

Abstract
Tumor necrosis factor-α (TNF-α) converting enzyme (TACE/ADAM-17) is responsible for the release of TNF-α, a potent proinflammatory cytokine associated with many chronic debilitating diseases such as rheumatoid arthritis. Among the four variants of mammalian tissue inhibitor of metalloproteinases (TIMP-1 to -4), TACE is specifically inhibited by TIMP-3. We set out to delineate the basis for this specificity by examining the solvent accessibility of every epitope on the surface of a model of the truncated N-terminal domain form of TIMP-3 (N-TIMP-3) in a hypothetical complex with the crystal structure of TACE. The epitopes suspected of interacting with TACE were systematically transplanted onto N-TIMP-1. We succeeded in transforming N-TIMP-1 into an active inhibitor for TACE (Kiapp 15 nM) with the incorporation of Ser4, Leu67, Arg84, and the TIMP-3 AB-loop. The combined effects of these epitopes are additive. Unexpectedly, introduction of "super-N-TIMP-3" epitopes, defined in our previous work, only impaired the affinity of N-TIMP-1 for TACE. Our mutagenesis results indicate that TIMP-3-TACE interaction is a delicate process that requires highly refined surface topography and flexibility from both parties. Most importantly, our findings confirm that the individual characteristics of TIMP could be transplanted from one variant to another.
Keywords: N-TIMP-3, N-TIMP-1 as scaffold, functional epitopes, binding affinity, TACE interactions
Tumor necrosis factor-α converting enzyme (TACE/ADAM-17) is a member of the membrane-anchored, zinc-dependent ADAM (a disintegrin and metalloproteinase) proteinase family (Black et al. 1997). The enzyme is responsible for the cleavage and release of the potent proinflammatory cytokine, tumor necrosis factor-α (TNF-α) in many inflammatory diseases such as rheumatoid arthritis and Crohn's disease (Louis 2001; Ohta et al. 2001). Like other members of the ADAM proteinase family, TACE is composed of several domains. Besides the enzymatically active catalytic domain, TACE also contains a prodomain at its N-terminus and several distinctive domains at the C-terminus. These C-terminal domains are commonly referred to as the disintegrin, epidermal growth factor (EGF)-like, Crambin-like, transmembrane, and cytoplasmic domains.
The catalytic domain truncate of TACE was crystallized by Maskos et al. in 1998, and the structure resolved at 2.0 Å revealed a configuration that resembled that of the snake venom/adamalysin metalloproteinases (MMPs; Maskos et al. 1998). The enzyme is composed of a main "upper" molecular body and a small "lower" subdomain in the shape of an oblate ellipsoid. The catalytic zinc resides at the center of the catalytic cleft, coordinated by three highly conserved histidine (His-405, -409, -415) and a glutamate (Glu-406) residue in a typical zinc-binding motif (HEXXHXXGXXH). The S1` and S3` pockets are situated immediately to the right of the catalytic zinc, divided unequally in size by a "stem" created by hydrophobic contact between the Leu-348 and Ala-439 side chains. The S1` specificity pocket is slightly smaller than its S3` neighbor, and both pockets are essentially hydrophobic in nature.
To date, there are four known variants of mammalian tissue inhibitor of metalloproteinases (TIMP-1 to -4), but only TIMP-3 is capable of inhibiting TACE (Amour et al. 1998). Without exception, all of the TIMP molecules are ∼180 residues in length and consist of six disulfide bonds. Structural studies with TIMP-1 and -2 confirmed that the N-terminal two-thirds of the molecule are folded into a compact five-stranded β-barrel reminiscent of the oligosaccharide/oligonucleotide-binding moiety (Williamson et al. 1994; Gomis-Rüth et al. 1997; Musket et al. 1998; Tuuttila et al. 1998). Given the striking similarity between the tertiary structures of TIMP-1 and -2, it is generally believed that TIMP-3 should demonstrate a more or less identical configuration. However, no structural investigation has been carried out on TIMP-3 to date, because the protein is notoriously difficult to purify. Efforts to extract TIMP-3 from mammalian cells have always been hindered by the preponderance of the protein to associate itself with the extracellular matrix (Yu et al. 2000). Moreover, TIMP-3 is also known to cause apoptosis in the host cells overexpressing the protein (Ahonen et al. 1998; Bond et al. 2000, 2002). Expression of full-length TIMP-3 in a prokaryotic system, on the other hand, only resulted in inclusion bodies that are recalcitrant to refolding (M. Lee and G. Murphy, unpubl.).
Our laboratory is interested in developing TIMP-3 to be a potential therapeutic agent against TACE-related diseases. Hence, we are keen to delineate the structural and functional relationships between TIMP-3 and TACE. We previously demonstrated that the refolded N-terminal domain form of TIMP-3 (N-TIMP-3) retained full in vitro inhibitory activities against TACE (Lee et al. 2001, 2002a, 2002b). In the present study, we set out to investigate the basis of the specificity of TACE for TIMP-3 by devising a novel approach that entailed dissecting a model of N-TIMP-3 into modules of independent epitopes. The epitopes suspected of interacting with TACE were systematically transplanted onto an N-TIMP-1 molecule. The contribution of each epitope was quantitatively monitored throughout the kinetic analysis of mutants. Our findings reveal that the functional epitopes of N-TIMP-3 that are responsible for TACE inhibition can be transplanted onto N-TIMP-1. Most interestingly, our results clearly establish the strategic importance of the TIMP-3 AB-loop in defining the potency of the inhibitor.
Results
Refolding patterns of N-TIMP-1 and N-TIMP-3: The rationale of using N-TIMP-1 as the scaffold
Despite attempts to improve the refolding efficiency, the yield of N-TIMP-3 is still significantly lower than those of N-TIMP-1 and N-TIMP-2 (Kashiwagi et al. 2001; Lee et al. 2001). In our laboratory, the final yield of N-TIMP-3 is only 10%–15% of those for N-TIMP-1 and -2 (results not shown). We started this project by evaluating the effects of guanidinium chloride (GnHCl) concentrations on NTIMP-1 and N-TIMP-3 refolding dynamics. Ideally, the molecule that serves as the scaffold for our mutagenesis project should display close similarity to N-TIMP-3 in tertiary structure, be relatively easy to refold with minimal intermediates, and be capable of withstanding the incorporation of N-TIMP-3 epitopes without compromising its own.
