

Abstract
A complex pathway involving many molecular chaperones has been proposed for the folding, assembly, and maintenance of a high-affinity ligand-binding form of steroid receptors in vivo, including the glucocorticoid receptor. To better understand this intricate folding and assembly process, we studied the folding of the ligand-binding domain of the glucocorticoid receptor in vitro. We found that this domain can be refolded into a compact, highly structured state in vitro in the absence of chaperones. However, the presence of zwitterionic detergent is required to maintain the domain in a soluble form. In this state, the protein is dimeric and has considerable helical structure as shown by far-UV circular dichroism. Further investigation of the properties of this in vitro refolded state show that it is stable and resistant to denaturation by heat or low concentrations of chemical denaturants. A detailed analysis of the unfolding equilibria using three different structural probes demonstrated that this state unfolds via a highly populated dimeric intermediate state. Together, these data clearly show that the ligand-binding domain of the glucocorticoid receptor does not require chaperones for folding per se. However, this in vitro refolded state binds the ligand dexamethasone only weakly (Kd = 45 μM) compared to the in vivo assembled receptor (Kd = 3.4 nM). We suggest that the role of Hsp90 and associated chaperones is to bind to, and stabilize, a specific conformational state of the receptor which binds ligand with high affinity.
Keywords: Steroid, chaperone, assembly, Hsp90, folding
The glucocorticoid receptor is a member of a large family of ligand-inducible transcription factors which can act as activators or repressors of gene transcription. Members of the family include the steroid receptors estrogen, androgen, and progesterone receptors, as well as the glucocorticoid receptor. Nuclear receptors such as retinoic acid, vitamin D3, thyroid hormone, and peroxisome proliferator-activated receptors are also members of this extensive family. These receptors are soluble and not associated with the membrane, and are present in the cell in either the cytoplasm or nucleus. All members of this family have a domain structure which consists of an N-terminal transactivation domain (AF1) of variable length which is not highly conserved, a highly conserved DNA-binding domain (DBD), the structures of which have been solved and are zinc fingers, a conserved ligand-binding domain (LBD), and a small C-terminal transactivation domain (AF2) (Wurtz et al. 1996).
The structures of the ligand-binding domains of several steroid receptors have been solved by x-ray crystallography, including the progesterone receptor (PR) (Fig. 1A ▶; Tanenbaum et al. 1998; Williams and Sigler 1998) and the estrogen receptor (ER) (Fig. 1D ▶) (Brzozowski et al. 1997). In addition, the structures of the ligand-binding domains of the nuclear receptors retinoic acid RAR and RXR (Fig. 1C ▶) (Bourguet et al. 1995, 2000; Klaholz and Moras 1998; Klaholz et al. 1998), thyroid hormone receptor (TR) (Wagner et al. 1995; Feng et al. 1998; Ribeiro et al. 1998), vitamin D3 receptor (VDR) (Rochel et al. 2000), and peroxisome proliferator-activated receptor (PPAR) (Nolte et al. 1998) have been reported. The ligand-binding domain of the ultraspiracle protein, which is an invertebrate homolog of mammalian RXR, was recently published (Clayton et al. 2001). All members of the superfamily have a three-layer helical sandwich-type structure (Fig. 1 ▶), and the ligand occupies a buried, hydrophobic site. The C-terminal helix, helix 12, acts as a lid over the ligand-binding site and is repositioned on ligand binding. There is no clearly accessible entry or exit site for ligand. The exact position of helix 12 is critical for coactivator binding and transcriptional activity (Wurtz et al. 1996; Moras and Gronemeyer 1998). All crystal structures reported to date are dimers (Fig. 1D ▶), and the activated holoreceptors are known to be functional as hetero- or homodimers in vivo. The dimer interface is conserved within the superfamily, except for the ligand-binding domain of the progesterone receptor, which shows a different dimer interface (Williams and Sigler 1998). It is not known whether this is an artefact of crystallization conditions and packing or the functional interface in vivo.
Fig. 1.
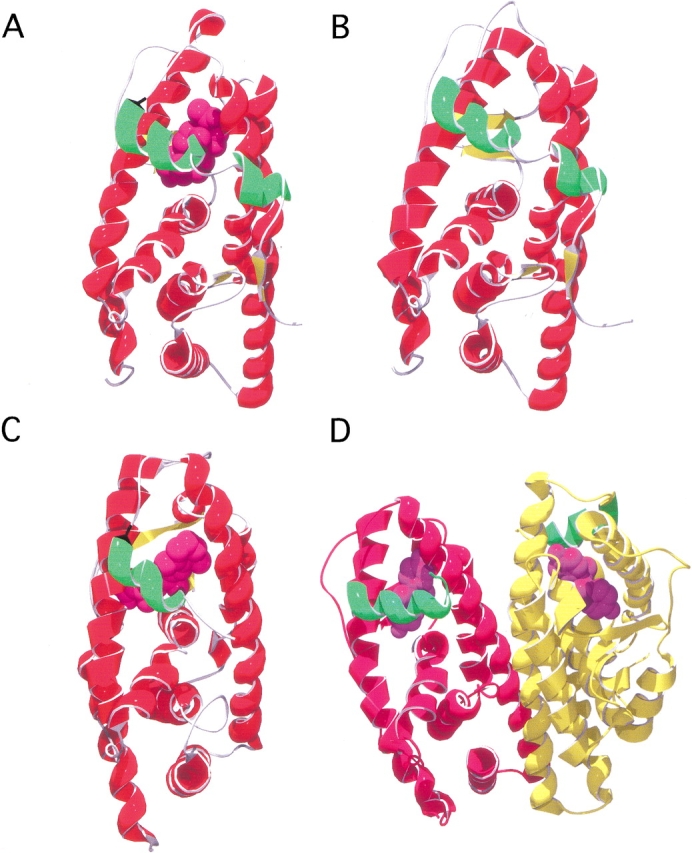
Representative structures of the ligand-binding domain of steroid and nuclear receptors. In (A–C), only the monomer is shown for simplicity; however, these structures are dimeric in the crystalline state. Helix 12, the N-terminal helix which caps the ligand-binding pocket, is indicated (green), and their respective ligands are shown in spacefilling (purple). (A) Ligand-binding domain of the progesterone receptor (PDB code 1A28; Williams and Sigler 1998) bound with progesterone. (B) Model of the ligand-binding domain of the glucocorticoid receptor; see Materials and Methods. (C) Ligand-binding domain of the retinoic acid receptor (RXR) (PDB code 2LBD; Renaud et al. 1995) bound to all-trans retinoic acid. (D) Dimeric form of the ligand-binding domain of the estrogen receptor in complex with diethylstilbestrol (PDB code 3ERD; Shiau et al. 1998).
