al. 1998.
b Positive energy of the crystal structure of the ligand is due to close contacts (see text and Harel et al. [1991] for details).
Generally, in docking calculations several consecutive trials are made for the same system. If a ligand has to maneuver over a large piece of the protein surface to find its proper location, then the probability of finding the energy minimum is much smaller than in the case of docking to a well-defined binding site on the protein, as the searching space is considerably smaller in that case. The necessity of using numerous trials in the latter cases is probably not as critical as in our studies. Virtually no information is available about the number of trials and the number of energy evaluations necessary for blind docking jobs. Therefore, a systematic scan of parameters (trials and energy evaluations) was made to test their influence on the ability of AutoDock to reproduce complex structures. After each job, a uniform evaluation procedure was performed (see Materials and Methods), the results of which are summarized in Tables 2 and 3.
Table 2.
Results of blind docking of the investigated systems using rigid ligands
| Classb | Subclassc | |||||||||
| Systema | No. of trials | No. of evaluations ×106 | Serial no. | Edocked (min) kcal/mole | RMSD (min) Å | N | Edocked (avg.) kcal/mole | RMSD (avg.) Å | N | CPU (h) |
| A1 | 100 | 10 | 1 | −7.94 | 0.371 | 98 | −7.93 | 0.318 | 98 | 8 |
| A2 | 500 | 5 | 1 | −7.94 | 0.379 | 456 | −7.93 | 0.322 | 455 | 19 |
| A3 | 100 | 50 | 1 | −7.94 | 0.368 | 100 | −7.94 | 0.366 | 100 | 38 |
| B | 100 | 50 | 1 | −9.16 | 1.080 | 86 | −9.155 | 1.081 | 86 | 72 |
| C | 100 | 50 | 1 | −12.43 | 0.415 | 57 | −12.43 | 0.415 | 57 | 44 |
| Cw | 100 | 50 | 1 | −15.26 | 0.217 | 75 | −15.26 | 0.216 | 75 | 44 |
| D | 100 | 50 | 1 | −11.03 | 0.653 | 66 | −11.03 | 0.653 | 66 | 58 |
| E | 100 | 50 | 1 | −11.55 | 0.0 | 88 | −11.55 | 0.011 | 88 | 56 |
| (0.676)d | ||||||||||
| F | 100 | 50 | 1 | −12.58 | 0.293 | 91 | −12.58 | 0.291 | 91 | 11 |
| G | 100 | 50 | 1 | −10.53 | 0.681 | 71 | −10.53 | 0.683 | 71 | 89 |
| H | 100 | 50 | 1 | −10.16 | 0.573 | 91 | −10.15 | 0.531 | 91 | 54 |
| Hw | 100 | 50 | 1 | −11.53 | 0.309 | 76 | −11.53 | 0.305 | 76 | 51 |
| I | 100 | 50 | 1 | −13.93 | 0.338 | 99 | −13.93 | 0.340 | 99 | 64 |
| J | 100 | 50 | 1 | −11.18 | 0.156 | 57 | −11.18 | 0.156 | 57 | 36 |
a Subscripted numbers denote different types of jobs at the same system.
b The energy minima of the first classes are the ones of the jobs too (see also Materials and Methods for explanation of the evaluation of the docking experiments).
c For definition of term subclass, see Materials and Methods.
d As the crystal structure had close contacts and positive energy (see Table 1), the energy minimum structure of job 3 was used as a reference. The RMSDs were calculated for the crystallographic ligand structure as well and given in brackets.
Table 3.
Results of blind docking of the investigated systems using flexible ligands
| Classb | Subclassc | |||||||||
| Systema | No. of trials | No of evaluations ×106 | Serial no. | Edocked (min) kcal/mole | RMSD (min) Å | N | Edocked (avg.) kcal/mole | RMSD (avg.) Å | N | CPU (h) |
| B1 | 100 | 10 | 1 | −10.66 | 9.008 | 6 | — | — | — | 30 |
| 2 | −9.79 | 2.458 | 12 | −9.39 | 1.005 | 2 | ||||
| B2 | 500 | 5 | 1 | −10.70 | 9.037 | 25 | — | — | — | 62 |
| 2 | −9.79 | 2.421 | 68 | −8.88 | 1.075 | 14 | ||||
| B3 | 100 | 50 | 1 | −10.47 | 8.514 | 6 | — | — | — | 124 |
| 2 | −9.82 | 2.433 | 18 | −8.95 | 1.065 | 1 | ||||
| C1 | 100 | 10 | 1 | −11.52 | 0.712 | 9 | −11.11 | 0.847 | 8 | 10 |
| C2 | 500 | 5 | 1 | −11.53 | 0.716 | 41 | −11.18 | 0.905 | 39 | 52 |
| C3 | 100 | 50 | 1 | −11.53 | 0.723 | 38 | −11.51 | 0.740 | 37 | 103 |
| Cw1 | 100 | 10 | 1 | −14.32 | 0.739 | 6 | −13.10 | 0.989 | 6 | 27 |
| Cw2 | 500 | 5 | 1 | −14.38 | 0.680 | 17 | −14.14 | 0.845 | 17 | 40 |
| Cw3 | 100 | 50 | 1 | −14.41 | 0.706 | 22 | −14.08 | 0.775 | 22 | 80 |
| D1 | 100 | 10 | 1 | −8.41 | 1.362 | 18 | −8.32 | 1.402 | 7 | 19 |
| D2 | 500 | 5 | 1 | −8.41 | 1.367 | 67 | −8.27 | 1.448 | 20 | 57 |