Figure 1a and b ▶ illustrate the refolding patterns of N-TIMP-1 and N-TIMP-3 over the denaturant (GnHCl) concentrations. It is clear from these figures that the refolding pathway of N-TIMP-3 is different from that of N-TIMP-1, and N-TIMP-3 refolding might be dominated by energetically unstable intermediates. In our opinion, the presence of such intermediates renders the efficiency of N-TIMP-3 refolding unfavorable. N-TIMP-1, on the other hand, seems to follow a different, possibly simpler refolding pathway that might be devoid of such hindrance.
Fig. 1.
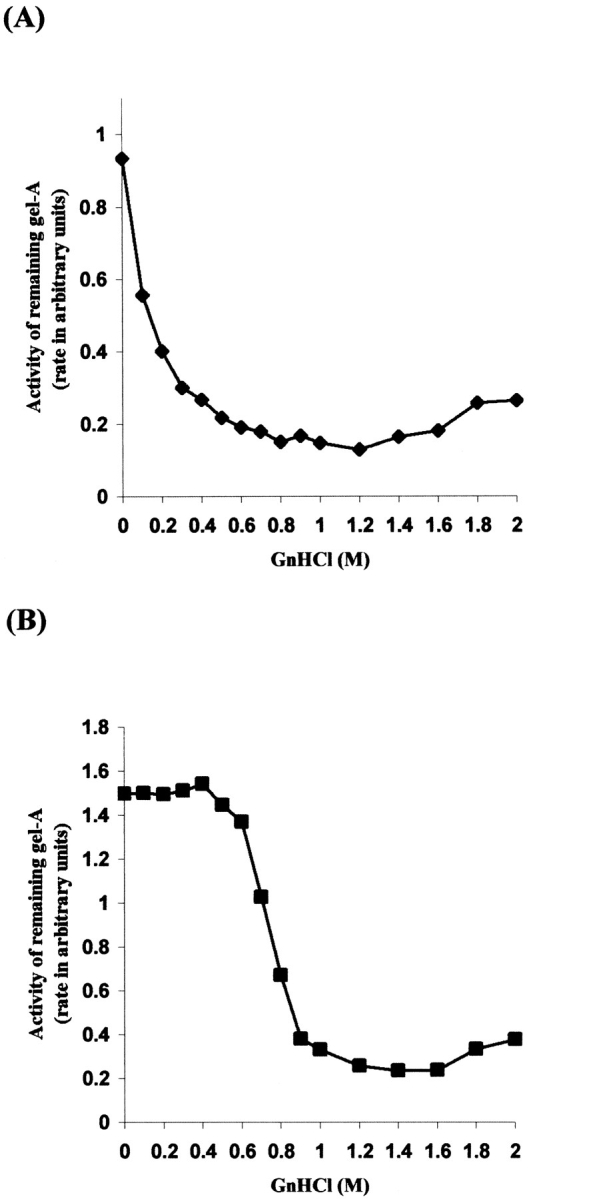
Refolding profiles of N-TIMP-1 and N-TIMP-3 in various GnHCl concentrations. N-TIMP-1 and N-TIMP-3 were refolded under a wide range of GnHCl concentrations. The amount of active inhibitor present was determined by incubating an equal volume of the dialysed refolding solution with gel-A. (A) N-TIMP-1: Typical profile of a protein that follows the "two-state transition" refolding pathway (B): N-TIMP-3: Intermediates dominate the refolding pathway up to 0.5 M GnHCl. The y-axis represents the activity of remaining gel-A after 3-h incubation with the dialysed inhibitors. Note that (B) was published in our previous paper (Lee et al. 2002b).
N-TIMP-3 model construction
The structures of TIMP-1 and TIMP-2 have been resolved either by crystallization or protein NMR (Williamson et al. 1994; Muskett et al. 1998; Tuuttila et al. 1998). We constructed our N-TIMP-3 model using the templates of TIMP-1 and TIMP-2 available from the Protein Data Bank (PDB). In the present study, we decided against speculating on the functional epitopes of N-TIMP-3 by sequence alignment with TIMP-1 and -2, because the primary sequence of TIMP-3 differs from those of TIMP-1 and -2 by as much as 60%. Moreover, structural elucidation of TIMP-1 and -2 reveals that many of the side chains are firmly buried in the core of the molecule and therefore unlikely to take part in TACE interactions. Instead, we chose to subject our N-TIMP-3 model to hypothetical docking with the crystal structure of TACE (PDB identity # 1BKC). From the solvent accessibility of the residues, we deduced the amino acids that are most likely to be responsible for interacting with TACE. These residues were "transplanted" onto N-TIMP-1 either by single-point site-directed mutagenesis or by replacing sections of N-TIMP-1 with the corresponding residues from N-TIMP-3 (Fig. 2 ▶).
Fig. 2.

N-TIMP-3 model (green ribbon) in association with the crystal structure of TACE-cat: Illustration of the positions of the functional epitopes. (a) Frontal view of N-TIMP-3-TACE complex. (b) Aerial view of N-TIMP-3-TACE complex. N-TIMP-3 functional epitopes involved in TACE interaction are shown as stick: Ser4 (red), the AB-loop (-Gly32-Pro-Phe-Gly-Thr36-) (orange), Leu67 (yellow), and Arg84 (navy blue). In the AB-loop, only the side chains of Pro33, Phe34, and Thr36 are shown and labeled. The catalytic zinc (Zn) at the center of the active-site cleft is colored in magenta. For ease of identification, all TACE-cat labels are in italic, and N-TIMP-3 labels are in normal text.
Identification of N-TIMP-3 functional epitopes and the mutagenesis strategy of N-TIMP-1
Group 1: N-terminal epitopes (Val4Ser, Val4Met, Thr10Asp mutants)
At the very N-terminus of N-TIMP-3, the most obvious epitopes that should interact with TACE are no doubt the Thr2 and Ser4 residues (Figs. 2,3 ▶ ▶). It is generally accepted that these two amino acids interact with TACE by docking into the S1` and S3` active-site pockets of the enzyme. Because N-TIMP-1 also has threonine at the second position, we decided to replace only the Val4 residue with serine (hence Val4Ser; Fig. 3 ▶). Concurrently, we also created a Val4Met mutant because our previous mutagenesis results showed that the affinity of N-TIMP-3 against TACE could be significantly enhanced by replacing Ser4 with a bulky, hydrophobic residue such as methionine (Lee et al. 2002b). Asp10 is prominently located on the first α-helix of N-TIMP-3. The side chain of the residue points away from the molecule, possibly to the surface of TACE in a would-be enzyme-inhibitor complex. In our opinion, this negatively charged side chain could be an important residue that determines the acceptability of an inhibitor. The corresponding residue in N-TIMP-1 is Thr10, and this amino acid was therefore replaced by an aspartate residue (Thr10Asp).