Molecular dynamic simulations of ligand exit from RARγ suggest that the exit channel is on the same side of the ligand-binding pocket as helix 12. Significant rearrangements are observed during the escape phase and involve the small β-sheet, the Omega-loop and helix H6. After the escape phase, further conformational changes primarily affect the C-terminal helices H11–H12 and the Omega-loop (Blondel et al. 1999). NMR studies on the apo- and holo-forms of the ligand-binding domain of PPAR-γ show that the apo-form is in a conformationally mobile state, and that agonist binding is associated with a marked stabilization of the structure (Johnson et al. 2000). The mobile region was mapped and found to be extensive, including the ligand-binding and cofactor-binding sites. It was proposed that activation of this nuclear receptor results from a shift in population of a dynamic ensemble of conformations, rather than a two-state switch from inactive to active conformation (Johnson et al. 2000).
Steroid receptors exist in the cell in inactive but activatable states complexed with several molecular chaperones and coproteins. A model describing the sequential transient interactions of the steroid receptor with chaperones and the pathway to a high-affinity ligand-binding state was proposed (Smith 1993; Smith et al. 1995; Pratt and Toft 1997; Kosano et al. 1998; Cheung and Smith 2000). In this model, (for review, see Buchner 1999), the steroid receptor initially interacts with the Hsp40/Hsp70/HIP chaperone complex and is then passed on to an Hsp90-containing complex (Chen et al. 1996). In vivo studies have shown that Hsp90 is not only required for the glucocorticoid receptor to achieve a high-affinity ligand binding which is capable of activation, but it is continuously required to maintain that state (Picard et al. 1990; Nathan and Lindquist 1995; Nathan et al. 1997) as the receptor cycles between free and chaperone-bound states. (Smith 1993). Once steroid hormone binds to the receptor, the resulting conformational change prevents its interaction with the chaperone system. Interestingly, and despite structural similarity with the steroid receptors, there is little evidence that the nuclear receptors such as RXR, PPAR, etc., follow the same pathway, and they are not found associated with molecular chaperones in vivo.
Many aspects of the assembly process remain unclear. For example, what is the conformation of the steroid receptor free in solution, or in association with Hsp70, or with Hsp90? What are the exact roles of the chaperones and associated proteins? Why do steroid receptors require this complex pathway to assemble correctly, yet the nuclear receptors do not? To begin to address these and other questions, we investigated the folding and stability of the ligand-binding domain of the glucocorticoid receptor (GR-LBD). It was previously shown that the ligand-binding domain, and not the DNA-binding domain, interacts with Hsp90 (Howard et al. 1990).
Using recombinantly expressed GR-LBD purified under unfolding conditions, here we show, for the first time, that GR-LBD can be refolded to a compact and highly structured state in vitro in the absence of any molecular chaperones. We characterize the structure and stability of this state and show that it is likely to be native-like.
Results
Comparison of the modeled structure of the GR-LBD with other steroid and nuclear hormone receptor ligand-binding domains
The structure of the GR-LBD based on homology modeling with the ligand-binding domain of the progesterone receptor is shown in Figure 1B ▶. As expected, the structure is close to that of other members of the steroid and nuclear hormone receptor superfamily. In our model, helix 12 is capping the ligand-binding site, despite the fact that ligand was not included in the modeling and energy minimization process. This may well be artefactual, resulting from the fact that ligand-bound progesterone receptor was used as a starting point in the calculations. In general, in most of the ligand-bound structures, helix 12 acts as a lid over the ligand-binding site effectively burying the ligand. Exceptions to this are the estradiol-bound form of the estrogen receptor (1A52) solved by Sigler and coworkers (Tanenbaum et al. 1998) and the apo-form of the RXRα (1LBD) (Bourguet et al. 1995). In these structures, helix 12 makes no contacts with the main body of the ligand-binding domain; however, this unusual conformation of helix 12 may be due to crystal packing. Relative to the holo forms of the LBDs, few apo structures have been solved. Those that have include the apo form of RAR (see above) and the heterodimer formed between RAR and RXR (1DKF) (Bourguet et al. 2000). In this structure, helix 12 caps the ligand-binding site but has high B-factors indicating that it is somewhat disordered. There is also evidence from NMR studies on the apo- and holo-form of the ligand-binding domain of PPAR that suggests that helix 12 is largely unstructured in the apo form (Johnson et al. 2000).
Determination of oligomeric state
The oligomeric state of GR-LBD was determined by gel filtration chromatography. A Superdex G200 column (Pharmacia) was used and calibrated using alcohol dehydrogenase, BSA, carbonic anhydrase, and cytochrome b. The elution profile of these proteins in 50 mM Tris HCl, 150 mM NaCl, pH 8.0, in the absence and presence of detergent (0.1% (w/v) Zwittergent 3–12) is shown in Figure 2A and B ▶, respectively. Standard curves under these conditions are shown as inserts. The presence of 0.1% (w/v) Zwittergent 3–12 does not have a significant effect on the elution volumes of the standard proteins. Figure 2C ▶ shows the elution profile for the ligand-binding domain of the glucocorticoid receptor in the presence and absence of detergent. In the absence of detergent, the protein elutes in the void volume, indicating that it has a high molecular mass and is likely to be a soluble aggregate. In the presence of detergent, however, the protein elutes in a volume of 13.8 mL with a KAV of 0.34. This corresponds to a molecular mass of 66.5 kD, which is that expected for a dimer of GR-LBD.
Fig. 2.
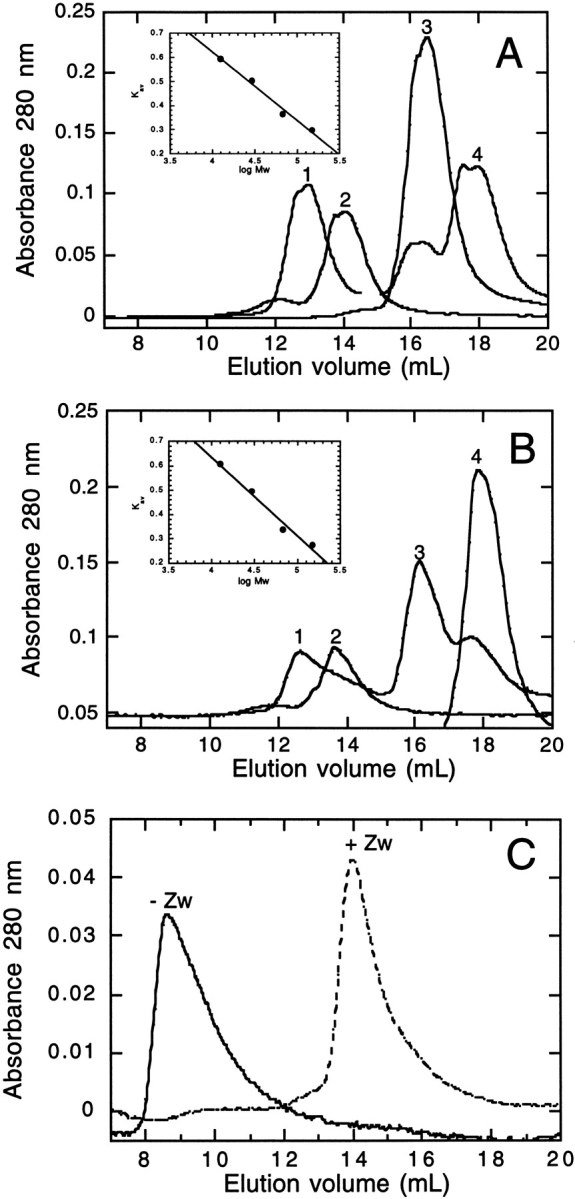
Determination of the oligomeric state: gel filtration chromatography results. (A) Elution profile of alcohol dehydrogenase (1), BSA (2), carbonic anhydrase (3), and cytochrome b (4) in 50 mM TrisHCl, 150 mM NaCl, pH 8.0. Inset shows the calibration curve. KAV is calculated using equation 1. (B) Elution profile of alcohol dehydrogenase (1), BSA (2), carbonic anhydrase (3), and cytochrome b (4) in 50 mM TrisHCl, 150 NaCl, pH 8.0 in 0.1% Zwittergent 3–12. Insert shows the calibration curve calculated as shown above. (C) Elution profile of GR-LBD in 50 mM TrisHCl, 150 mM NaCl in the absence (solid line), or presence of 0.1% Zwittergent 3–12 (dashed line).
