

Abstract
The solution structure of the copper-free state of a monomeric form of superoxide dismutase (153 amino acids) was determined through 13C and 15N labeling. The protein contained two mutations at the native subunit–subunit interface (F50E and G51E) to obtain a soluble monomeric species and a mutation in the active site channel (E133Q). About 93% of carbon atoms, 95% of nitrogen atoms, and 96% of the protons were assigned. A total of 2467 meaningful NOEs and 170 dihedral angles provided a family of 35 conformers with RMSD values of 0.76 ± 0.09 Å for the backbone and 1.22 ± 0.13 Å for all heavy atoms. The secondary structure elements, connected by loops, produce the typical superoxide dismutase Greek key fold, formed by an eight-stranded β-barrel. The comparison with the copper-bound monomeric and dimeric structures shows that the metal ligands have a conformation very close to that of the copper-bound forms. This feature indicates that the copper-binding site is preorganized and well ordered also in the absence of the copper ion. The active-site channel shows a sizable increase in width, achieving a suitable conformation to receive the copper ion. The histidines ring NH resonances that bind the copper ion and the region around the active-site channel experience, as found from 15N relaxation studies, conformational exchange processes. The increased width of the channel and the higher mobility of the histidine rings of the copper site in the copper-free form with respect to the holoprotein is discussed in terms of the process of copper insertion.
Keywords: Copper-free superoxide dismutase, solution structure, NMR, Protein mobility
The study of the process involving copper trafficking and delivery to each specific apo-protein target, represents an active field of research. Several copper chaperone proteins have been described and their role in copper trafficking investigated (Valentine and Gralla 1997; O'Halloran and Culotta 2000; Rosenzweig et al. 2000; Huffman and O'Halloran 2001). A protein called CCS acts as copper chaperone to eukaryotic copper, zinc SOD (Culotta et al. 1997; Rae et al. 1999, 2001; Schmidt et al. 2000). SOD is a dimeric protein in which each identical subunit contains one copper and one zinc ion. The two subunits experience extensive contacts, mainly hydrophobic in nature, even if some hydrophilic interactions further stabilize the dimer (Parge et al. 1992; Rae et al. 2001). The crystal structure of CCS from yeast shows that the protein is composed by three domains (Schmidt et al. 1999). The first domain resembles the copper chaperone Atx1 (Rosenzweig et al. 1999; Arnesano et al. 2001; Rae et al. 2001). The second domain is largely homologous to SOD, with which it is supposed to interact (Hall et al. 2000; Lamb et al. 2000; Torres et al. 2001). The third domain has been proposed to be involved in the copper donation to SOD.
Some monomeric forms of SOD have been obtained by introducing charged residues at the hydrophobic interface of the dimer (Bertini et al. 1994; Banci et al. 1999). In one of these, the hydrophobic Phe 50 and Gly 51 residues were substituted by two hydrophilic Glu groups (M2SOD) (Bertini et al. 1994). A further mutation at position 133 (Glu to Gln) was introduced (M2E133QSOD) to increase the activity of the monomeric species to mimic the behavior of the native dimeric protein (Getzoff et al. 1992; Banci et al. 1995). A further monomeric species, in which the overall charge was maintained, was obtained by mutating four residues at the protein–protein interface, (M4SOD) (Banci et al. 1997). The solution structures of these copper(I)-containing monomeric forms were solved through NMR spectroscopy (Banci et al. 1998) as well as that of the wild-type dimeric protein (Banci et al. 2002). For both protein forms, the X-ray structures are also available (Parge et al. 1992; Ferraroni et al. 1999). Each subunit is constituted by the classical β-barrel, made of eight antiparallel β strands, connected by seven turns and loops. The metal ligand residues are located in loops IV and VII. In the reduced form, the copper(I) ion is coordinated by three histidines (His 46, His 48, and His 120), and zinc by three histidines (His 63, His 71, and His 80) and by an Asp residue (Asp 83), the latter belonging to the fifth β-strand. In the oxidized forms, His 63 forms a bridge between the two metal ions by becoming a histidinate group (Parge et al. 1992; Graden et al. 1994). The copper ion is solvent exposed, whereas the zinc is, when copper is present, completely buried inside the protein. The copper sits at the bottom of the active site channel, which is formed on one side by loop IV and on the other by the electrostatic loop VII that also contains a short α-helix.