| D3 | 500 | 50 | 1 | −8.43 | 1.314 | 88 | −8.36 | 1.362 | 31 | 570 |
| E1 | 100 | 10 | 1 | −13.20 | 0.178 | 16 | −13.08 | 0.254 | 5 | 27 |
| (4.908)d | ||||||||||
| E2 | 500 | 5 | 1 | −13.13 | 0.226 | 46 | −12.52 | 0.710 | 10 | 67 |
| (4.961)d | ||||||||||
| E3 | 100 | 50 | 1 | −13.33 | 0.000 | 17 | −13.27 | 0.148 | 7 | 138 |
| (4.930)d | ||||||||||
| F1 | 100 | 10 | 1 | −10.07 | 0.675 | 12 | −9.92 | 0.707 | 10 | 29 |
| F2 | 500 | 5 | 1 | −9.99 | 0.521 | 35 | −9.64 | 0.830 | 11 | 78 |
| F3 | 100 | 50 | 1 | −10.15 | 1.308 | 13 | −9.97 | 0.787 | 5 | 155 |
| G1 | 100 | 10 | — | −11.03 | 28.048 | 2 | — | — | — | 47 |
| G2 | 500 | 5 | 1 | −11.41 | 1.960 | 14 | −11.02 | 1.581 | 4 | 119 |
| G3 | 100 | 50 | 1 | −12.28 | 1.852 | 6 | −12.275 | 1.740 | 2 | 235 |
| H1 | 100 | 10 | 1 | −14.48 | 17.496 | 19 | — | — | — | 27 |
| 4 | −10.96 | 4.733 | 5 | — | — | — | ||||
| H2 | 500 | 5 | 1 | −14.31 | 17.541 | 95 | — | — | — | 56 |
| 4 | −11.78 | 5.036 | 10 | — | — | — | ||||
| H3 | 100 | 50 | 1 | −14.40 | 17.613 | 29 | — | — | — | 114 |
| 4 | −11.62 | 4.251 | 6 | — | — | — | ||||
| Hw1 | 100 | 10 | 1 | −11.60 | 1.855 | 9 | −10.87 | 1.533 | 5 | 21 |
| Hw2 | 500 | 5 | 1 | −11.21 | 1.681 | 42 | −10.66 | 1.330 | 18 | 57 |
| Hw3 | 100 | 50 | 1 | −11.86 | 3.701 | 20 | −11.12 | 1.556 | 16 | 162 |
| I1 | 100 | 10 | 1 | −11.74 | 7.543 | 6 | — | — | — | 43 |
| I2 | 500 | 5 | 1 | −11.63 | 7.256 | 9 | — | — | — | 213 |
| I3 | 500 | 50 | 1 | −11.86 | 7.418 | 23 | — | — | — | 1017 |
| J1 | 100 | 10 | 1 | −10.88 | 7.590 | 3 | — | — | — | 41 |
| J2 | 500 | 5 | 1 | −12.03 | 1.584 | 10 | −12.03 | 1.584 | 1 | 102 |
| J3 | 100 | 50 | 1 | −12.41 | 1.545 | 5 | −12.38 | 1.523 | 2 | 161 |
See notes for Table 2.
The most important requirement of a blind docking calculation is its ability of distinguishing the real binding site on the protein from nonspecific and/or energetically unfavorable ones. Ideally, the crystal structure (or a structure with very low root mean square deviation [RMSD]) should be predicted as having the lowest energy (Edocked). Because the AutoDock program package allows the use of ligands with fixed and flexible torsions, both types were involved in our investigations.
Rigid ligands
Table 2 contains the results of blind docking of rigid ligands, in which the ligand had the conformation it has in the experimental complex. Therefore, the docking algorithm only has to optimize the position and orientation of the ligand molecule. In all cases, the minimum energies Edocked of the first classes and the average ones of subclasses (see Materials and Methods for definition of the term "subclass" in this study) were lower than or close to the energies Edocked calculated for the original crystal structures (Table 1). The reasons for this are the finite grid spacing and the limited accuracy of the force field. For the simplest system, benzamidine-trypsin (A), not much difference was found between the three types of jobs (with different number of trials and energy evaluations, see Materials and Methods). Hence, we performed only one type of the three jobs (100 trials and 50 × 106 energy evaluations per trial) for the other systems.
The RMSD values of the first class and its subclass were generally <0.7 Å. For system B, the RMSD value was >1.0 Å. In that case, a piece of the central part of the cypA-binding loop of the p24 (HIV capsid) protein was used as a ligand. This particular fragment of the loop proved to be too small to mimic perfectly the fit of the central part of the loop of p24 on the cypA molecule. With the use of an additional amino acid (His) of the p24-loop in the fragment to be docked, a more accurate fit was obtained (system G), indicating that it may be possible to predict protein interactions by blind docking of a fragment. However, one should be careful with the selection of the appropriate fragment of a protein for such a modeling as both the presence and the absence of a single amino acid could, in principle, result in different binding modes or even different binding positions.