Circular dichroism
To determine how structured the in vitro refolded GR-LBD was in detergent, we performed far-UV circular dichroism experiments. The far-UV CD spectra shows minima at 208 and 222 nm typical of a highly helical protein (Fig. 3 ▶). The structures of the ligand-binding domains of estrogen and progesterone receptors contain between 62% and 70% α-helix [ProMotif; CATH database (Orengo et al. 1997)]. Thus, it is likely that the in vitro refolded GR-LBD in detergent adopts a conformation similar to the native structure.
Fig. 3.

Far-UV circular dichroism spectra. Refolded GR-LBD in 20 mM phosphate pH 8.0, 150 mM NaCl, 0.1% Zwittergent 3–12 at 25°C (filled circles) and 95°C (open circles) and in 6.4 M GdnHCl (filled triangles).
For the stability and folding studies, we investigated the stability of the protein with respect to thermal and chemical denaturation. Figure 3 ▶ shows the far-UV spectra for GR-LBD at 25°C and 95°C. There is a slight reduction in the signal at 208 and 222 nm at 95°C, indicating that the protein has lost some structure at this temperature. However, the protein is highly stable with respect to thermal denaturation and does not fully unfold even at high temperatures. The partial thermal unfolding observed was found to be fully reversible (data not shown). Figure 3 ▶ also shows the far-UV spectrum in the presence of 6.4 M GdnHCl. Under these highly denaturing conditions, the protein has completely unfolded and there is little indication of significant residual secondary structure. Chemical denaturation using guanidine hydrochloride was therefore used to study the stability and folding of in vitro refolded GR-LBD.
Chemical denaturant-induced unfolding under equilibrium conditions
The folding and stability of the GR-LBD were investigated using a combination of biophysical techniques. Far-UV CD (see above) showed that the protein fully unfolds in high concentrations of guanidinium chloride. A number of different probes of the folded/unfolded state were employed to characterize the unfolding transitions.
Fluorescence spectroscopy
Figure 4A ▶ shows the fluorescence emission spectra of GR-LBD in 0 and 7.11 M GdnHCl. There is a large decrease in fluorescence and shift of the λmax from 335 to 355 nm on addition of 7.11 M GdnHCl, confirming the far-UV CD results that show that the protein is fully unfolded under these conditions. The maximum difference in fluorescence between the folded and unfolded states is at 328 nm. This wavelength was used to monitor the fluorescence of the protein as a function of guanidinium chloride concentration (Fig. 4B ▶). Two transitions are observed: the first has a midpoint between 1.5 and 3 M GdnHCl, and the second transition has a midpoint between 4 and 6 M GdnHCl and is dependent upon protein concentration (see below). The presence of two transitions during equilibrium unfolding suggests that GR-LBD unfolds and populates a stable intermediate state. All experiments were performed at 10°C, as the protein is soluble and the unfolding transitions fully reversible under these conditions.
Fig. 4.

Guanidinium chloride-induced unfolding of GR-LBD. (A) Fluorescence emission spectra of refolded GR-LBD in 50 mM TrisHCl pH 8.0, 10 mM DTT, 0.1% Zwittergent 3–12 in 0 M (closed circles) and 7.11 M GdnHCl (open circles) at 10°C. (B) Fluorescence at 330 nm as a function of denaturant concentration at 1 μM GR-LBD in 50 mM TrisHCl, 150 mM NaCl, 0.1% Zwittergent 3–12 at 10°C. (C) Far-UV CD ellipticity at 222 nm as a function of denaturant concentration at 3 μM GR-LBD in 20 mM sodium phosphate, pH 8.0, 5 mM DTE, 0.1% Zwittergent 3–12 at 10°C. (D) Elution volume of GR-LBD (filled circles) on a G200 10/30 Pharmacia column as a function of denaturant concentration. The buffer was 50 mM TrisHCl, pH 8.0, 10 mM DTT, 0.1% Zwittergent 3–12. The experiment was run at 4.9°C in buffer containing 150 mM NaCl. For calibration purposes, the elution volume of cytochrome c (closed square), carbonic anhydrase (closed triangle), and bovine serum albumin (open circle) in 7 M GdnHCl are also shown.
Circular dichroism
Figure 4C ▶ shows the far-UV CD signal at 222 nm as a function of denaturant concentration. Two transitions are observed with midpoints in the same range as those observed in the fluorescence experiment. Higher protein concentrations were used here and this affects the position of the second transition (discussed below). These results confirm that GR-LBD unfolds via a stable intermediate state under equilibrium conditions.
Gel filtration chromatography
The hydrodynamic state of the protein during equilibrium denaturation was investigated by analytical gel filtration chromatography (Fig. 4D ▶). A large increase in the hydrodynamic volume occurs between 2.5 and 4 M GdnHCl, which we attribute to the partial unfolding of the native dimer to a less structured and less compact dimer. Although this transition occurs at slightly higher concentrations of denaturant than the first transition observed in the fluorescence and CD experiments, this is due to the fact that the gel filtration experiments were not performed under the same set of conditions. A lower temperature (5°C) was used and salt (150 mM) added to reduce nonspecific interactions of the protein with the column matrix. A fluorescence experiment performed under these conditions showed a shift in the midpoint of the first transition to a higher concentration of denaturant, consistent with the results obtained from the gel filtration experiment (data not shown). The second transition, between partially folded dimer and unfolded monomer, is not observed, as the change in hydrodynamic radii between these two states is small. These data are consistent with a model in which the dimeric, folded protein initially unfolds to an intermediate state which is dimeric and which represents a slightly expanded form of the native state. At higher concentrations of denaturant, the protein completely unfolds and becomes monomeric.
