

Abstract
The mitochondrial prohibitin complex consists of two subunits (PHB1 of 32 kD and PHB2 of 34 kD), assembled into a membrane-associated supercomplex of approximately 1 MD. A chaperone-like function in holding and assembling newly synthesized mitochondrial polypeptide chains has been proposed. To further elucidate the function of this complex, structural information is necessary. In this study we use chemical crosslinking, connecting lysine side chains, which are well scattered along the sequence. Crosslinked peptides from protease digested prohibitin complexes were identified with mass spectrometry. From these results, spatial restraints for possible protein conformation were obtained. Many interaction sites between PHB1 and PHB2 were found, whereas no homodimeric interactions were observed. Secondary and tertiary structural predictions were made using several algorithms and the models best fitting the spatial restraints were selected for further evaluation. From the structure predictions and the crosslink data we derived a structural building block of one PHB1 and one PHB2 subunit, strongly intertwined along most of their length. The size of the complex implies that approximately 14 of these building blocks are present. Each unit contains a putative transmembrane helix in PHB2. Taken together with the unit building block we postulate a circular palisade-like arrangement of the building blocks projecting into the intermembrane space.
Keywords: Crosslinking, mass spectrometry, prohibitin complex, PHB complex, structure prediction
The two structurally related proteins PHB1 and PHB2, previously referred to as prohibitin proteins, localize to mitochondria in mammals, plants, and yeast (Ikonen et al. 1995; Coates et al. 1997; Snedden and Fromm 1997; Berger and Yaffe 1998; Steglich et al. 1999; Nijtmans et al. 2002). In yeast, levels of PHB1 and PHB2 have shown to be interdependent, and have been shown to physically associate with each other to form a large multimeric complex in the mitochondrial inner membrane of mammals and yeast (Snedden and Fromm 1997; Steglich et al. 1999; Nijtmans et al. 2000). The molecular mass of the so-called PHB complex is estimated to be 1 MD by migration in Blue Native Electrophoresis (BNE) experiments. PHB constituent proteins are ubiquitously expressed in mammalian tissues, and have been highly conserved through evolution, suggesting a vital function among eukaryotes. To date, various functions have been attributed to both PHB proteins, including cell cycle regulation, receptor-mediated signaling at the cell surface, aging, apoptosis, and a role in the assembly of mitochondrial respiratory chain complexes (for review, see Nijtmans et al. 2002). Although the now clear localization of the PHB complex to the mitochondria has already provided the context where to place its function, the exact role of these proteins still remains unclear. Based on recent results we propose that the PHB complex forms a novel type of membrane-associated holdase/unfoldase chaperone for the stabilization of mitochondrial proteins. Because of the ability of the PHB complex to stabilize newly translated mitochondrial gene products via direct interaction it was proposed that the PHB complex could form a barrel structure where proteins could be contained (Nijtmans et al. 2000). In the yeast, Saccharomyces cerevisiae, the PHB1 and PHB2 subunits have molecular weights of 32 and 34 kD, respectively, varying very little among other eukaryotes. Assuming a molecular mass of 1 MD, between 12 and 16 subunits of each PHB1 and PHB2 would be needed to form such a complex. Beyond the interdependence of PHB1 and PHB2 to form the PHB complex, which suggests that they probably associate in a 1:1 ratio (further substantiated by the equal staining intensity of the protein bands upon blue native 2D electrophoresis) (Nijtmans et al. 2000), nothing is known about the structure of the complex. More detailed structural information of the PHB complex is required to further elucidate the molecular mechanism of action of the PHB complex. Resolving the structure of a protein with unknown function will give valuable clues to its role in vivo. Three-dimensional protein structures have been traditionally determined by X-ray crystallography and NMR spectroscopy. X-ray crystallography is limited to the availability of analyte crystals and NMR to small protein complexes. These techniques require relatively large amounts of pure protein, and this becomes especially labor-intensive in the case of membrane proteins.