The energy minima of all jobs were in the subclasses of the first classes, namely, the structures with low RMSD values with respect to the crystal structure. One can therefore conclude that blind docking is a reliable technique for finding the binding site of rigid ligands up to at least 30 heavy atoms. In many pharmaceutical applications, the ligand is almost rigid or has a well-defined conformation (Sotriffer et al. 2000).
Flexible ligands
The real challenge for docking is the use of flexible ligand molecules, that is, those with rotatable torsion angles. The results of these types of dockings are listed in Table 3. In all cases, the energies (Edocked) of the first classes (subclasses) were lower than or close to Edocked calculated for the original crystal structures (Table 1). We found this for the rigid ligands as well (Table 2), but here the effect is somewhat larger in some cases, owing to the extra degrees of freedom. In other cases, the rigid ligands do find lower energies (e.g., Cw) and lower RMSD, most likely because one effectively has more energy evaluations for searching the right conformation.
Efficiency and robustness
For small peptides such as Ile-Val (system C and Cw) and Gly-Tyr (system D), RMSD values <1.0 Å (Figs. 1a, 2a ▶ ▶) and 1.5 Å (Fig. 1b ▶), respectively, were achieved. Moreover, the lowest energy conformations of 100 out of 500 docking trials were the members of the subclasses in all cases. The occupancies (N) of the classes and their subclasses were lower, than those of C, Cw, and D in Table 2. This is owing to the larger number of degrees of freedom of the ligand: The search algorithm was used not only for finding the correct binding position/orientation but also for searching the conformation of the ligand in this case. Therefore, the number N of successful docking trials was lower. For Ile-Val, both solvated (Cw) and water-free (C) targets were used. The presence of water molecules right above the binding site did not hinder docking to the site: The occupancies N were smaller, but the Edocked values were lower than in the case of "dry" protein. This trend was found also for the crystal energies and the Edocked values of rigid ligands owing to interactions with the water molecules (Tables 1, 2). In system B, the real binding position of the loop was found only in the second class. This limited success is caused by the length of the peptide fragment that was used as a ligand (for details, see Rigid Ligands section). Tripeptides, such as Gly-Ala-Trp (system E; Fig. 2b ▶) and Ace-Ala-Pro-Tyr (system F; Fig. 1c ▶), were also docked successfully to their respective pockets (Table 3). The Gly-Ala-Trp (γ-chymotrypsin) system is a difficult task for docking calculations. The crystal structure of this complex (Harel et al. 1991) contains an average structure of a covalently bound ligand and a nonbonded complex. Obviously, the molecular mechanics-based docking technique is developed for only nonbonded interactions. Despite the problematic crystal structure, the hydrophobic pocket (the most important part of the binding site, see Fig. 2b ▶) of γ-chymotrypsin was identified reproducibly. Together with system B, the latter example proves the robustness of the method: The binding location was found even when only part of the ligand or of the site was defined properly. In the complex of SG protease and Ace-Pro-Ala-Pro-Tyr (system I), the first Pro residue of the peptide has no specific contacts with the protein (and there was only a small energy difference between the crystal structures of systems F and I; see Table 1), and hence, docking was not as successful as with the truncated peptide Ace-Ala-Pro-Tyr (Fig. 1c ▶). However, because the rigid ligand docks with 2 kcal/mole lower energy than the flexible one, and with low RMSD with respect to the crystal structure, we should conclude that the poor results with system I are owing to insufficient searching.
Fig. 1.
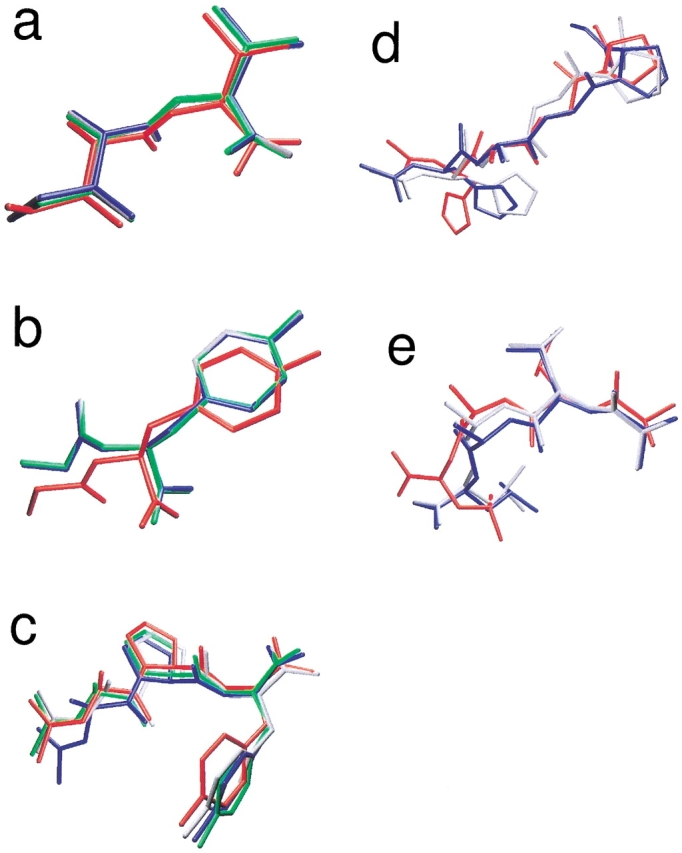
Comparison of the original, crystallographic position (in red) of the different peptides: system C (a), system D (b), system F (c), system G (d), and system J (e). The crystal positions are compared to the energy minima of each job type (green indicates job type 1; grey, job type 2; blue, job type 3; see Table 3). A good correspondence was obtained in all cases.