Protein concentration dependence of the unfolding transitions
To verify this model, we performed a series of unfolding experiments with varying protein concentrations, using intrinsic tryptophan fluorescence to monitor the unfolding reaction. The first transition was found to be independent of protein concentration (data not shown), whereas the second transition was found to be dependent on protein concentration. Fluorescence data were normalized to give the percentage of the intermediate state as a function of GdnHCl concentration (Fig. 5A ▶). This plot clearly shows the difference in the second unfolding transition measured at 0.25 and 6 μM GR-LBD. The midpoint of the unfolding transition increases with increasing [GR-LBD] (Fig. 5B ▶). From these data, the free energy of unfolding in water, ΔGH2OU−I can be calculated (Fig. 5C ▶). These data are consistent with the model, in which the second unfolding transition occurs between a partially folded dimeric intermediate and unfolded monomer.
Fig. 5.
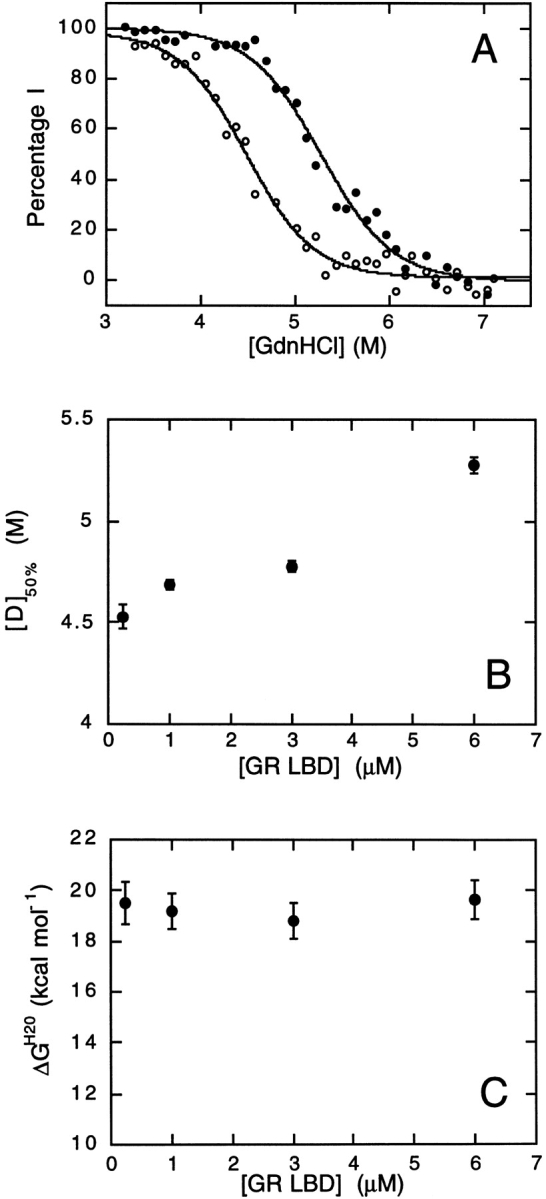
Protein concentration dependence of the second unfolding transition. (A) Concentration of the intermediate state (%I) as a function of denaturant concentration at 0.25 μM GR-LBD (open circles) and 6 μM GR-LBD (closed circles). The midpoints of the unfolding transitions are 4.49 ± 0.04 M and 5.28 ± 0.04 M for 0.25 and 6 μM GR-LBD, respectively. (B) Data transformed to show the midpoint of the second unfolding transition [D]50% as a function of protein concentration [GR-LBD]. (C) Free energy of unfolding, calculated using equation 6, as a function of protein concentration dependence.
GdnHCl-induced unfolding in the presence of ligand
The unfolding transitions were further investigated by determining the effect of the ligand, dexamethasone (Dex), on the unfolding curves. The presence of 200 μM Dex greatly reduced the fluorescence yield, either by quenching of the fluorescence or by an inner-filter effect. The change in fluorescence of the first unfolding transition becomes difficult to observe under these conditions; however, the second unfolding transition can still be seen (data not shown). Fitting the second transition in the presence and absence of ligand to equation 5, estimates of the midpoints of these transitions can be made. The values for mU-I and [D]50% are 2.34 ± 0.16 kcal mol−1 M−1 and 5.00 ± 0.03 M in the absence of ligand and 1.90 ± 0.30 kcal mol−1 M−1 and 5.26 ± 0.08 M, in the presence of 200 μM Dex, respectively. There is a slight increase in the midpoint of unfolding in the presence of ligand. This could mean that the dimeric intermediate state binds ligand; however, the relatively high concentration of ligand and small increase in midpoint indicate that binding, if it is occurring, is very weak.
Ligand-binding experiments
The ligand Dex is a tight-binding ligand of the glucocorticoid receptor in vivo with a Kd of 3.4 nM in COS7 cells (Zhang and Danielsen 1995). Binding of Dex to in vitro refolded GR-LBD in the present study was measured using the change in intrinsic tryptophan fluorescence on binding (Fig. 6A ▶). A correction factor is introduced which compensates for the inner-filter effect resulting from the absorption of incident light at 295 nm by dexamethasone. This was measured using a protein which does not bind dexamethasone as a standard, chymotrypsin inhibitor 2 (Fig. 6A ▶). Using a single-binding site model, data can be fitted to equation 10. The dissociation constant for the binding of Dex to the GR-LBD in 0 M GdnHCl is 46 ± 2 μM.
Fig. 6.
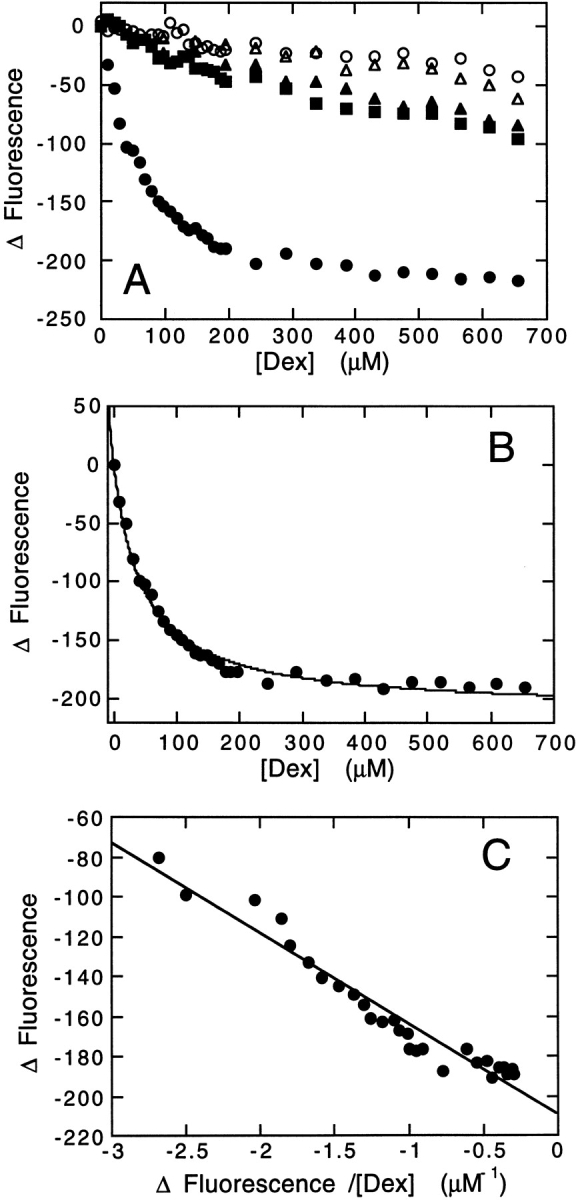
Ligand-binding measurements. (A) Change in fluorescence of 1 μM GR-LBD at 336 nm (0 M GdnHCl) or 350 nm (3, 7 M GdnHCl) as a function of dexamethasone concentration in 0 M (filled circles), 3 M (filled squares), and 7 M (filled triangles) GdnHCl. Change in fluorescence of 1 μM chymotrypsin inhibitor (CI2) at 330 nm as a function of dexamethasone concentration in 0 M (open circles) and 7 M (open triangles) GdnHCl. Baseline fluorescence was measured in parallel incubations with increasing volumes of ethanol. (B) Change in fluorescence of 1 μM GR-LBD at 350 nm in 0 M GdnHCl as a function of dexamethasone concentration after correction for inner-filter effects. This is the same data as that shown in A (filled circles). The solid line shows the best fit of the data to equation 8. (C) Data from B transformed into an Eadie Plot. The solid line shows the best fit of data to equation 9.