Fig. 3.

Amino acid sequence alignment of human N-TIMP-1 and N-TIMP-3. The consensus residues that form the scaffold of a TIMP molecule are in bold. N-TIMP-1 residues chosen for mutagenesis in this study are boxed for ease of identification.
Group 2: AB-loop epitopes (AB-loop transplant, Tyr35Glu, Tyr35Thr mutants)
The AB-loop is another section that stood out in our model. In our N-TIMP-3-TACE complex, the loop folds over the rim of the "surface-bulge" created by residues Gln320 to Phe323 on the long "intermediate" α-helix (hB) at the "upper left"-hand side of the TACE catalytic zinc (Fig. 2 ▶; Maskos et al. 1998). The most likely mode of interaction at this site, in our opinion, is hydrophobic bonding. To establish efficient contact with this "surface-bulge", however, the entire AB-loop is required to orient itself toward the direction of the enzyme. Further, we suspected that the N-TIMP-1 AB-loop needs to be "lengthened" by one residue in order to reach its target effectively. Hence, we truncated the AB-loop of N-TIMP-1 (-Thr32-Thr-Leu-Tyr35-) and replaced it with that of N-TIMP-3 (-Gly32-Pro-Phe-Gly-Thr36-; Fig. 4 ▶). We also created two N-TIMP-1 point mutants (i.e., Tyr35Glu and Tyr35Thr) to test whether replacing Tyr35 with a negatively charged (Glu) or smaller, hydrophilic (Thr) residue could result in better interaction with TACE.
Fig. 4.

The "AB-loop" and "middle CD-loop" transplant mutants. The transplanted amino acids are highlighted in bold. The residues are numbered according to the original sequences of N-TIMP-1 and N-TIMP-3, respectively.
Group 3: CD-loop epitopes (Met66Ser, Glu67Ser, Val69Leu, Val69Met, and "middle CD-loop" transplant mutants)
Another area of N-TIMP-3 that might come into close contact with TACE, in our hypothetical cocrystal structure, is the CD-loop. In our model, one or several of the residues located at the beginning part of the loop, that is, from Ser64 to Leu67 (just before the Cys1-Cys68 disulfide bridge) should dock into the S2 pocket at the "nonprimed" side of the catalytic zinc. Hence, we mutated the corresponding Met66 and Glu67 residues in N-TIMP-1 to serine (Met66Ser and Glu67Ser mutants). At the same time, we substituted Val69 with leucine (Val69Leu) because we believed a longer side chain at this position should, in theory, allow better hydrophobic bonding with the S2 pocket. Val69Met, on the other hand, was created because our previous mutagenesis studies with N-TIMP-3 indicated that increasing the hydrophobicity of the Leu67 residue led to better inhibition against TACE (Lee et al. 2002b).
The four amino acids after the Cys1-Cys68 disulfide bridge, namely Lys71, Leu72, Glu73, and Val74, cooperatively constituted the "middle" part of the N-TIMP-3 CD-loop (Figs. 2,3 ▶ ▶). The side chains of these residues seem to be pointing outward, most likely towards the lower domain of TACE. Although it is difficult to predict the exact residues of TACE that will come into contact with these residues, we anticipate that this region of the N-TIMP-3 molecule influences the binding efficiency of the inhibitor. Thus, we also decided to replace the equivalent section of N-TIMP-1 with the corresponding residues from N-TIMP-3 (hence the "middle CD-loop" transplant mutant; Fig. 4 ▶).
Group 4: Lys88Met, Lys88Arg mutants
There are many interesting features worth noting in the TACE structure, one of the remarkable characteristics being the eminent positions of some "knobs" protruding out from the area bordering the active-site cleft of the enzyme (Maskos et al. 1998). Especially noteworthy are the Met345 and Tyr390 side chains at the upper edge of the stem that sits in between the S1` and S3` pockets. In our N-TIMP-3 model, residue Arg84 is located on the "upper" surface of the molecule (Fig. 2 ▶). This residue is envisaged to form the interface of a TACE-N-TIMP-3 binary complex, in all probability right above the Met345 and Tyr390 side chains. Although the area above the Met345/Tyr390 knobs is generally nonpolar in appearance, there is a small "patch" constituted by Thr387-Asn389 that appeared to be negatively charged (Fig. 2 ▶). We believed the side chain of Arg84 could establish contact with this "patch" via electrostatic bonding. Accordingly, we mutated the equivalent residue on N-TIMP-1 (i.e., Lys88) to methionine (Lys88Met) and arginine (Lys88Arg) to evaluate our hypothesis. If our prediction is correct, both mutations should bring about better inhibitory activity against TACE.
First-generation mutants: Screening and identification of "good" and "bad" epitopes
Wild-type N-TIMP-3 derived mutants (category II in Table 1)
Table 1.