Fig. 2.
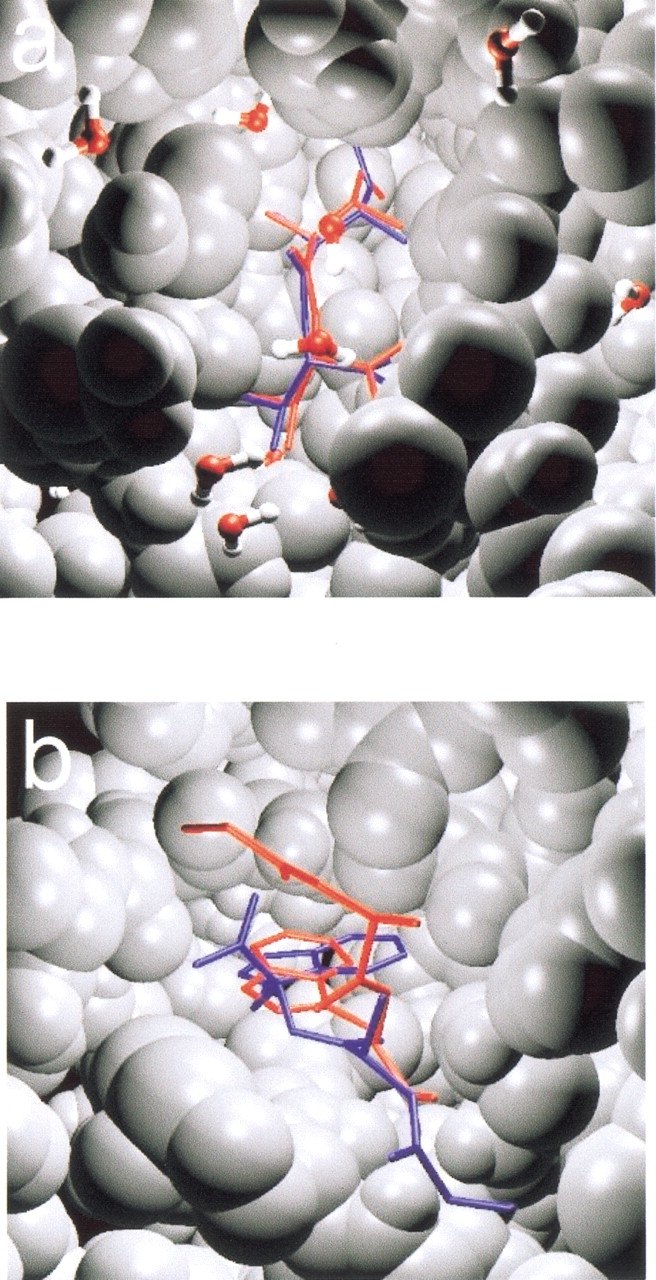
(a) The Ile-Val dipeptide (system Cw) located below the crystallographic water molecules after the docking. (b) The aromatic side-chain of the Gly-Ala-Trp tripeptide (system E) found the hydrophobic pocket on the protein surface (in van der Waals representation). For clarity, only the crystallographic (red) conformation and the one of job type 3 (blue) were used.
The Ace-Ala-Gly-Pro-NMe tripeptide (B) proved to be too small to perfectly mimic the binding loop of p24 protein (Fig. 3 ▶); a different conformation was found with lower energy than the crystal structure. In contrast, the Ace-His-Ala-Gly-Pro-Nme tetrapeptide was more successful (system G; see Fig. 1d ▶): The subclass of the first class contained the conformer closest to the crystal structure, in two out of three jobs. The parameters of the first type of jobs (100 trials/10 × 106 energy evaluations) were not sufficient for finding the site of this larger peptide. The class/subclass populations were also lower than in the case of the above-mentioned shorter peptides: the minimization problem became more difficult. Ala-Leu-Ala-Leu is a simple peptide (system J; see Fig. 1e ▶) but has the largest number of flexible torsions among our test systems. The docked conformers closest to the crystallographic one were reproducibly found in the first class and had the lowest energies in the second and third jobs, similarly to system G.
Fig. 3.

The Ace-His-Ala-Gly-Pro-NMe tetrapeptide (system G, red) mimics the central part of the cypA-binding loop (cyan) of the N-terminal domain of the HIV p24 capsid protein after a docking trial of 50 million energy evaluations. See also Fig. 1d ▶ for a close-up of the crystallographic position of the fragment of the loop and the docked results. The cypA molecule is represented in blue.