Discussion
Folding of the ligand-binding domain of the glucocorticoid receptor in vivo and in vitro
A model was proposed in which the folding and assembly of steroid receptors into a high-affinity ligand-binding conformation is mediated by a variety of molecular chaperones and coproteins (Smith 1993; Smith et al. 1995; Pratt and Toft 1997; Kosano et al. 1998; Buchner 1999; Cheung and Smith 2000). Why such a complex pathway is necessary is unknown. This is especially surprising given that the structurally homologous nuclear receptors do not appear to require such assistance. Currently, nothing is known about the conformation of the steroid receptors as they are assembled into high-affinity ligand-binding states by chaperones and other proteins, and the role of many of the accessory proteins including Hsp90 is not well understood.
To start to dissect this complex assembly process, with the ultimate goal of establishing both the conformational state of the receptor as it passes through the chaperone machinery and the role of each component of the machinery in the assembly process, we have investigated the folding, stability, and ligand binding of an in vitro refolded form of the GR-LBD.
Characterization of the structure of an in vitro refolded form of GR-LBD
GR-LBD was expressed recombinantly in a bacterial expression system. A purification protocol was developed involving the unfolding and subsequent refolding of the protein in vitro in the absence of molecular chaperones. Although a nondenaturing detergent is necessary to maintain the protein in a soluble, nonaggregated state, the protein refolds in vitro to a dimeric and compact, highly structured state, capable of binding ligand, even in the absence of chaperones.
The structure of the in vitro refolded GR-LBD was characterized using a variety of biophysical techniques. The structure is highly helical, as shown by its characteristic far-UV CD signal with minima at 208 and 222 nm (Fig. 3 ▶). In addition, the fluorescence emission spectrum of the folded protein in water shows a λmax at 335 nm, typical of tryptophan residues that are largely protected from solvent in a nonpolar environment. In addition, gel filtration studies indicate that the protein is compact and dimeric (Fig. 2C ▶). Although the ligand-binding domain is dimeric in all of the crystal structures, it has never been clearly established whether the ligand-binding domains are dimeric in solution. Indeed, NMR studies of PPAR have shown that this nuclear receptor is monomeric in solution. The in vitro refolded protein binds ligand, albeit significantly more weakly than in vivo (Fig. 6B ▶). The binding constant for the association of dexamethasone with in vitro refolded GR-LBD is 46 ± 2 μM, compared with a value of 3.4 nM measured in vivo in COS7 cells (Zhang and Danielsen 1995). We can exclude the possibility that the low affinity is a result of expressing the isolated ligand-binding domain, because fusion constructs containing this domain were shown to be regulated by ligand in vivo (Picard et al. 1988).
Characterization of the stability of the in vitro refolded form of GR-LBD
The stability of the in vitro refolded GR-LBD was studied by unfolding experiments using the chemical denaturant guanidine hydrochloride, which reversibly unfolds GR-LBD at 10°C. Two unfolding transitions were observed using different probes. Intrinsic tryptophan fluorescence, which is a good probe of tertiary structure, and far-UV CD, a good probe of secondary structure, were both employed. Results were consistent (Fig. 4B,C ▶), and a model is proposed for the unfolding of GR-LBD in which the dimeric, folded protein initially unfolds to an intermediate state which is dimeric and which represents a slightly expanded form of the folded state. At higher concentrations of denaturant, the protein completely unfolds and becomes monomeric (Fig. 4C ▶).
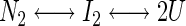 |
The midpoint of unfolding of the second transition was found to be dependent on protein concentration (Fig. 5 ▶), indicating a transition between folded (or in this case, partially folded) oligomer and unfolded monomer.
Together, the results of both structural and stability studies suggest that the in vitro refolded protein is similar in structure to the ligand-binding domains of other steroid receptors (Fig. 1 ▶), as it is a highly structured compact dimer that cooperatively unfolds via a dimeric intermediate.
Comparison of results of in vitro experiments with the model for the folding and assembly of the GR in vivo
In vivo, it was clearly established that the glucocorticoid receptor requires several different molecular chaperones and coproteins in order to maintain a high-affinity ligand-binding state (Nathan et al. 1997). Many of these chaperones and coproteins have also been shown to be essential for the reconstitution of GR and PR in rabbit reticulocyte lysates (for reviews, see Pratt and Toft 1997; Pratt 1998; Cheung and Smith 2000; Smith 2000). However, it is not clear why the GR and other steroid receptors require such a complex pathway, especially as the nuclear hormone receptors do not appear to have any such requirements. One possibility is that the chaperones are required to aid the folding of the protein by preventing aggregation. Given the propensity of the in vitro refolded GR-LBD to form large oligomers in solution in the absence of detergent (Fig. 2C ▶), this may be one role for chaperones such as Hsp90. Because many molecular chaperones do not convey steric information onto the proteins that they interact with during their folding, it would be predicted that under conditions where aggregation is suppressed (e.g., in the presence of detergent) the GR-LBD should be able to refold in the absence of chaperones to a high-affinity binding state. However, as shown in Figure 6 ▶, the in vitro refolded GR-LBD binds ligand with much lower affinity than in vivo, where it is bound to Hsp90, revealing a difference in the conformation in free solution compared to in association with Hsp90. There are several possibilities to account for the low affinity of the in vitro refolded dimeric LBD: (1) in the absence of hormone and chaperones, the domain forms an alternative highly stable conformation that has low affinity for ligand; (2) the domain is not fully folded but in an intermediate state on the folding pathway to the high-affinity state; (3) only the monomeric domain binds ligand with high affinity; or (4) the conformation of helix 12 is such that it cannot cap the binding site and trap the ligand. Hence, Hsp90 and associated chaperones could activate the receptor by rearranging the conformation of helix 12, maintaining the receptor in a monomeric state prior to ligand binding or directing the folding of the receptor to a specific conformational state which can bind ligand with high affinity.