The inhibitory constants (Kiapp) of N-TIMP-1 mutants against TACE-cat and TACE-long
| Kiapp (nM) | |||
| 1st generation mutants | TACE-cat | TACE-long | |
| I. | Wt N-TIMP-1 and N-TIMP-3 | ||
| Wt N-TIMP-1 | 356 ± 87 | N/A | |
| Wt N-TIMP-3 | 0.22 ± 0.07 | 1.75 ± 0.20 | |
| II. | Wt N-TIMP-3 derived mutants | ||
| 1. | V4S | 216 ± 44 | N/A |
| 2. | V4S + T10D | 452 ± 41 | N/A |
| 3. | V4S + T10D + V69L | 198 ± 25 | N/A |
| 4. | V4S + T10D + Y35T + V69L | 178 ± 32 | 503 ± 122 |
| 5. | V4S + Y35E + V69L | 102 ± 14 | 183 ± 39 |
| 6. | Y35T + V69L | 212 ± 30 | N/A |
| III. | Super N-TIMP-3 derived mutants | ||
| 1. | V4M + T10D | 387 ± 71 | N/A |
| 2. | V4M + T10D + V69L | 158 ± 23 | 473 ± 96 |
| 3. | V4M + T10D + Y35T + V69L | 232 ± 31 | N/A |
| 4. | V4M + T10D + V69M | 926 ± 208 | N/A |
| IV. | Loop mutants | ||
| A. Mutants on AB-loop | |||
| 1. | Y35E | 445 ± 33 | N/A |
| 2. | Y35T | 740 ± 86 | N/A |
| 3. | AB-loop transplant | 67 ± 17 | 452 ± 86 |
| B. Mutants on CD-loop | |||
| 1. | M66S | 838 ± 375 | N/A |
| 2. | E67S | 871 ± 226 | N/A |
| 3. | V69L | 183 ± 33 | N/A |
| 4. | V69M | 931 ± 210 | N/A |
| 5. | Middle CD-loop transplant | 752 ± 167 | N/A |
| C. Mutants on β-strand D | |||
| 1. | K88M | 183 ± 29 | N/A |
| 2. | K88R | 235 ± 195 | N/A |
| 2nd generation mutants Combination of "good" and "neutral" epitopes | |||
| 1. | V4S + Y35E + V69L + K88M | 60 ± 14 | 150 ± 21 |
| 3rd generation mutants Combination of "only good epitope" mutants | |||
| 1. | V4S + AB-Loop + V69L | 15 ± 3 | 50 ± 5 |
| 2. | V4S + AB-Loop + V69L + K88M | 23 ± 5 | 34 ± 4 |
The majority of the low affinity mutants are not capable of inhibiting TACE-long. TACE-cat, on the contrary, is much more easily inhibited. A similar trend was observed in N-TIMP-3 mutants, as described in our previous paper (Lee et al. 2002a). Wt, wild-type; N/A, not able to determine the Kiapp values.
The first-generation mutants are composed of single and multiple mutants that incorporated the N-TIMP-3 epitopes that, in our view, are directly involved in TACE interactions. The binding affinities of the resultant mutants are listed in Table 1. It is rather obvious that Val4Ser and Val69Leu mutations singularly as well as cooperatively enhanced the binding affinities with TACE-cat (i.e., the truncated catalytic domain form of TACE). Introduction of these epitopes into N-TIMP-1 consistently improved the Kiapp values. Their effects are remarkably apparent when we compared the Kiapp of wild-type N-TIMP-1 (Kiapp 356 nM) with these four mutants: the V4S single mutant (Kiapp 216 nM), the V4S+T10D double mutant (Kiapp 452 nM), the V4S+T10D+V69L triple mutant (Kiapp 198 nM), and the V4S+Y35E+V69L triple mutant (Kiapp 102 nM). In short, all of the N-TIMP-1 mutants that bear the Val4Ser and Val69Leu mutations exhibited improved affinities against TACE. Thr10Asp, conversely, seemed to have a negligible or slightly negative effect on TACE inhibition (Ki app : V4S, 216 nM; V4S+T10D, 452 nM).
"Super-N-TIMP-3"-derived mutants (category III)
Without exception, all N-TIMP-1 mutants harboring epitopes from our "super-N-TIMP-3" (i.e., Val4Met and Val69Met) were poor inhibitors. In our previous study, the binding tightness of N-TIMP-3 could be significantly enhanced by replacing the residues at these loci with amino acids of a bulkier and more hydrophobic nature (Lee et al. 2002b). Applying the same tactics to N-TIMP-1 obviously only impaired the already poor affinity. Indeed, the value of the V4M+T10D+V69M triple mutant (Kiapp 926 nM) was so high that it was barely measurable with our fluorimetric technique.
Tyr35 and AB-loop transplant mutants (category IVA)
Only the AB-loop "transplant" mutant exhibited clear and substantial improvement in TACE inhibition. The Kiapp of 67 nM was a definite improvement in comparison with the wild-type N-TIMP-1 (Kiapp 356 nM). In contrast, the binding affinity of both Tyr35Glu and Tyr35Thr mutants decreased substantially (Kiapp 450–740 nM) compared to the wild-type inhibitor.
CD-loop mutants (category IVB)
Among the four single mutants created at this site (Met66Ser, Glu67Ser, Val69Leu, and Val69Met), only Val69Leu showed significant improvement in Kiapp, as explained earlier (Table 1, category II). The others, including the "middle CD-loop" transplant mutant, were distinctively poorer, with Kiapp values ranging from 750 nM to nearly 1 μM.
Lys88 mutants (category IV,C)
An arginine (Lys88Arg) or methionine (Lys88Met) in the position of Lys88 provided similar Kiapp values with TACE-cat (Kiapp : Lys88Met and Lys88Arg 180–240 nM ), and both mutants are slightly better than the wild-type N-TIMP-1 (Kiapp : 356 nM) in TACE inhibition.
Second-generation mutants: Combination of suspected "good" and "neutral" epitopes
The second-generation mutant, V4S+Y35E+V69L+K88M, was created for the sole purpose of ascertaining the authenticity of the suspected "good" epitopes, namely the Val4Ser, Val69Leu, and Lys88Met mutations. We combined these three "good" epitopes and a "neutral" one (i.e., Tyr35Glu) into a single mutant to confirm the reproducibility of their effects on binding affinity. Undeniably, the Kiapp value of the resultant mutant (60 nM) was much better than that of any other single mutant from the previous generation (Table 1).
Third-generation mutants: Combination of only "good" epitopes
Following our success with the second-generation mutants, we decided to assimilate all known "good" epitopes into two final constructs, each bearing only the "good" epitopes. Two multiple mutants were made, that is, V4S+AB-loop+V69L and V4S+AB-loop+V69L+K88M. Without doubt, the affinities of these mutants were superior to any mutants described so far (Kiapp 15 to 23 nM). The contribution of Lys88Met epitope seems to be slightly inconsistent with TACE-cat and TACE-long (i.e., the longer form of TACE that includes the catalytic domain and the cysteine-rich domains at its C-terminus). In the case of TACE-cat, incorporation of this residue decreased the binding efficiency slightly (Kiapp : V4S+AB-loop+V69L, 15 nM; V4S+AB-loop+V69L+K88M, 23 nM). TACE-long, in contrast, exhibited improved affinity with the inclusion of this epitope (Kiapp : V4S+AB-loop+V69L, 50 nM; V4S+AB-loop+V69L+K88M, 34 nM; Table 1). Figure 5 ▶ illustrates the contrast in Kiapp profiles between the wild-type N-TIMP-1 and the V4S+AB-loop+V69L mutant with regard to TACE-cat inhibition.