Effect of changes in parameters
We have systematically investigated the effects of the number of trials (runs), the number of energy evaluations, and the size of the population of the genetic algorithm on the quality of the docked complexes (RMSD and Edocked values of the subclasses) and on the probability of finding the binding position and mode (N values of Tables 2, 3). In Figure 4a ▶, we have plotted the number N in the first class and subclass as a function of the number of trials for system D (flexible). Both lines are roughly constant, indicating (as expected) that the fraction of correct sites does not depend on the number of trials. In contrast, the number of energy evaluations (Fig. 4b ▶) does have an influence: Below 10 million energy evaluations per trial, fewer correct binding sites are found. Based on these results, it does not seem worthwhile to use >20 million energy evaluations per trial for systems like system D. This is also expected, as it takes a certain number of energy evaluation to converge to the correct energy minimum, but more members of the population will converge to the lowest energy conformation. With more energy evaluations, the system remains in the same conformation. Finally, we performed a test with the size of the population for the genetic algorithm. The population size was increased while keeping the total number of energy evaluations constant (at 5 × 106 energy evaluations and 100 trials). We see that below 50 (the AutoDock default value), the number of correct sites increases fast with the population size. But even after 50, the number of correct sites keeps increasing, indicating that it can be beneficial to use a value well above the default. Although we have no numbers for a population size of >300, it should be expected that the number of correct sites will start to diminish at some stage as the total number of energy evaluations per member of the population (the number of generations) becomes too low to find the minimum. A further test of our recommendations with system F showed that the number of correct sites (class) increases from nine to 22 when going from a population size of 100 to 250, that is, roughly linear. With a population size of 500, we find the correct site is found only in 18 cases.
Fig. 4.
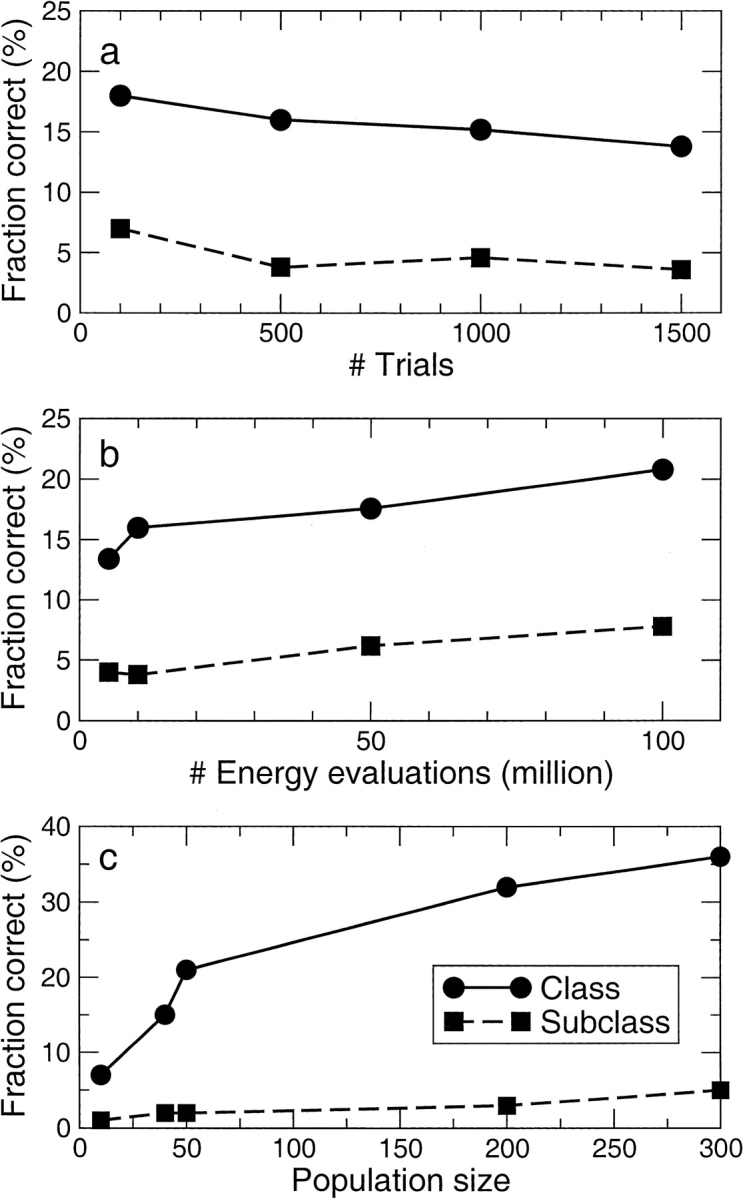
Representation of the effect of different parameters on the fraction of jobs with correct results. (a) Effect of the number of trials (runs) at 50 × 106 energy evaluations and Lamarckian genetic algorithm (LGA) population size of 50. (b) Effect of the number of energy evaluations per trial at 500 trials (runs) and LGA population size of 50. (c) Effect of the LGA population size at 5 × 106 energy evaluations and 100 trials, that is, at constant computational cost.
Limitations
The Val-Lys dipeptide (systems H and Hw) is a small but quite flexible molecule. The side-chain of Lys and the C-terminal group points toward the solvent in the crystal structure, and the atoms have high B-factors, whereas the hydrophobic side-chain of Val and the N-terminal group are buried inside the protein. There is a considerable energy difference between the crystal structures of Val-Lys in system H ("dry" thermolysin target) and Hw (thermolysin target covered by crystal waters), because of the role of the waters in the definition of the binding site. Docking jobs of system H placed the real binding pocket in the fourth class (with higher energies and large RMSD values), whereas when using the crystal waters (Hw), the binding position was ranked at the first place with the lowest energy, and the RMSD values were also significantly lower. This finding shows that solvent molecules can be important in the finding of binding sites, and for some cases, it may therefore be necessary to use explicit water molecules to explore the binding site (Minke et al. 1999).