The studies presented here represent an ideal starting point for studies of the interactions of chaperones and coproteins with the ligand-binding domain of the glucocorticoid receptor in vitro. Such studies should illuminate the pathway by which these receptors, and possibly many other target proteins of Hsp90, assemble into their active conformations.
Materials and methods
Homology modeling of the ligand-binding domain of the human glucocorticoid receptor
A model structure of the ligand-binding domain of the human glucocorticoid receptor (GR-LBD) was generated by comparative homology modeling using the x-ray crystal structure of the ligand-binding domain of the progesterone receptor complexed with progesterone (PDB code 1A28; Williams and Sigler 1998). This structure was used as a template within the SWISS-MODEL suite of programs (Guex and Peitsch 1997).
Construction of GR-LBD expression vector
The GR-LBD coding region (1503–2334 bp) was amplified using the polymerase chain reaction from a cDNA clone (gratefully received from Prof. R.M. Evans, Salk Institute), and ligated as a BamHI-EcoRI fragment into pTrcHis-A (Invitrogen). The LBD DNA was subcloned into pHisThio (M. Proctor, Centre for Protein Engineering, Cambridge) as a BamHI-HindIII fragment to generate the plasmid pSHM26. This plasmid expresses the ligand-binding domain as a fusion with E. coli thioredoxin engineered to contain an N-terminal hexa-histidine purification tag. All constructs were fully sequenced.
Expression, purification, and refolding of GR-LBD
Overnight cultures of the E. coli strain C41(DE3) (Miroux and Walker 1996) harboring pSHM26 were used to inoculate 2xTY cultures containing 100 μg mL−1 ampicillin (Sigma) incubating at 37°C. When the absorbance of the cultures at 600 nm reached 0.6, expression was induced with a final concentration of 0.5 mM isopropyl-thio-β-D-galactoside (HT-Technologies). After a further 4 h at 37°C, the cells were harvested, resuspended in 1/25 of the original culture volume of lysis buffer: 50 mM Tris HCl, pH 8.0, 150 mM NaCl, 0.5% (v/v) Triton X-100 (Sigma), and lysed by sonication on ice. Cell debris was removed by centrifugation and the pellets were resuspended and sonicated again in an equal volume of lysis buffer. The supernatants were loaded onto a 15-mL iminodiacetic acid agarose column (Sigma) precharged with nickel ions and equilibrated in 50 mM Tris HCl, pH 8.0. To remove a putative 60 kD molecular chaperone, the column was washed with 2 column volumes of 10 mM MgATP in 50 mM Tris HCl, pH 8.0. Unbound material was washed with 20 column volumes of 50 mM Tris HCl, pH 8.0. Hexa-histidine thioredoxin-GR-LBD fusion was eluted from the column with 250 mM imidazole, 50 mM Tris, pH 8.0. The GR-LBD was cleaved from the fusion by incubation with 10 units mg−1 bovine thrombin (Sigma) overnight at 4°C and then separated from the thioredoxin by precipitation with sodium chloride (final concentration 2 M) at room temperature for 1 h and centrifugation at 17,000 rpm in a Sorvall SS-34 rotor for 30 min at 4°C. The pellets were resolublized in 8 M GdnHCl, 50 mM Tris HCl, pH 8.0, 2.5 mM DTT at a final protein concentration of less than 1 mg mL−1 and stirred at room temperature for 45 min. Any remaining precipitated protein was removed by centrifugation at 17,000 rpm in a Sorvall SS-34 rotor at room temperature for 10 min prior to refolding of the GR-LBD by buffer exchange on a G200 Sepharose HR26/60 column (Pharmacia) preequilibrated in 50 mM Tris, pH 8.0, 150 mM NaCl, 10 mM DTT, 0.1% (w/v) Zwittergent 3–12 (Calbiochem). A flow rate of 5 mL min−1 was used. A concentration of Zwittergent 3–12 of 0.1% (w/v) was found to be optimal in refolding trials and more effective than other reagents used to aid refolding such as glycerol, low concentrations of the denaturants urea and guanidine hydrochloride, and other detergents such as Triton X-100 and CHAPS. The purity of the refolded protein was determined by SDS-PAGE under reducing and nonreducing conditions, mass spectrometry, and N-terminal protein sequencing (data not shown). Purified protein was concentrated, flash frozen, and stored in aliquots at −80°C. Protein concentration was determined spectrophotometrically using a molar extinction coefficient, 43960 cm−1 M−1 at 280 nm, which is for monomeric protein unless otherwise stated.
Determination of the oligomeric state
Analytical size exclusion experiments were performed on a Sephadex G200 HR10/30 (Pharmacia) column equilibrated in 50 mM Tris HCl, pH 8.0, 150 mM NaCl with or without the inclusion of Zwittergent 3–12 at a final concentration of 0.1% (w/v). The relative elution volume of 200 μL of injected 10 μM GR-LBD was compared with molecular weight standards (Sigma). The relative elution volume was calculated as:
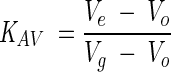 |
(1) |
where Ve is the elution volume, Vo is the void volume determined by elution of Blue Dextran 2000 (Sigma), and Vg is the geometric column volume.
Equilibrium guanidine hydrochloride denaturation
Stock solutions of 8 M guanidine hydrochloride (Fluka Biochemicals) were prepared volumetrically. A range of guanidine hydrochloride concentrations (0–7.2 M) were prepared using a Hamilton Microlab dispenser. Next, 100 μL of a stock solution of GR-LBD in 450 mM Tris HCl, pH 8.0, 90 mM DTT (Melford Laboratories), 0.9% (w/v) Zwittergent 3–12 was added to 800 μL denaturant aliquots and incubated at 10°C for greater than 2 h or overnight prior to measurement. The final protein concentration ranged from 0.25 to 6 μM.