Fig. 5.

Comparison of the inhibition (Kiapp) profiles of wild-type N-TIMP-1 (Kiapp 356 nM) and V4S + AB-loop + V69L triple mutant (Kiapp 15 nM) against TACE-cat. The triple mutant that incorporated the V4S, AB-loop, and V69L epitopes is much more potent than the wild-type inhibitor. The final concentrations of TACE-cat in the reactions are 0.20 nM.
The inhibitory profiles of TACE-cat and TACE-long with N-TIMP-1 mutants
We demonstrated in our earlier study that TACE-long is much more recalcitrant towards N-TIMP-3 inhibition than TACE-cat (Lee et al. 2002a). One of the reasons for the discrepancy, in our opinion, could be the steric hindrance exerted by the cysteine-rich domains of TACE-long toward an incoming TIMP molecule. Here, TACE-long also displayed a similar pattern of resilience toward the N-TIMP-1 mutants. In general, the affinities of our N-TIMP-1 mutants with TACE-long are significantly lower than those with TACE-cat. The Kiapp of TACE-long with most of our N-TIMP-1 mutants exceeded 1 μM, and thus were beyond the observable limit of our fluorimetric assays (Table 1).
Association profiles (kon) of third-generation mutants against TACE-cat and TACE-long
Figure 6a ▶ depicts the association profiles (kon) of V4S+AB-loop+V69L mutant (Kiapp 15 nM) against TACE-cat at different concentrations. We also subjected TACE-long to a similar analysis with its best inhibitor, namely the V4S+AB-loop+V69L+K88M quadruple mutant. The association profile is shown in Figure 6b ▶. Also included are the inhibition profiles of CT1399, a hydroxamate inhibitor that has similar binding affinity with TACE-cat (Kiapp 15 nM) as the N-TIMP-1 mutant (Fig. 6c ▶), as well as one of our previous N-TIMP-3 mutants, Thr2Tyr (Kiapp 1.3 nM) (Fig. 6d ▶; Lee et al. 2002b). Without doubt, the V4S+AB-loop+V69L mutant is capable of inhibiting TACE-cat, but in a manner not totally similar to that of a slow, tight-binding inhibitor. Instead, the association seems to be simple and rapid, with equilibrium achieved in a relatively short period.
Fig. 6.

Comparison of the association rate (kon) profiles of (a) N-TIMP-1 triple mutant, V4S + AB-loop + V69L against TACE-cat (Kiapp 15 nM), (b) N-TIMP-1 quadruple mutant, V4S + AB-loop + V69L + K88M against TACE-long (Kiapp 34 nM), (c) the hydroxamate inhibitor CT1399 (Kiapp 15 nM) against TACE-cat, and (d) one of our previous N-TIMP-3 mutants, Thr2Tyr against TACE-cat (Kiapp 1.3 nM). The time courses of TACE activities were monitored for up to 8000 sec. The final concentrations of inhibitors are shown beside each curve. The arrows indicate the instants where the inhibitors are added to the reaction mixtures. Note that the Thr2Tyr mutant was described in our previous publication and on the present occasion, steady-state might not have been achieved between the enzyme and inhibitor (Lee et al. 2002b).
Discussion
Although members of the TIMP family share at least 30% sequence homology, only TIMP-3 is capable of inhibiting TACE. Kinetic analysis carried out in our laboratory shows that all of the critical elements needed for TACE inhibition reside in the N-terminal domain of the TIMP-3 molecule (Lee et al. 2001, 2002b). This unique quality of TIMP-3 presents the biochemist with an exceptional opportunity to delineate the structure activity relationship between TIMP and metalloproteinases. Our previous experience with N-TIMP-3 engineering showed that the protein is highly intolerant of mutagenesis. In fact, many of our N-TIMP-3 mutants were resistant to in vitro refolding even after extensive modifications in refolding conditions (Lee et al. 2002b). Hence, we decided to seek an alternative strategy in order to bypass this difficulty. We are interested in developing TIMPs to be agents able to control MMP and ADAM activities in a focused manner. As a result, we decided to transplant N-TIMP-3 epitopes onto N-TIMP-1 as a means to evaluate the viability of our new strategy in TIMP engineering. N-TIMP-1 was chosen as the scaffold because the molecule has less unfavorable intermediates in its refolding pathways, and is therefore most likely to withstand the incorporation of alien residues without comprising its conformational integrity.
One of the most significant findings of this work is the revelation of the dominant role of the AB-loop. Our findings suggest that the AB-loop contributes appreciably more towards TACE inhibition than any other epitope. Due to the presence of two glycine residues at both ends of the AB-loop (-Gly32-Pro-Phe-Gly-Thr36-), we suspect the loop to be flexible and, most probably, without a sturdy conformation. In our opinion, it is precisely this unique quality of flexibility that allows the loop to tilt itself towards the "surface-bulge" of the enzyme. Further detailed mutagenesis and structural studies are undoubtedly necessary to confirm this theory. As predicted, insertion of the five residues from the N-TIMP-3 AB-loop into the space of four residues (N-TIMP-1 AB-loop: Thr32-Thr33-Leu34-Tyr35) does not destabilize the tertiary conformation of the N-TIMP-1 scaffold.
Val69Leu is the only mutant in the vicinity of the CD-loop that contributes positively towards TACE inhibition. In our TACE-N-TIMP-3 hypothetical complex, Leu67 is located right before the Cys1-Cys68 disulfide bridge. This residue should interact directly with the S2 pocket at the "left-hand" side of the catalytic zinc. Here, our kinetic results demonstrate that, despite its rather shallow appearance, the S2 pocket is one of the so-called "alternative pockets" that could be as important as the S1` and S3` pockets when it comes to TACE inhibition. We believe that these "alternative" pockets should be taken into consideration in the development of a synthetic anti-TACE chelating inhibitor in the future.
Close scrutiny of the TACE-cat structure shows that the topography of the areas surrounding the catalytic cleft is dominated by several distinctive "knobs" and "grooves". We believe that the "groove" situated in between the Met345 and Tyr390 "knobs" is another one of these "alternative pockets" that could be exploited for the design of a better inhibitor. Our hypothesis is strengthened by the successful prediction of the positive outcome of Lys88 to arginine/methionine mutations. Both mutants displayed enhanced affinity with TACE despite the contrast in the physical attributes of their side chains.