Another limitation of blind docking is the size of the system or the computer time one can spend to perform enough trials (Tables 2, 3). An example for this limitation is system I, which has the largest ligand (Ace-Pro-Ala-Pro-Tyr) used in our investigations. Although blind docking was successful using the truncated Ace-Ala-Pro-Tyr form of this ligand (system F), the longer tetrapeptide was docked with limited success only (large RMSD values for the first rank) to the target. Additionally, the weak interactions between Pro1 and the protein makes this peptide a difficult ligand for docking. Finally, the possibility of accurately docking a ligand to a target depends critically on the quality of the target structure, and AutoDock, like other docking programs, has as of yet no way to treat flexible target, although a work-around using multiple target structures has been tried with success (Österberg et al. 2002).
Recommendations
Based on our results (Tables 2,3; Fig. 4 ▶), we recommend the use of a fairly large population size (e.g., 250) and at least 10 million energy evaluations per trial for blind docking of flexible peptide ligands to proteins. The number of trials should be ≥100. For rigid ligands, more modest requirements will do; the default population size of 50 and 50 trials should suffice in most cases. Obviously, a finer grid size would be advantageous for the quality of docking results (Hetényi et al. 2002); however, the memory requirements of the algorithm scale as the inverse third power of the grid spacing make it difficult to go much beyond our current grid spacing.
Conclusions
In the present work, we have used a docking algorithm for combined binding site and binding mode search. We named the method blind docking, and it represents a novel application of docking programs. The latest version of the AutoDock package proved to be efficient and robust in finding the binding pockets and binding orientations of the ligands, even for problematic protein-peptide complexes. Using a systematic test of parameters of the docking calculations, we have shown that the results are reproducible. In some cases, we found that the binding position could be located using fragments of ligands. The latter finding hints at the possibility of a combination of blind docking with fragment docking (Friedman et al. 1994) and to studies of protein-protein interactions. The limitations of our approach are basically the same as those for directed docking: The target molecule is rigid, and the accuracy of the force field parameters is limited. Furthermore, the computer time requirements are still considerable (Tables 2, 3).
Blind docking can be a useful tool for exploration of possible binding sites of drugs on their target proteins, if the active site of the drug is unknown. It is particularly attractive that a single method can be used for binding site search and accurate docking of ligands.
Materials and methods
Preparation of ligand and target molecules
For system A, the original structure files of the ligand and target molecules supplied with the test set of the AutoDock program package were used. For the other tests with rigid ligands, the crystallographic structures were used after the addition of polar hydrogen atoms. For the tests with flexible peptides, starting conformations of the ligand molecules were built and optimized with the aid of the TINKER program package (Pappu et al. 1998). AutoTors (an AutoDock tool) was used to define the torsions of the ligand molecules for the docking algorithm. The numbers of released torsion angles are listed in Table 1. In addition to amide and ring torsions, all torsions were released for flexible ligands. AutoTors was used to create a united atom representation of the ligands when necessary. Generally, all water molecules and ions were removed from the original Protein Data Bank files. In case of systems Cw and Hw, the original 151 and 157 water molecules, respectively, were used. The positions of the hydrogen atoms of the water molecules were optimized using a short molecular dynamics simulation with GROMACS (Lindahl et al. 2001) and treated as a part of the target during docking. Protein molecules were equipped with polar hydrogen atoms, and AMBER charges (Cornell et al. 1995) were used for protein as well as ligand. Atomic solvation parameters and fragmental volumes were assigned using Addsol (an AutoDock tool).
Docking procedure
Mass-centered grid maps were generated with 0.55 Å spacing by the AutoGrid program for the whole protein target. Lennard-Jones parameters 12–10 and 12–6 (supplied with the program package) were used for modeling H-bonds and Van der Waals interactions, respectively. The distance-dependent dielectric permittivity of Mehler and Solmajer (1991) was used for the calculation of the electrostatic grid maps. The Lamarckian genetic algorithm (LGA) and the pseudo-Solis and Wets methods were applied for minimization using default parameters (Table 4, except as indicated). The number of generations was set to 10 million in all trials (runs), and the stopping criterion was therefore defined by the total number of energy evaluations. Random starting positions on the entire protein surface, random orientations, and torsions (flexible ligands only) were used for the ligands. Three different sets of jobs were performed, (1) 100 trials, 10 × 106 energy evaluations; (2) 500 trials, 5 × 106 energy evaluations; and (3) 100 trials and 50 × 106 energy evaluations (except D and I, 500 trials). The computational cost in energy evaluations of the different job types is then (1) 109, (2) 2.5 × 109, and (3) 5 × 109 (D and I: 25 × 109). For all job types, the populations in the genetic algorithm was 50. Some jobs with different parameters were performed, as indicated in the Results section.
Table 4.