Fluorescence studies
Fluorescence emission spectra were recorded from 300 to 400 nm in a thermostated cuvette holder at 10°C on an SLM Aminco Bowman Series 2 luminescence spectrometer using an excitation wavelength of 280 nm. Both the excitation and emission band passes were set at 4 nm. The fluorescence data were analyzed using the nonlinear regression program Kaleidagraph (version 3.0 Synergy Software, PCS) assuming GR-LBD unfolds through a dimeric intermediate:
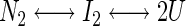 |
Consequently, the equilibrium constant (KU-I) for the second transition, during which dissociation and unfolding of the dimer occurs, is dependent on the total protein concentration:
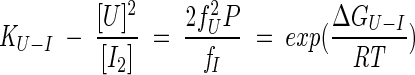 |
(2) |
where P is the total monomeric protein concentration, the fraction of protein unfolded (fu) is [U]/P, the fraction in the intermediate dimer (fI) is 2[I2]/P, R is the gas constant, and T is the temperature in Kelvin. The free energy of unfolding, ΔGU-I, has been found experimentally to be related to the concentration of denaturant [D] by the following equation (Tanford 1968; Pace 1986):
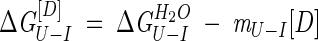 |
(3) |
where mU−I is a constant that is proportional to the increase in the degree of exposure of the protein on denaturation. As the fraction of unfolded protein, fU, can be calculated from the observed fluorescence F:
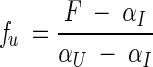 |
(4) |
were F is the observed fluorescence, αI and αU are the fluorescence baselines at intermediate (I) and high (D) denaturant concentrations, equation (2) can be solved in terms of the observed fluorescence and [D] to derive the following equation to fit the fluorescence data set:
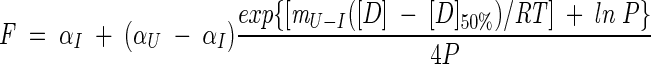 |
(5) |
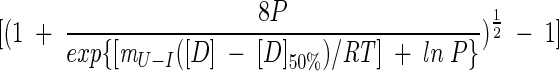 |
where [D]50% is the midpoint of unfolding for the second transition. Hence, the free energy of unfolding in water, ΔGU−IH2O, can be calculated from equation (3) at [D]50% where fU is 0.5:
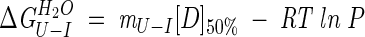 |
(6) |
Circular dichroism
Far-UV CD spectra were recorded on a Jasco J720 spectropolarimeter with 0.1 mm pathlength cell in a thermostated cuvette holder maintained at 10°C or 25°C. A protein concentration of 3–6 μM GR-LBD was used in 20 mM sodium phosphate buffer, pH 8.0 containing 5 mM DTE, 0.1% (w/v) Zwittergent 3–12, and a range of GdnHCl concentrations prepared as described above. The Zwittergent was found not to contribute to the signal.
Size exclusion chromatography
Samples of GR-LBD incubated in a range of GdnHCl concentrations, as described above, were analyzed on a Superose 12 HR10/30 column (Pharmacia) equilibrated in buffer containing the same concentration of denaturant with the addition of 150 mM NaCl, at 4.9°C. Molecular weight standards were denatured overnight in 50 mM Tris HCl, pH 8.0, 10 mM DTT, 7.11 M GdnHCl prior to analysis.
Ligand-binding studies
Binding of increasing concentrations of dexamethasone (10 mM stock in 100% ethanol) to 1 μM GR-LBD was measured by fluorescence emission at 336 nm using an SLM Aminco Bowman Series 2 luminescence spectrometer and an excitation wavelength of 295 nm with excitation and emission band pass of 4 nm. For GR-LBD in the presence of GdnHCl, an emission wavelength of 350 nm was used. Baseline fluorescence was measured in parallel incubations with increasing volumes of ethanol. To correct for the apparent decrease in fluorescence of GR-LBD due to the inner-filter effect of absorption of incident light at 295 nm by dexamethasone, the change in fluorescence of 1 μM of chymotrypsin inhibitor 2 (CI2) was similarly measured at the emission wavelength of 330 nm. The ratio of CI2 fluorescence observed in the presence of ligand (Fobs) to that in absence of ligand (Fcorr) was fitted to the simple equation:
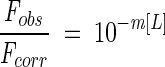 |
(7) |
where [L] is ligand concentration and m is a constant. Using a single-binding site model where the concentration of the GR-LBD-ligand complex is directly proportional to the change in fluorescence (ΔF), the data were fitted to the following equations:
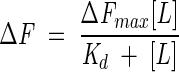 |
(8) |
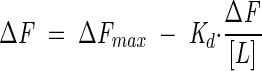 |
(9) |
where Kd is the dissociation constant and [L] is the concentration of ligand.
Acknowledgments
We thank Prof. R.M. Evans for the gift of full-length human glucocorticoid receptor cDNA. S.E.J. is a Royal Society University Research Fellow. S.H.M. is a BBSRC Postdoctoral Fellow. We also gratefully acknowledge Sebastien Poget for running the mass spectrometry experiments, and Mike Weldon, Protein and Nucleic Acid Facility, Biochemistry Department, Cambridge University for performing the N-terminal sequencing. We thank the Cambridge Centre for Molecular Recognition and Prof. Alan Fersht, Centre for Protein Engineering for access to facilities and instrumentation.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.5000102.
References
- Blondel, A., Renaud, J.P., Fischer, S., Moras, D., and Karplus, M. 1999. Retinoic acid receptor: A simulation analysis of retinoic acid binding and the resulting conformational changes. J. Mol. Biol. 291 101–115. [DOI] [PubMed] [Google Scholar]
- Bourguet, W., Ruff, M., Chambon, P., Gronemeyer, H., and Moras, D. 1995. Crystal-structure of the ligand-binding domain of the human nuclear receptor RXR-alpha. Nature 375 377–382. [DOI] [PubMed] [Google Scholar]
- Bourguet, W., Vivat, V., Wurtz, J.M., Chambon, P., Gronemeyer, H., and Moras, D. 2000. Crystal structure of a heterodimeric complex of RAR and RXR ligand-binding domains. Mol. Cell 5 289–298. [DOI] [PubMed] [Google Scholar]
- Brzozowski, A.M., Pike, A.C.W., Dauter, Z., Hubbard, R.E., Bonn, T., Engstrom, O., Ohman, L., Greene, G.L., Gustafsson, J.A., and Carlquist, M. 1997. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature 389 753–758. [DOI] [PubMed] [Google Scholar]
- Buchner, J. 1999. Hsp90 & Co.—A holding for folding. Trends Biochem. Sci. 24 136–141. [DOI] [PubMed] [Google Scholar]
- Chen, S., Prapapanich, V., Rimerman, R.A., Honore, B., and Smith, D.F. 1996. Interactions of p60, a mediator of progesterone receptor assembly, with heat shock proteins Hsp90 and Hsp70. Mol. Endocrinol. 10 682–693. [DOI] [PubMed] [Google Scholar]
- Cheung, J. and Smith, D.F. 2000. Molecular chaperone interactions with steroid receptors: An update. Mol. Endocrinol. 14 939–946. [DOI] [PubMed] [Google Scholar]