Also, despite the increasing number of determined macromolecular structures, structural genomics and protein modeling are still limited to known folds or structures. Although PHB proteins are highly conserved in evolution, their low sequence similarity to known protein folds makes them difficult to model.
As an alternative approach to structure determination we have generated amino acid distance information using crosslinking technology and mass spectrometry for the identification of crosslinks (Bennett et al. 2000b). Although the atomic distance information provided by crosslinks might be of low resolution, a limited number of distance constraints (Young et al. 2000) can be of valuable help to solve the tertiary structure of a macromolecule. Our crosslinking results suggest that the PHB complex is built from heterodimers, where PHB1 and PHB2 run parallel and intertwined for a long distance through their primary structure, raising the concept of a PHB "unit cell." We predict the unit cells to be organized into a large palisade shaped macromolecular complex.
Results
Isolation of the complex
Highly purified mitochondria from yeast cells overexpressing the PHB complex were extracted for blue native electrophoresis as described in Materials and Methods. After the first native dimension the predominant high molecular weight band corresponding to the PHB complex was cut out. The stability of the complex after electroelution was tested by reapplying the unmodified electroeluted complex to two-dimensional blue native electrophoresis. Approximately 90% of the electroeluted complex remained stable, as seen with Western analysis of its migration as a high molecular weight entity (Fig. 1A ▶).
Fig. 1.

Reelectrophoresis of the electroeluted PHB complex. The PHB complex was cut out of a first dimension BNE gel and electroeluted as described in Materials and Methods. (A) Control incubation of an aliquot of complex in crosslink buffer for the same duration as a crosslinking experiment. After incubation the complex was run again on a native first dimension and subsequently on a denaturing (SDS) second dimension. Protein was visualized by an anti-PHB1 antibody. Approximately 90% of the loaded amount of protein is still in intact complexes running at 1 MD. (B) Crosslinking with sBID. An aliquot of the electroeluted complex was incubated with 1 mM sBID as described in Materials and Methods. Western analysis after BNE2D revealed bands running at 1 MD in the first dimension (size of the PHB complex) that dissociated into 70-kD bands in the second dimension (size of PHB dimers).
Crosslinking
Crosslinks were introduced into the PHB complex with the bifunctional lysine reactive crosslinkers DTSP and sBID as described in Materials and Methods. For crosslinking in solution, the PHB complex was electroeluted from the gel slices. In other experiments DTSP was applied directly to the gel pieces.
After crosslinking the complex still migrated in BNE at 1 MD. In the second (SDS) dimension the band at 32 kD was replaced by a new band at approximately 70 kD (illustrated for sBID in Fig. 1B ▶). Peptide mass fingerprinting showed this new band to contain both PHB1 and PHB2 (data not shown). Figure 2 ▶ shows the aligned sequences of the homologous PHB polypeptides. Also indicated are the positions of the many lysine-residues, scattered along the peptide chains, providing numerous crosslink opportunities.
Fig. 2.

Aligned sequences of PHB1 and PHB2. The region expected to be membrane spanning in PHB2 and membrane associated in PHB1 is underlined. Lysine residues (possible targets of the crosslinkers used in this study) are boxed. It can be seen that lysines are well distributed along the chains.
After crosslinking the complex was digested with trypsin in solution, or in gel pieces cut out after second dimension denaturing electrophoresis. Peptide mixtures were mass-mapped by MALDI-TOF and ESI-QTOF mass spectrometry. Subsequently, MS data were scanned for digest fragments that were modified with the used chemical crosslinker. These analyses were supported by a custom-made software tool called FindLink (see Materials and Methods). The crosslinks found in this study are summarized in Table 1. To validate our identification method we performed low-energy CID MSMS of some proposed crosslinked peptides and confirmed the identification. This is illustrated in Figure 3 ▶ for the fragmentation of the DTSP crosslinked peptide in which PHB1 K204 links to PHB2 K233. For DTSP-linked peptides reduction and alkylation of the disulfide bridge in the crosslinker and retrieval of the alkylated products (Bennett et al. 2000a) also yielded additional confirmations.