An intriguing finding from this study is the apparent failure of Val4Met and Val69Met mutants to improve TACE inhibition. Indeed, both Val4Met and Val69Met epitopes decreased the affinities of the resulting mutants considerably. We ruled out the likelihood that the incorporation of methionine into these loci has somehow led to misfolded proteins, as our titration assays with gelatinase-A showed no discernible variation in specific activities between these two mutants and the others (results not shown). On the contrary, we believed that the introduction of a large and hydrophobic residue such as methionine renders the local conformation too rigid and unwieldy for "fine-tuning". It has been shown by NMR and crystal structure analysis that TIMP-1 undergoes an "induced fit" process during association with MMP (Wu et al. 2000). The N-terminus and the CD-loop are two malleable parts of the TIMP molecule that are coerced to sustain moderate to dramatic "reorientation" during this process. We surmise that incorporation of methionine residue at these loci could hinder this process of reorientation and consequently lead to poor inhibitory potency.
The effects of combining the functional epitopes are additive. Close examination of wild-type N-TIMP-1 (Ki app 356 nM), the single mutant bearing the V4S (Ki app 216 nM), V69L (Ki app 183 nM), and AB-loop (Ki app 60 nM) epitopes as well as the third-generation mutant, V4S+AB-loop+V69L (Ki app 15 nM) revealed that the binding affinities of N-TIMP-1 were additively enhanced by the systematic introduction of the epitopes. We attempted to calculate the association rates (kon) of the V4S+AB-loop+V69L mutant with TACE-cat using equations developed for slow, tight-binding inhibitor (Morrison and Walsh 1995) but to no avail, as our data failed to fit the trajectory for such inhibitors (results not shown). Figure 6 ▶ shows that the association curves of the third-generation N-TIMP-1 mutants do not strictly echo the characteristics of a slow, tight-binding inhibitor, such as the N-TIMP-3 mutant Thr2Tyr. On the other hand, a Lineweaver-Burk plot with increasing inhibitor concentrations ruled out the prospect of a Michaelis-Menten-type classical competitive inhibitor. Instead, we envisage that the N-TIMP-1 mutant forms a rapid but loose binary complex with TACE. Due to the lack of malleability in certain part(s) of the N-TIMP-1 molecule, this loose binary complex fails to progress to the advanced "isomerization" stage whereby a tight complex is brought into being (Morrison and Walsh 1995).
In summary, we have characterized and defined a number of N-TIMP-3 functional epitopes that are critical in TACE inhibition. Transplantation of these epitopes onto N-TIMP-1 resulted in a moderate inhibitor with the Kiapp value of 15 nM. However, it should be kept in mind that the outcome of this kind of "epitope prediction approach" is highly subjective, even when the structure of the molecule is known. Owing to the limits of computer modeling, there are still many uncertainties in our N-TIMP-3 model that require clarification from a proper crystal structure. We believe that our mutagenesis results could be further improved with the advent of a TIMP-3-TACE cocrystal structure. We are in the process of developing a high-yield N-TIMP-3 mutant that could be used for cocrystallization with TACE.
Materials and methods
Refolding plots of N-TIMP-1 and N-TIMP-3
The effects of GnHCl on N-TIMP-3 refolding were described in our previous publication (Lee et al. 2002b). In the present study, ∼50 mg of N-TIMP-1 inclusion bodies was initially dissolved in 1.5 mL solubilization buffer (6 M GnHCl, 50 mM Tris-HCl, pH 7.5, 10 mM dithiothreitol) for 30 min at room temperature. Eighty μL of supernatant was aliquoted into 5 mL refolding buffer (Tris-HCl, pH 8.8; oxidized and reduced glutathione 0.5 mM : 0.2 mM) in Bijou vials containing 0 to 2 M of GnHCl. Refolding was allowed at room temperature for 8 h before the samples were dialysed overnight against 35 L of 50 mM Tris-HCl buffer, pH 7.8. After the removal of insoluble precipitates by centrifugation, 100 μL of each dialysed sample was incubated with 80 pM gel-A for 3 h. The amount of active N-TIMP-1 in the samples was determined by examining the activity of remaining gel-A, that is, the rate of QF-24 (Mca-Pro-Leu-Gly-Leu-Dpa-Ala-Arg-NH2) proteolysis.
N-TIMP-3 model construction
A model of N-TIMP-3 was created by the automated knowledge-based comparative protein modeling server Swiss-Model version 3.5 (Glaxo Smith-Kline). With a conformational comparison between an energetically minimized, free N-TIMP-3 model (based on the templates of free human TIMP-2, PDB identity # 1BR9 or 2TMP) and a hypothetically MMP-bound, restrained model (based on the templates of human TIMP-1 in complex with stromelysin, PDB identity # 1UEA), we showed that the discrepancies reside exclusively at the areas surrounding the AB-loop and the CD-loop, as well as the very N-terminus of the protein. In our view, the parts of N-TIMP-3 that will undergo the most significant conformational changes upon TACE binding are the AB- and CD-loops. Thus, we decided to create single mutants as well as transplanted loop mutants at these loci. The 3D molecular structures were visualized with WebLab Viewer (Accelrys), and the interatomic distance calculations were performed with Rasmol (created by Roger Sayle).
TACE-cat, TACE-long enzymes, and reagents
TACE-cat (also known as TACE-473) is the truncated catalytic domain of TACE that spans residues −215 to −473. TACE-long (also known as TACE-651) is the longer form of the enzyme that encompasses the catalytic domain as well as the disintegrin, epidermal growth factor (EGF)-like, and Crambin-like domains (i.e., residues −215 to −651). Both enzymes were kind gifts from Glaxo Smith Kline Research and Development. The expression and purification of the enzymes are described elsewhere (Milla et al. 1999). All chemicals and reagents used in this work were purchased from Sigma-Aldrich. Restriction enzymes for molecular biology work were obtained from New England Biolabs.