Docking parameters
| Translation step | 2 Å |
| Quaternion step | 50° |
| Torsion step | 50° |
| Translation reduction factor | 1/cycle |
| Quaternion reduction factor | 1/cycle |
| Torsion reduction factor | 1/cycle |
| No. of top individuals that automatically survive | 1 |
| Rate of gene mutation | 0.02 |
| Rate of crossover | 0.8 |
| No. of generations for picking worst individual | 10 |
| Mean of Cauchy distribution for gene mutation | 0 |
| Variance of Cauchy distribution for gene mutation | 1 |
| No. of iterations of Solis and Wets local search | 300 |
| No. of consecutive successes before changing ρ | 4 |
| No. of consecutive failures before changing ρ | 4 |
| Size of local search space to sample | 1 |
| Lower bound on ρ | 0.01 |
| Probability of performing local search on an individual | 0.06 |
Evaluation of results of docking jobs
A two-step procedure was used for classification of the results of each job. First, the docked conformations of the ligand peptides were listed in increasing energy order. Subsequently, the ligand conformation with lowest energy was used as a reference, and all conformations with a centre of mass to centre of mass distance of <3 Å from the reference were taken to belong to the first class. Once a ligand was assigned to a class, it was not used again for other (energetically less favorable) classes. Then the process was repeated for all hitherto unclassified conformations until all conformations were put in a class.
Second, within each class, the positional RMSD of the nonhydrogen atoms of each ligand structure with respect to the crystal ligand structure was calculated. Structures having RMSD <2 Å were placed in subclasses of the classes. Therefore, in this study, subclasses are defined as the subset of a class that contains the ligand conformations that are structurally closest to the crystallographic ones. That is, ligands placed in the subclass of the first class of a job have the best energies and the best correspondence with the crystal structure. Note that a subclass of any class may be empty if no structure with low RMSD with respect to the crystal structure is found.
In real blind docking jobs (in which the crystallographic position of the ligand on the target is unknown, and no further experimental information is available), one can only assume that the energy minimum represents the best model for the real ligand binding site and binding mode. In that case, one can cluster the results directly by calculating RMSD with the energy minimum. Here we seek to distinguish trials that find the binding site (class) and trials that find both the binding site (class) and the binding mode (subclass).
The VMD program (Humphrey et al. 1996) was used for graphical interpretation and representation of results. Image rendering was performed using Raster 3D (Merritt and Bacon 1997).
Acknowledgments
Tripep AB (Huddinge, Sweden) is acknowledged for financial support. We thank Prof. Janos Hajdu for critical reading of the manuscript.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0202302.
References
- Almog, O., Gallagher, T., Tordova, M., Hoskins, J., Bryan, P., and Gilliland, G.L. 1998. Crystal structure of calcium-independent subtilisin BPN` with restored thermal stability folded without the prodomain. Proteins 31 21–32. [DOI] [PubMed] [Google Scholar]
- Brady, Jr., G.P. and Stouten, P.F.W. 2000. Fast prediction and visualization of protein binding pockets. J. Comput. Aided Mol. Des. 14 383–401. [DOI] [PubMed] [Google Scholar]
- Budin, N., Majeux, N., and Caflisch, A. 2001. Fragment-based flexible ligand docking by evolutionary optimization. Biol. Chem. 382 1365–1372. [DOI] [PubMed] [Google Scholar]
- Cornell, W.D., Cieplak, P., Bayly C.I., Gould, I.R., Merz, Jr., K.M., Ferguson, D.M., Spellmeyer, D.C., Fox, T., Caldwell, J.W., and Kollman, P.A. 1995. A second generation force field for the simulation of nucleic acids, proteins, and organic molecules. J. Am. Chem. Soc. 117 5179–5197. [Google Scholar]
- Friedman, A.R., Roberts, V.A., and Tainer, J.A. 1994. Predicting molecular interactions and inducible complementarity: Fragment docking of Fab-peptide complexes. Proteins 20 15–24. [DOI] [PubMed] [Google Scholar]
- Gamble, T.R., Vajdos, F.F., Yoo, S., Worthylake, D.K., Houseweart, M., Sunquist, W.I., and Hill, C.P. 1996. Crystal structure of human cyclophilin A bound to the amino-terminal domain of HIV-1 capsid. Cell 87 1285–1294. [DOI] [PubMed] [Google Scholar]
- Harel, M., Su, C.-T., Frolow, F., Silman, I., and Sussman J.L. 1991. γ-Chymotrypsin is a complex of γ-chymotrypsin with its own autolysis products. Biochemistry 30 5217–5225. [DOI] [PubMed] [Google Scholar]
- Hendlich, M., Rippmann, F., and Barnickel, G. 1997. LIGSITE: Automatic and efficient detection of potential small molecule-binding sites in proteins. J. Mol. Graph. Model. 15 359–363. [DOI] [PubMed] [Google Scholar]
- Hetényi, C., Körtvélyesi, T., and Penke, B. 2002. Mapping of the possible binding sequences of two β-sheet breaker peptides on beta amyloid peptide of Alzheimer's disease. Bioorg. Med. Chem. 10 1587–1593. [DOI] [PubMed] [Google Scholar]