Table 1.
Crosslinks identified by mass spectrometry
| Experimenta | MH+expb | Error (ppm) | Fragment 1c | Fragment 2c | Crosslinkd |
| 1,3,6 | 1117.55 | 32 | PHB1: 73–76 TKPK | PHB2: 102–105 AKPR | PHB1K74 PHB2K103 |
| 1,3,6 | 1485.71 | 9 | PHB2: 11–21 YAKAFQKQLSK | PHB2K13 PHB2K17 | |
| 1,2,3,4,6,7,8 | 1646.83 | 32 | PHB1: 152–159 IRKELSTR | PHB2: 102–105 AKPR | PHB1K154 PHB2K103 |
| 1,3,4,6,7,8 | 1777.93 | 1 | PHB1: 147–154 EIISQKIR | PHB2: 175–179 EKVSR | PHB1K151 PHB2K176 |
| 1,2,3,4,5,6,7,8 | 1874.99 | 4 | PHB1: 147–160 EIISQKIRKELSTR | PHB1K151 PYB1K154 | |
| 9,10,11 | 1888.06 | 6 | PHB1: 147–160 EIISQKIRKELSTR | PHB1K151 PHB1K154 | |
| 1,2,3,4,5,6,7,8 | 2398.20 | 4 | PHB1: 200–209 FLVEKAEQER | PHB2: 227–235 AAFVVDKAR | PHB1K204 PHB2K233 |
| 1,2,3,4,6,7,8 | 2442.26 | 10 | PHB1: 73–85 TKPKSIATNTGTK | PHB2: 264–270 DYVELKR | PHB1K76 PHB2K269 |
| 1,3,4,6 | 3212.70 | 25 | PHB1: 189–204 QIAQQDAERAKFLVEK | PHB2: 262–270 SRDYVELKR | PHB1K199 PHB2K269 |
a 1 DTSP crosslink in blue native gel; 2, DTSP crosslink in solution; 3, DTSP MALDI-TOF + FindLink; 4, DTSP ESI-QTOF-MS + FindLink; 5, DTSP ESI-QTOF-MSMS; 6, reductive alkylation of DTSP + MALDI-TOF; 7, reductive alkylation of DTSP + ESI-QTOF-MS; 8, reductive alkylation of DTSP + ESI-QTOF-MSMS; 9, BID crosslink in solution; 10, BID crosslink + MALDI-TOF + FindLink; 11, BID crosslink + ESI-QTOF-MSMS.
b Measured m/z (z = 1) for MALDI-TOF or charge deconvoluted ESI-QTOF-MS.
c Protein chain, peptide fragment, residue numbers, and peptide sequence. Lysine residues involved in the crosslink are underlined.
d Positions of the crosslinked lysines.
Fig. 3.

ESI-QTOF-MSMS analysis of a DTSP crosslinked peptide. P1K204 is linked to P2K233. The singly charged peptide was seen in MALDI-TOF at m/z 2398.2 (deviation from calculated mass = 14 ppm). In ESI a triply charged ion was seen at m/z 800.1 and fragmented by low-energy CID as described in Materials and Methods. (A) Structure of the crosslinked peptide: the lysine residues are linked by their ɛ-amino groups through DTSP. Observed fragment ions are indicated (Roepstorff and Fohlman nomenclature). (B) ESI-QTOF-MSMS spectrum: fragment ions corresponding to the ions marked in (A) are annotated.
As expected, in addition to the crosslinks, we found a large number of surface-labeled peptides (where lysine has reacted with one end of the crosslinker and the other reactive moiety has been hydrolyzed).