- Clayton, G.M., Peak-Chew, S.Y., Evans, R.M., and Schwabe, J.W. 2001. The structure of the ultraspiracle ligand-binding domain reveals a nuclear receptor locked in an inactive conformation. Proc. Natl. Acad. Sci. 98 1549–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, W.J., Ribeiro, R.C.J., Wagner, R.L., Nguyen, H., Apriletti, J.W., Fletterick, R.J., Baxter, J.D., Kushner, P.J., and West, B.L. 1998. Hormone-dependent coactivator binding to a hydrophobic cleft on nuclear receptors. Science 280 1747–1749. [DOI] [PubMed] [Google Scholar]
- Guex, N. and Peitsch, M.C. 1997. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 18 2714–2723. [DOI] [PubMed] [Google Scholar]
- Howard, K.J., Holley, S.J., Yamamoto, K.R., and Distelhorst, C.W. 1990. Mapping the Hsp90 binding region of the glucocorticoid receptor. J. Biol. Chem. 265 11928–11935. [PubMed] [Google Scholar]
- Johnson, B.A., Wilson, E.M., Li, Y., Moller, D.E., Smith, R.G., and Zhou, G.C. 2000. Ligand-induced stabilization of PPAR gamma monitored by NMR spectroscopy: Implications for nuclear receptor activation. J. Mol. Biol. 298 187–194. [DOI] [PubMed] [Google Scholar]
- Klaholz, B.P. and Moras, D. 1998. A structural view of ligand binding to the retinoid receptors. Pure Appl. Chem. 70 41–47. [Google Scholar]
- Klaholz, B.P., Renaud, J.P., Mitschler, A., Zusi, C., Chambon, P., Gronemeyer, H., and Moras, D. 1998. Conformational adaptation of agonists to the human nuclear receptor RAR gamma. Nat. Struct. Biol. 5 199–202. [DOI] [PubMed] [Google Scholar]
- Kosano, H., Stensgard, B., Charlesworth, M.C., McMahon, N., and Toft, D. 1998. The assembly of progesterone receptor-Hsp90 complexes using purified proteins. J. Biol. Chem. 273 32973–32979. [DOI] [PubMed] [Google Scholar]
- Miroux, B. and Walker, J.E. 1996. Over-production of proteins in Escherichia coli: Mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J. Mol. Biol. 260 289–298. [DOI] [PubMed] [Google Scholar]
- Moras, D. and Gronemeyer, H. 1998. The nuclear receptor ligand-binding domain: Structure and function. Curr. Opin. Cell Biol. 10 384–391. [DOI] [PubMed] [Google Scholar]
- Nathan, D.F., and Lindquist S., 1995. Mutational analysis of Hsp90 function—Interactions with a steroid-receptor and a protein-kinase. Mol. Cell. Biol. 15 3917–3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan, D.F., Vos, M.H., and Lindquist, S. 1997. In vivo functions of the Saccharomyces cerevisiae Hsp90 chaperone. Proc. Natl. Acad. Sci. 94: 12949–12956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolte, R.T., Wisely, G.B., Westin, S., Cobb, J.E., Lambert, M.H., Kurokawa, R., Rosenfeld, M.G., Willson, T.M., Glass, C.K., and Milburn, M.V. 1998. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature 395 137–143. [DOI] [PubMed] [Google Scholar]
- Orengo, C.A., Michie, A.D., Jones, S., Jones, D.T., Swindells, M.B., and Thornton, J.M. 1997. CATH—A hierarchic classification of protein domain structures. Structure 5 1093–1108. [DOI] [PubMed] [Google Scholar]
- Pace, C.N. 1986. Methods Enzymol. 131 266–279. [DOI] [PubMed] [Google Scholar]
- Picard, D., Khursheed, B., Garabedian, M.J., Fortin, M.G., Lindquist, S., and Yamamoto, K.R. 1990. Reduced levels of Hsp90 compromise steroid-receptor action in vivo. Nature 348 166–168. [DOI] [PubMed] [Google Scholar]
- Picard, D., Salser, S.J., and Yamamoto, K.R. 1988. A movable and regulable inactivation function within the steroid binding domain of the glucocorticoid receptor. Cell 54 1073–1080. [DOI] [PubMed] [Google Scholar]
- Pratt, W.B. 1998. The Hsp90-based chaperone system: Involvement in signal transduction from a variety of hormone and growth factor receptors. Proc. Soc. Exp. Biol. Med. 217 420–434. [DOI] [PubMed] [Google Scholar]
- Pratt, W.B. and Toft, D.O. 1997. Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr. Rev. 18 306–360. [DOI] [PubMed] [Google Scholar]
- Renaud, J.P., Rochel, N., Ruff, M., Vivat, V., Chambon, P., Gronemeyer, H., and Moras, D. 1995. Crystal-structure of the RAR-gamma ligand-binding domain bound to all-trans retinoic acid. Nature 378 681–689. [DOI] [PubMed] [Google Scholar]
- Ribeiro, R.C.J., Apriletti, J.W., Wagner, R.L., Feng, W.J., Kushner, P.J., Nilsson, S., Scanlan, T.S., West, B.L., Fletterick, R.J., and Baxter, J.D. 1998. X-ray crystallographic and functional studies of thyroid hormone receptor. J. Steroid Biochem. Mol. Biol. 65 133–141. [DOI] [PubMed] [Google Scholar]
- Rochel, N., Wurtz, J.M., Mitschler, A., Klaholz, B., and Moras, D. 2000. The crystal structure of the nuclear receptor for vitamin D bound to its natural ligand. Mol. Cell 5 173–179. [DOI] [PubMed] [Google Scholar]
- Shiau, A.K., Barstad, D., Loria, P.M., Cheng, L., Kushner, P.J., Agard, D.A., and Greene, G.L. 1998. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell 95 927–937. [DOI] [PubMed] [Google Scholar]
- Smith, D.F. 1993. Dynamics of heat shock protein 90-progesterone receptor binding and the disactivation loop model for steroid receptor complexes. Mol. Endocrinol. 7 1418–1429. [DOI] [PubMed] [Google Scholar]
- Smith, D.F. 2000. Chaperones in progesterone receptor complexes. Semin. Cell Dev. Biol. 11 45–52. [DOI] [PubMed] [Google Scholar]
- Smith, D.F., Whitesell, L., Nair, S.C., Chen, S., Prapapanich, V., and Rimerman, R.A. 1995. Progesterone receptor structure and function altered by geldanamycin, an Hsp90-binding agent. Mol. Cell. Biol. 15 6804–6812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanenbaum, D.M., Wang, Y., Williams, S.P., and Sigler, P.B. 1998. Crystallographic comparison of the estrogen and progesterone receptor’s ligand binding domains. Proc. Natl. Acad. Sci. 95 5998–6003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanford, C. 1968. Protein denaturation. Adv. Protein Chem. 23 121–282. [DOI] [PubMed] [Google Scholar]
- Wagner, R.L., Apriletti, J.W., McGrath, M.E., West, B.L., Baxter, J.D., and Fletterick, R.J. 1995. A structural role for hormone in the thyroid-hormone receptor. Nature 378 690–697. [DOI] [PubMed] [Google Scholar]
- Williams, S.P. and Sigler, P.B. 1998. Atomic structure of progesterone complexed with its receptor. Nature 393 392–396. [DOI] [PubMed] [Google Scholar]
- Wurtz, J.M., Bourguet, W., Renaud, J.P., Vivat, V., Chambon, P., Moras, D., and Gronemeyer, H. 1996. A canonical structure for the ligand-binding domain of nuclear receptors. Nat. Struct. Biol. 3 87–94. [DOI] [PubMed] [Google Scholar]
- Zhang, S. and Danielsen, M. 1995. 8-Br-cAMP does not convert antagonists of the glucocorticoid receptor into agonists. Recent Prog. Horm. Res. 50 429–435. [DOI] [PubMed] [Google Scholar]