- Holland, D.R., Hausrath, A.C., Juers, D., and Matthews, B.W. 1995. Structural analysis of zinc substitutions in the active site of thermolysin. Protein Sci. 4 1955–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey, W., Dalke, A., and Schulten, K. 1996. VMD: Visual molecular dynamics. J. Mol. Graphics 14 33–38. [DOI] [PubMed] [Google Scholar]
- James, M.N.G., Sielecki, A.R., Brayer, C.D., Delbaere, L.T.J., and Bauer, C.-A. 1980. Structures of product and inhibitor complexes of Streptomyces griseus protease A at 1.8 Å resolution. J. Mol. Biol. 144 43–88. [DOI] [PubMed] [Google Scholar]
- Klepeis, J.L., Ierapetritou, M.G., and Floudas, C.A. 1998. Protein folding and peptide docking: A molecular modeling and global optimization approach. Comp. Chem. Eng. 22 S3–S10. [Google Scholar]
- Lindahl, E., Hess, B., and van der Spoel, D. 2001. GROMACS 3.0: A package for molecular simulation and trajectory analysis. J. Mol. Model. 7 306–317. [Google Scholar]
- Marquart, M., Walter, J., Deisenhofer, J., Bode, W., and Huber, R. 1983. The geometry of the reactive site and of the peptide groups in trypsin, trypsinogen and its complexes with inhibitors. Acta Cryst. B 39 480–490. [Google Scholar]
- Mehler, E.L. and Solmayer, T. 1991. Electrostatic effects in proteins: Comparison of dielectric and charge models. Protein Eng. 4 903–910. [DOI] [PubMed] [Google Scholar]
- Menziani, M.C., De Rienzo, F., Cappelli, A., Anzini, M., and De Benedetti, P.G. 2001. A computational model of the 5-HT3 receptor extracellular domain: search for ligand binding sites. Theor. Chem. Acc. 106 98–104. [Google Scholar]
- Merritt, E.A. and Bacon, D.J. 1997. Raster3D: Photorealistic molecular graphics. Methods Enzymol. 277 505–524. [DOI] [PubMed] [Google Scholar]
- Minke, W.E., Diller, D.J., Hol, W.G.J., and Verlinde, C.L.M.J. 1999. The role of waters in docking strategies with incremental flexibility for carbohydrate derivatives: Heat-labile enterotoxin, a multivalent test-case. J. Med. Chem. 42 1778–1788. [DOI] [PubMed] [Google Scholar]
- Morelli, X.J., Palma, P.N., Guerlesquin, F., and Rigby, A. 2001. A novel approach for assessing macromolecular complexes combining soft-docking calculations with NMR-data. Protein Sci. 10 2131–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris, G.M., Goodsell, D.S., Huey, R., and Olson, A.J. 1996. Distributed automated docking of flexible ligands to proteins: Parallel applications of AutoDock 2.4. J. Comp. Aided. Mol. Des. 10 293–304. [DOI] [PubMed] [Google Scholar]
- Morris, G.M., Goodsell, D.S., Halliday, R.S., Huey, R., Hart, W.E., Belew, R.K., and Olson, A.J. 1998. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comp. Chem. 19 1639–1662. [Google Scholar]
- Neurath, A.R., Jiang, S., Strick, N., Lin, K., Li, Y.-Y., and Debnath, A.K. 1996. Bovine β-lactoglobulin modified by 3-hydroxyphtalic anhydride blocks the CD4 cell receptor for HIV. Nature Med. 2 230–234. [DOI] [PubMed] [Google Scholar]
- Österberg, F., Morris, G.M., Sanner, M.F., Olson, A.J., and Goodsell, D.S. 2002. Automated docking to multiple target structures: Incorporation of protein mobility and structural water heterogeneity in AutoDock. Proteins 46 34–40. [DOI] [PubMed] [Google Scholar]
- Pang, Y.-P., Perola, E., Xu, K., and Prendergast, F.G. 2001. EUDOCK: A computer program for identification of drug interaction sites in macromolecules and drug leads from chemical databases. J. Comp. Chem. 22 1750–1771. [DOI] [PubMed] [Google Scholar]
- Pappu, R.V., Hart, R.K., and Ponder, J.W. 1998. Analysis and application of potential energy smoothing and search methods for global optimization. J. Phys. Chem. B 102 9725–9742. [Google Scholar]
- Rees, D.C. and Lipscomb, W.N. 1983. Crystallographic studies on apocarboxypeptidase A and the complex with glycyl-l-tyrosine. Proc. Natl. Acad. Sci. 80 7151–7154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoichet, B.K. and Kuntz, I.D. 1993. Matching chemistry and shape in molecular docking. Protein Eng. 6 723–732. [DOI] [PubMed] [Google Scholar]
- Sotriffer, C.A., Flader, W., Cooper, A., Rode, B.M., Linthicum, D.S., Liedl, K.R., and Varga, J.M. 1999. Ligand binding by antibody IgE Lb4: Assessment of binding site preferences using microcalorimetry, docking and free energy simulations. Biophys. J. 76 2966–2977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotriffer, C.A., Ni, H., and McCammon, J. A. 2000. Active site binding modes of HIV-1 integrase inhibitors. J. Med. Chem. 43 4109–4117. [DOI] [PubMed] [Google Scholar]
- Stigler, R.-D., Hoffmann, B., Abagyan, R., and Schneider-Mergener, J. 1999. Soft docking an L and a D peptide to an anticholera toxin antibody using internal coordinate mechanics. Structure 7 663–670. [DOI] [PubMed] [Google Scholar]
- Verdonk, M.L., Cole, J.C., Watson, P., Gillet, V., and Willett, P. 2001. SuperStar: Improved knowledge-based interaction fields for protein binding sites. J. Mol. Biol. 307 841–859. [DOI] [PubMed] [Google Scholar]