Foremost it is noteworthy that all but two (PHB1 K151-K154 and PHB2 K13 -K17, which are four residue spanning crosslinks, amounting to one α-helical turn) crosslinks observed are between residues in a PHB1 and a PHB2 chain. We have neither observed any crosslinks from one PHB1 molecule to another PHB1, nor from a PHB2 molecule to another PHB2. This leads directly to the hypothesis that the complex consists of heterodimeric PHB1–PHB2 building blocks.
From the observation of the crosslink pairs PHB1K74–PHB2K103, PHB1K151–PHB2K176, and PHB1K204–PHB2K233, which align in primary and secondary structure, we hypothesize both chains to run intertwined for long stretches. This fits the results in that it would give extensive possibilities for interchain crosslinking, and render the chances of residues within the same chain to be within crosslinking distance less likely.
As can be seen in Figure 4 ▶ crosslinks are found predominantly in the C-terminal helical region. In the central beta sheets only PHB1K74 links to PHB2K103. This latter PHB2K103 is also close to PHB1K154, as is PHB1K76 with PHB2K269.
Fig. 4.

Predicted secondary structure of the PHB proteins. Solid blocks indicate α-helices, filled arrows indicate β-sheets. Alignment is the same as in Figure 2 ▶. Crosslinks found in this study (see also Table 1) are indicated. Most crosslinks connect residues in PHB1 to residues in PHB2.
Integrating predicted secondary structure and observed crosslinks
The lack of suitable model templates with high homology to prohibitins and other members of the so-called Band 7 protein family prompted us to use prediction methods as a starting point. To check for possible trans-membrane regions, we used the TMHMM algorithm (Krogh et al. 2001). The suggested evidence for a possible transmembrane helix for PHB2 (positions 37–59) seems adequate (data not shown). The homologous helical site in PHB1 may not quite fulfill the requirements for transmembrane spanning helices. Yet, the similarity to PHB2 is so high that in our model it is considered membrane associated. As we did not find crosslinks that contribute structural data in the region N terminal to the putative membrane stretches, we cannot experimentally derive a structure in this region.
To predict possible fold formation, we used the algorithms Jpred2 (Cuff and Barton 1999, 2000) and PSSM (Kelley et al. 2000) at default settings. From the output a model for both chains was built, in which we assigned similar secondary structure to aligned portions of both molecules. This structure is depicted in Figure 4 ▶. It is clear that the proteins belong to the mixed α/β-fold family, a very broad group of proteins with many possible fold formations.
The PSSM fold recognition algorithm subsequently suggested homology of the C-terminal domain of prohibitin to the PSSM superfamily with founder ID c1fioa, consisting of the four-helix bundle proteins syntaxin 1A and T-Snare protein SSO1. For PHB2 this structure ranked first, scoring a PSSM E-value of 0.513; for PHB1, this structure ranked third, scoring an E-value of 1.81. Although these values are not convincing in itself (confidence interval approximately 50%), many of the crosslinks found in this study were superimposable on this template, while other possible solutions were ruled out because we could not fit in the observed crosslinks in the corresponding 3D models. This further increases the confidence intervals, although it cannot be yet expressed in numbers. To our knowledge this is the first report on a hitherto unknown protein structure where computed structural predictions were substantiated via crosslinks as distance restraints. Still, all precautions that are to be taken with structural modeling are valid, and the model presented should be viewed as a best approach given current limitations. Therefore, the model is not refined to high resolution, but only a fold designation of the predicted secondary structure is presented.
The output of the structural prediction algorithms was imported into 3D viewers. The suggested regions of homology, comprising two α helices and a connecting β strand were selected and positioning was constrained by the span of the crosslinks found. We used a length of 8 Å (± 1 Å) for the length of DTSP, as computed by Green (Green et al. 2001). It is plausible that the four-helix bundle present in Snares is represented by a two-helix contribution from both PHB1 and PHB2. Taken together, a model depicted in Figure 5 ▶ is constructed. In this model a β-sheet region (containing the crosslink between PHB1K74 and PHB2K103) is close to the membrane and the four-helix bundle protrudes further into the mitochondrial intermembrane space.