N-TIMP-1 mutagenesis
Wild-type N-TIMP-1 cDNA (spanning residues Cys1 to Glu126) in pRSET-c expression vector (Invitrogen) was used as the template for mutagenesis in this work. For the ease of downstream purification, we fused the cDNA to a 6 × histidine tag at the C-terminus. Different strategies were employed for making N-TIMP-1 mutants, depending on the location and complexity of the mutation process. Generally, all mutations close to the N- and C-termini were created by PCR amplification of wild-type N-TIMP-1 using oligonucleotides incorporating the desired mutations. For more sophisticated loop mutants (e.g., AB loop mutant), we adopted the Transformer™ Site-Directed Mutagenesis System (Clontech) whereby two primers (a mutation primer and a toggle primer) were involved. Second- and third-generation N-TIMP-1 mutants were constructed by subcloning DNA fragments generated from various restriction enzyme digestions that covered different epitopes, in order to create the multiple mutations. All DNA constructs mentioned in this work were sequenced to confirm the incorporation of the right epitopes.
Expression, refolding, and purification of N-TIMP-1 mutants
N-TIMP-1 constructs were expressed in E. coli BL21(DE3) pLys supercompetent cells. The protocol for N-TIMP-1 refolding was modified from one reported elsewhere (Huang et al. 1996). Soluble refolded N-TIMP-1 was extracted with 50 mL Ni-NTA agarose (Invitrogen), washed, and eluted with 200 mM imidazole before final dialysis in 50 mM Tris-HCl, pH 7.8 containing 25% glycerol.
Activities of N-TIMP-1 mutants
All of the N-TIMP-1 mutants described in this work refold without much difficulty. We observed no significant discrepancy in the final yields between the wild-type and the mutant N-TIMP-1 proteins. Typically, we obtained more than 40 mL of active N-TIMP-1 (concentration exceeding 10 μM) from 250 mL of E. coli cultures overexpressing the inclusion bodies. The inhibitory activities of the mutants were determined by titration against a known amount of gelatinase-A, as described in our previous report (Lee et al. 2001). The refolding efficiency of N-TIMP-1 mutants was examined by comparing the protein concentrations (Bio-Rad protein assay kit) with the inhibitory activities against gel-A.
Measurements of binding affinity (Kiapp) and association rate constants (kon) with TACE-cat and TACE-long
N-TIMP-1 mutants were titrated against gel-A primarily because the metalloproteinase is readily inhibited by all variants of mammalian TIMP (TIMP-1 to -4). For Kiapp measurements, 0.2 nM of TACE-cat and TACE-long were preincubated with increasing amounts of N-TIMP-1 mutants. Preincubation was allowed for 3 h at room temperature prior to steady-state (Vs) measurement. The final volume of the incubation mixtures in all Kiapp studies was 2.5 mL unless otherwise stated. FAB buffer (10 mM CaCl2, 50 mM Tris-HCl, pH 7.5, 0.05% Brij-35, 1% DMSO, 0.02% NaN3) was used as diluent in the enzyme assays. Quenched fluorescent peptide QF-45 (Mca-Ser-Pro-Leu-Ala-Gln-Ala-Val-Arg-Ser-Ser-Ser-Arg-Lys-Dnp-NH2) was the substrate for TACE-cat and TACE-long assays (final concentration 1 μM). Measurements of enzyme activities were initiated by adding substrate to incubation mixtures at 27°C. A Perkin-Elmer LS-50B spectrofluorometer with thermostatic cuvettes holders was used in kinetic studies throughout this work. The Kiapp values were calculated with the equation below (Morrison and Walsh 1995), using the computer program Grafit (Erithacus Software) to obtain an estimation of the values:
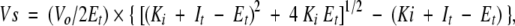 |
where Vs is the steady state rate enzyme, Vo is the rate in the absence of inhibitor, Et is the total enzyme concentration, and It is the total inhibitor concentration.
Association rate (kon) studies were initiated by adding N-TIMP-1, N-TIMP-3, or CT1399 (final concentrations ranged from 5 nM to 250 nM) to TACE-cat and TACE-long (0.1 nM) at 27°C. The rate of inhibition was monitored by continuous fluorometric assay until after 8000 sec.
Acknowledgments
This work was generously funded by the Arthritis Research Campaign (grant no. M0602), Medical Research Council, Wellcome Trust and Biotechnology and Biological Sciences Research Council, United Kingdom. Thanks also to Dr. J.D. Becherer, Glaxo Smith Kline, USA for provision of TACE enzymes.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
ADAM, a disintegrin and metalloproteinase
ADAM-TS, ADAM proteinase with thrombospondin domains
Dnp, 2,4-dinitrophenyl
Gel-A, gelatinase-A (MMP-2)
GnHCl, guanidinium chloride
Mca, (7-methoxycoumarin-4-yl) acetyl
MMP, matrix metalloproteinase
MT-1, membrane-type metalloproteinase-1
N-TIMP-1, N-terminal domain form of TIMP-1
N-TIMP-3, N-terminal domain form of TIMP-3
TACE, TNF-α converting enzyme
TACE-cat, the catalytic domain form of TACE
TACE-long, TACE incorporating the catalytic and the cysteine-rich domains
TIMP, tissue inhibitor of metalloproteinases
TNF-α, tissue necrosis factor-α
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0216202.