Fig. 5.
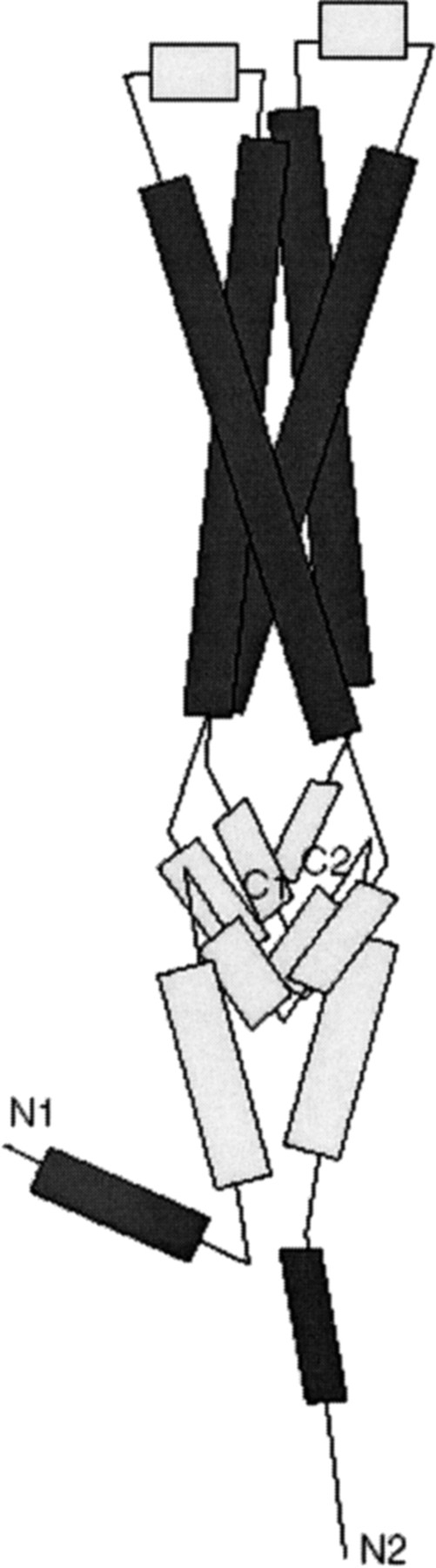
The model of a unit-cell building block. An elongated three segment assembly consisting of a membrane anchor, a central β-sheet and a protruding four α-helix bundle (similar to the four helix-bundles of Snare proteins) is postulated.
Discussion
In this study we have proposed a partial structure for the prohibitin complex. The requirement for both PHB1 and PHB2 to be present stoichiometrically to form a complex (Nijtmans et al. 2000) already indicates a strong codependence of both subunits. The striking finding that hardly any contacts within the same polypeptide chain are observed, nor any crosslink between two PHB1 chains or between two PHB2 chains leads to the concept of a PHB "unit cell." Although the lack of observation of crosslinks between two chains of the same type does not entirely rule out the possibility of contacts between them, the prevalence of links between chains of PHB1 and PHB2 suggest that these are in closest proximity. In this model the complex is built from heterodimers, which run parallel and intertwined for a long distance across the primary structure.
Prediction methods propose a trans-membrane helix for PHB2 (residues 37–59) (Krogh et al. 2001). By analogy, in our model the hydrophobic helix in the N terminus of PHB1 (residues 14–30) is expected to be membrane associated. Both chains are on the same side of the membrane (Nijtmans et al. 2002), as is exemplified by our finding of an extensive number of crosslinks between the two chains. A model in which an assembly of PHB1 is found on one side, and an assembly of PHB2 on the other side of the mitochondrial inner membrane is ruled out as has already been suggested (Nijtmans et al. 2002). A model best fitting our experimental results is presented in Figure 5 ▶. We propose this assembly to project into the mitochondrial intermembrane space based on previous observations (Steglich et al. 1999). It should be kept in mind that this structure is built based on computed predictions.
Combining the proposed function of the PHB complex as a holdase/unfoldase for the assembly of respiratory chain protein complexes (Nijtmans et al. 2002) with the elongated unit cell building block and the expected size of the PHB complex, we arrive at a tentative model of the super structure, as given in Figure 6 ▶. The molecular mass of the complex (1 MD), suggests an intact complex to be built from approximately 14 (range 12–16) of these unit cells. Although this will be a fixed number, the exact count does not affect the palisade shaped model. Large multimeric assemblies consisting of a number of identical structural units usually arrange in a circular fashion, which in this case lines the cavity to hold newly synthesized mitochondrial polypeptide chains.
Fig. 6.

A representation of the superstructure of the complex. The membrane associated elongated building blocks assemble in a ring shaped structure, probably acting as a holdase/unfoldase for newly synthesized mitochondrial proteins. (A) Dimeric building block (top view). (B) Proposed circular arrangement of the building blocks (top view). (C) A section of four building blocks and the mitochondrial inner membrane (side view).
Materials and methods
Strains and media
For overexpression of the PHB complex the Saccharomyces cerevisiae strain W303/1A(Δphb1Δphb2) transformed with the multicopy shuttle vector YEplac195 containing both PHB1 and PHB2 genes was used (see Nijtmans et al. 2000). Yeast was grown on WO minimal media (0.67% Yeast Nitrogen Base, 2% galactose) complemented with amino acids and lacking uracil for selection of the plasmid.
Preparation of mitochondrial fractions
Yeast cells were grown as described above. Highly purified mitochondria were obtained following the protocol described by Glick et al (Glick and Pon 1995), with slight modifications.
Blue native electrophoresis and electro-elution
Blue native gel electrophoresis was performed according to Schägger and von Jagow (1991). Electroelution was performed basically as described by Schägger (1995). In some instances the electroelution buffer was supplemented with 0.01% lauryl maltoside and 2.5% glycerol.
Secondary structure prediction
For predicting trans-membrane helices we used the TMHMM 2.0 algorithm (Krogh et al. 2001) available at http://www.cbs.dtu.dk/services/TMHMM-2.0/. Secondary structure was predicted using the Jpred2 (Cuff and Barton 1999, 2000) algorithm available at http://jura.ebi.ac.uk:8888/, and 3D-PSSM V2.6.0 (Kelley et al. 2000) available at http://www.bmm.icnet.uk/~3dpssm/. All programs were used at default settings.
Chemical crosslinking
Dithiobis (succinimidylpropionate) (DTSP) was purchased from Sigma. SulfoBID, a water-soluble analog of BID (Back et al. 2001), was synthesized as previously described for BID, with the replacement of N-hydroxysuccinimide by sulfo-N-hydroxysuccinimide (Pierce). The sulfonate groups introduced by this procedure render the crosslinker water soluble.
Crosslinking of the electroeluted complex was done at a protein concentration of 0.5 mg/mL in 20 mM sodium phosphate pH 7.8, 1.25% glycerol. Crosslinkers were added to a final concentration of 2 mM. The solution was kept at room temperature for 25 min.
Crosslinking in Blue native gel pieces was performed as follows. Pieces of gel were cut out and washed five times for 1 h with crosslinking buffer (50 mM tri-ethanolamine pH 8.0) at 4°C. Then an aliquot with a volume equal to the gel pieces of 4 mM DTSP in crosslinking buffer was added and incubated at room temperature for 30 min.
Tryptic digestion and sample preparation for mass spectrometry
Digestion of proteins and crosslinked products contained in polyacrylamide gel pieces was performed according to (Shevchenko et al. 1996) with sequencing grade trypsin (Roche). Peptides were collected in 20 mM NH4HCO3 and desalted and concentrated on ZipTip C18 (Millipore), and eluted in 10 μL 60% acetonitrile/1% HCOOH.