References
- Ahonen, M., Baker, A.H., and Kahari, V.M. 1998. Adenovirus-mediated gene delivery of tissue inhibitor of metalloproteinases-3 inhibits invasion and induces apoptosis in melanoma cells. Cancer Res. 11 2310–2315. [PubMed] [Google Scholar]
- Amour, A., Slocombe, P.M., Webster, A., Butler, M., Knight, C.G., Smith, B.J., Stephens, P.E., Shelly, C., Hutton, M., Knäuper, V., Docherty, A.J.P., and Murphy, G. 1998. TNF-α converting enzyme (TACE) is inhibited by TIMP-3. FEBS Lett. 435 39–44. [DOI] [PubMed] [Google Scholar]
- Black, R.A., Rauch, C.T., Kozlosky, C.J., Peschon, J.J., Slack, J.L., Wolfson, M.F., Castner, B.J., Stocking, K.L., Reddy, P., Srinivasan, S., Nelson, N., Boiani, N., Schooley, K.A., Gerhart, M., Davis, R., Fitzner, J.N., Johnson, R.S., Paxton, R.J., March, C.J., and Cerretti, D.P. 1997. A metalloproteinase disintegrin that releases tumor-necrosis factor-α from cells. Nature 385 729–732. [DOI] [PubMed] [Google Scholar]
- Bond, M., Murphy, G., Bennett, M.R., Amour, A., Knäuper, V., Newby, A.C., and Baker, A.H. 2000. Localization of the death domain of tissue inhibitor of metalloproteinase-3 to the N-terminus. J. Biol. Chem. 275 41358–41363. [DOI] [PubMed] [Google Scholar]
- Bond, M., Murphy, G., Bennett, M.R., Newby, A.C., and Baker, A.H. 2002. Tissue inhibitor of metalloproteinase-3 induces a Fas-associated death domain-dependent type II apoptotic pathway. J. Biol. Chem. 277 13787–13795. [DOI] [PubMed] [Google Scholar]
- Gomis-Rüth, F., Maskos, K., Betz, M., Bergner, A., Huber, R., Suzuki, K., Yoshida, N., Nagase, H., Brew, K., Bourenkov, G.P., Bartunik, H., and Bode, W. 1997. Mechanism of inhibition of the human matrix metalloproteinase stromelysin-1 by TIMP-1. Nature 389 77–81. [DOI] [PubMed] [Google Scholar]
- Huang, W., Suzuki, K., Nagase, H., Arumugam, S., Van Doren, S.R., and Brew, K. 1996. Folding and characterization of the amino-terminal domain of human tissue inhibitor of Metalloproteinases-1 (TIMP-1) expressed at high yield in E. coli. FEBS Lett. 384 155–161. [DOI] [PubMed] [Google Scholar]
- Kashiwagi, M., Tortorella, M., Nagase, H., and Brew, K. 2001. TIMP-3 Is a potent inhibitor of aggrecanase 1 (ADAM-TS4) and aggrecanase 2 (ADAM-TS5) J. Biol. Chem. 276 12501–12504. [DOI] [PubMed] [Google Scholar]
- Lee, M.H., Knäuper, V., Becherer, J.D., and Murphy, G. 2001. Full-length and N-TIMP-3 display equal inhibitory activities towards TNF-α convertase. Biochem. Biophys. Res. Comm. 280 945–950. [DOI] [PubMed] [Google Scholar]
- Lee, M.H., Verma, V., Maskos, K., Becherer, J.D., Knäuper, V., Dodds, P., Amour, A., and Murphy, G. 2002a. The C-terminal domains of TACE weaken the inhibitory action of N-TIMP-3. FEBS Lett. 520 102–106. [DOI] [PubMed] [Google Scholar]
- Lee, M.H., Verma, V., Maskos, K., Nath, D., Knäuper, V., Dodds, P., Amour, A., and Murphy, G. 2002b. Engineering N-terminal domain of TIMP-3 to be a better inhibitor against tumor necrosis factor-α converting enzyme. Biochem. J. 364 227–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis, E. 2001. The immuno-inflammatory reaction in Crohn's disease and ulcerative colitis: Characterization, genetics and clinical application. Focus on TNF-α. Acta Gastroenterol. Belg. 64 1–5. [PubMed] [Google Scholar]
- Maskos, K., Fernandez-Catalan, C., Huber, R., Bourenkov, G.P., Bartunik, H., Ellestad, G.A., Reddy, P., Wolfson, M.F., Rauch, C.T., Castner, B.J., Davis, R., Clarke, H.R.G., Peterson, M., Fitzner, J.N., Cerretti, D.P., March, C.J., Paxton, R.J., Black, R.A., and Bode, W. 1998. Crystal structure of the catalytic domain of human tumour necrosis factor-α-converting enzyme. Proc. Natl. Acad. Sci.. 95 3408–3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milla, M.E., Leesnitzer, M.A., Moss, M.L., Clay, W.C., Carter, H.L., Miller, A.B., Su, J.L., Lambert, M.H., Willard, D.H., Sheeley, D.M., Kost, T.A., Burkhart, W., Moyer, M., Blackburn, R.K., Pahel, G.L., Mitchell, J.L., Hoffman, C.R., and Becherer, J.D. 1999. Specific sequence elements are required for the expression of functional tumor necrosis factor α-converting enzyme (TACE). J. Biol. Chem. 274 30563–30570. [DOI] [PubMed] [Google Scholar]
- Morrison, J.F. and Walsh, C.T. 1995. The behavior and significance of slow binding enzyme inhibitors. Meth. Enzymol. 248 201–301. [DOI] [PubMed] [Google Scholar]
- Muskett, F.W., Frenkiel, T.A., Feeney, J., Freedman, R.B., Carr, M.D., and Williamson, R.A. 1998. High resolution structure of the N-terminal domain of tissue inhibitor of metalloproteinases-2 and characterization of its interaction site with matrix metalloproteinase-3. J. Biol. Chem. 273 21736–21743. [DOI] [PubMed] [Google Scholar]
- Ohta, S., Harigai, M., Tanaka, M., Kawaguchi, Y., Sugiura, T., Takagi, K., Fukasawa, C., Hara, M., and Kamatani, N. 2001. Tumour necrosis factor-α (TNF-α) converting enzyme contributes to production of TNF-α in synovial tissues from patients with rheumatoid arthritis. J. Rheumatol. 28 1756–1763. [PubMed] [Google Scholar]
- Tuuttila, A., Morgunova, E., Bergmann, U., Lindqvist, Y., Maskos, K., Fer-nandez-Catalan, C., Bode, W., Tryggvason, K., and Schneider, G. 1998. Three-dimensional structure of human tissue inhibitor of metalloproteinases-2 at 2.1 Å resolution. J. Mol. Biol. 284 1133–1140. [DOI] [PubMed] [Google Scholar]
- Williamson, R.A., Martorell, G., Carr, M.D., Murphy, G., Docherty, A.J., Freedman, R.B., and Feeney, J. 1994. Solution structure of the active domain of tissue inhibitor of metalloproteinases-2. A new member of the OB fold protein family. Biochemistry 33 11745–11159. [DOI] [PubMed] [Google Scholar]
- Wu, B., Arumugam, S., Gao, G., Lee, G., Semenchenko, V., Huang, W., Brew, K., and Van Doren, S.R. 2000. NMR structure of tissue inhibitor of metalloproteinases-1 implicates localized induced fit recognition of matrix metalloproteinases. J. Mol. Biol. 295 257–268. [DOI] [PubMed] [Google Scholar]
- Yu, W.S., Yu, S.C., Meng, Q., Brew, K., and Woessner, J.F. 2000. TIMP-3 binds to sulfated glycosaminoglycans of the extracellular matrix. J. Biol. Chem. 275 31226–31232. [DOI] [PubMed] [Google Scholar]