Mass spectrometry
For MALDI analyses 0.5 μL of peptides were mixed with 0.5 μL α-cyano hydroxycinnaminic acid (10 mg/mL in ethanol:acetonitrile 1:1), spotted on target plates and allowed to dry.
Reflectron MALDI-TOF mass spectra were recorded on a TofSpec 2EC mass spectrometer (Micromass) equipped with a 2 GHz digitizer. Electrospray MS and low-energy collision-induced dissociation (MSMS) analyses were performed on a Q-Tof (Micromass) mass spectrometer with a Z-Spray orthogonal ESI source. Fragmentation of peptides was performed using argon as a collision gas at a quadrupole pressure gauge reading of 4 * 10−5 mbar.
For direct infusions, peptides were introduced using gold-coated nano electrospray capillaries (New Objective).
After mass calibration (better than 40 ppm), the MALDI and electrospray MS spectra were charge deconvoluted using the MaxENT 3 algorithm (MaxENT Solutions); what resulted is the mass list of principle isotope ions with single charge.
Analysis of mass spectrometric data
For each experiment, the above-obtained MS data were scanned for digest fragments that were modified with the used chemical cross linker. A custom-made software tool called FindLink supported these analyses. The FindLink program generates a mass/fragment database, based on the input of the residue sequence of the proteins in the complex, the selectivity of the digest cleavages, and the amino acid residue chemical selectivity of the chemical crosslink reagent molecule. Database entries include all fragment candidates for surface label modifications, for intramolecular crosslinking within a fragment, and all fragment combinations for crosslinking within and between proteins in the complex. Each database entry is automatically matched within a definable mass tolerance with the experimentally obtained mass lists. The matches for surface labeling, intramolecular crosslinking within a single digest fragment, and intermolecular crosslinking between digest fragments within and between proteins in the complex are systematically documented as output of the analyses.
Acknowledgments
Louis Hartog is acknowledged for synthesis of sBID, and Esther Willems for expert technical assistance. The Q-TOF and MALDI-TOF mass spectrometers were largely funded by a grant from the Council for Medical Sciences of The Netherlands Organization for Scientific Research (NWO).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
PHB, prohibitin
DTSP, dithiobis(succinimidylpropionate)
sBID,sulfo-N-benzyliminodiacetoylhydroxysuccinimid
BNE, blue native gel electrophoresis
MALDI, matrix-assisted laser desorption ionization
ESI, electrospray ionization
TOF, time of flight
MS, mass spectrometry
MSMS, low energy collision experiments
CID, collision-induced dissociation
TMHMM, trans-membrane hidden Markov model
3D-PSSM, three-dimensional position-specific scoring matrix
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0212602.
References
- Back, J.W., Hartog, A.F., Dekker, H.L., Muijsers, A.O., de Koning. L.J., and de Jong, L. 2001. A new cross-linker for mass spectrometric analysis of the quaternary structure of protein complexes. J. Am. Soc. Mass Spectrom. 12 222–227. [DOI] [PubMed] [Google Scholar]
- Bennett, K.L., Kussmann, M., Bjork, P., Godzwon, M., Mikkelsen, M., Sorensen, P., and Roepstorff, P. 2000a. Chemical cross-linking with thiol-cleavable reagents combined with differential mass spectrometric peptide mapping—A novel approach to assess intermolecular protein contacts. Protein Sci. 9 1503–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett, K.L., Matthiesen, T., and Roepstorff, P. 2000b. Probing protein surface topology by chemical surface labeling, cross-linking, and mass spectrometry. Methods Mol. Biol. 146 113–131. [DOI] [PubMed] [Google Scholar]
- Berger, K.H. and Yaffe, M.P. 1998. Prohibitin family members interact genetically with mitochondrial inheritance components in Saccharomyces cerevisiae. Mol. Cell. Biol. 18 4043–4052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coates, P.J., Jamieson, D.J., Smart, K., Prescott, A.R., and Hall, P.A. 1997. The prohibitin family of mitochondrial proteins regulate replicative lifespan. Curr. Biol. 7 607–610. [DOI] [PubMed] [Google Scholar]
- Cuff, J.A. and Barton, G.J. 1999. Evaluation and improvement of multiple sequence methods for protein secondary structure prediction. Proteins Struct. Funct. Genet. 34 508–519. [DOI] [PubMed] [Google Scholar]
- ———. 2000. Application of multiple sequence alignment profiles to improve protein secondary structure prediction. Proteins Struct. Funct. Genet. 40 502–511. [DOI] [PubMed] [Google Scholar]
- Glick, B.S. and Pon, L.A. 1995. Isolation of highly purified mitochondria from Saccharomyces cerevisiae. Methods Enzymol. 260 213–223. [DOI] [PubMed] [Google Scholar]
- Green, N.S., Reisler, E., and Houk, K.N. 2001. Quantitative evaluation of the lengths of homobifunctional protein cross-linking reagents used as molecular rulers. Protein Sci. 10 1293–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonen, E., Fiedler, K., Parton, R.G., and Simons, K. 1995. Prohibitin, an antiproliferative protein, is localized to mitochondria. FEBS Lett. 358 273–277. [DOI] [PubMed] [Google Scholar]
- Kelley, L.A., MacCallum, R.M., and Sternberg, M.J.E. 2000. Enhanced genome annotation using structural profiles in the program 3D-PSSM. J. Mol. Biol. 299 499–520. [DOI] [PubMed] [Google Scholar]
- Krogh, A., Larsson, B., von Heijne, G., and Sonnhammer, E.L. 2001. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 305 567–580. [DOI] [PubMed] [Google Scholar]
- Nijtmans, L.G., de Jong, L., Artal Sanz, M., Coates, P.J., Berden, J.A., Back, J.W., Muijsers, A.O., van der Spek, H., and Grivell, L.A. 2000. Prohibitins act as a membrane-bound chaperone for the stabilization of mitochondrial proteins. EMBO J. 19 2444–2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijtmans, L.G.J., Artal Sanz, M., Grivell, L.A., and Coates, P.J. 2002. The mitochondrial PHB-complex: Roles in mitochondrial respiratory complex assembly, ageing and degenerative disease. Cell. Mol. Life Sci. 59 143–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schägger, H. 1995. Native electrophoresis for isolation of mitochondrial oxidative phosphorylation protein complexes. Methods Enzymol. 260 190–202. [DOI] [PubMed] [Google Scholar]
- Schägger, H. and Von Jagow, G. 1991. Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal. Biochem. 199 223–231. [DOI] [PubMed] [Google Scholar]
- Shevchenko, A., Wilm, M., Vorm, O., and Mann, M. 1996. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 68 850–858. [DOI] [PubMed] [Google Scholar]
- Snedden, W.A. and Fromm, H. 1997. Characterization of the plant homologue of prohibitin, a gene associated with antiproliferative activity in mammalian cells. Plant Mol. Biol. 33 753–756. [DOI] [PubMed] [Google Scholar]
- Steglich, G., Neupert, W., and Langer, T. 1999. Prohibitins regulate membrane protein degradation by the m-AAA protease in mitochondria. Mol. Cell. Biol. 19 3435–3442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young, M.M., Tang, N., Hempel, J.C., Oshiro, C.M., Taylor, E.W., Kuntz, I.D., Gibson, B.W., and Dollinger, G. 2000. High-throughput protein fold identification by using experimental constraints derived from intramolecular cross-links and mass spectrometry. Proc. Natl. Acad. Sci. 97 5802–5806. [DOI] [PMC free article] [PubMed] [Google Scholar]