The dismutation reaction is diffusion limited. The electrostatic loop in the active-site channel increases the diffusion rates of superoxide and drives it toward copper and Arg 143 (Getzoff et al. 1992). An H-bond network inside of the cavity, formed by the side chains of some residues belonging to loop VII, stabilizes the latter in the optimal conformation for increasing the diffusion rates of the superoxide radical inside the cavity (Fisher et al. 1994). In particular, Glu 133 is invariantly H-bonded to the hydroxyl group of Thr 137, and the backbone NH of Lys 136 is H-bonded to the backbone carbonyl of Glu 132.
The guanidinium group of Arg 143 has the same conformation in all of the X-ray structures of eukaryotic proteins, pointing toward copper at a distance suitable for the superoxide-copper interaction. The orientation of Arg 143 is stabilized by some hydrogen bonds involving its side chain and residues 57, 58, and 61. On the contrary, in the monomeric species M2E133QSOD and M4SOD (Banci et al. 1998, 1999), the guanidinium group of Arg 143 moves further from the copper ion and these relevant H-bonds are present only in a few conformers.
The conformation of the metal-binding site is well defined also in the solution structures. It is stabilized and quite conserved among the many characterized SODs by some key H-bonds (Parge et al. 1992) among these, quite relevant is that between the backbone NH of His 71 and CO of Thr 135, as it stabilizes the relative conformation of loop IV and loop VII. Other strong and conserved H-bonds involve the side chain of Asp 83 and the backbone NH of His 80, Hδ1 of His 43 and CO of His 120, Hδ1 of 120 and CO of Gly 141, and Hɛ2 of His 71 and the side chain of Asp 124. These H-bonds are always present in all the human SOD structures solved either in solution or in the crystal (Parge et al. 1986; 1992; Banci et al. 1998, 1999, 2002; Hart et al. 1998).
Within the frame of an extensive investigation of the copper transport mechanisms, we are pursuing the solution structure and the backbone mobility of the copper-free monomeric M2E133QSOD (EZnM2E133QSOD, hereafter; the former E means empty), to understand how the zinc ion preorganizes the protein conformation, and particularly the copper-binding site to receive the copper ion from the CCS chaperone. It is well established that, in vitro, zinc should bind to SOD before copper, (Beem et al. 1974; Lippard et al. 1977; Pantoliano et al. 1982; Bertini et al. 1998) although, to our knowledge, the order of the metal binding has not been determined in vivo. It is likely that a similar sequential behavior is occurring in the cell also. The present structure of the copper-free protein is relevant for the comprehension of the copper-binding process. Within this frame, this copper-free structure is compared with those of the metal-loaded species and the similarities and differences are discussed. This structure can have also a high significance with respect to a better understanding of the interaction of zinc ion with SOD. This is an important issue with respect to the involvement of SOD in ALS disease. The ALS-related SOD mutants experience a reduced affinity for zinc ion (Rabizadeh et al. 1995; Lyons et al. 1999, 2000; Goto et al. 2000). These mutants experience a new, toxic activity.
Results
NMR spectra
At pH 5, all of the zinc-coordinated histidines are expected to be protonated on the ring nitrogen not involved in the coordination with the metal ion, whereas the histidines of the copper-binding site are expected to be protonated on both nitrogen atoms as it occurs for His 43, which is not involved in bonds with any metal ion in the fully metallated monomeric and dimeric forms. Eight imidazole NH signals, two of which are very broad, were detected in a one-dimensional NMR experiment, by use of the WATERGATE pulse scheme for water suppression (Fig. 1B ▶). Their 1H and 15N chemical-shift assignments and the binding mode of each histidine were achieved through a 1H–15N HSQC experiment tailored for the detection of 2J 1H–15N couplings and from the analysis of NOESY maps. From this analysis, in addition to His 63, His 71, and His 80, which coordinate zinc, also His 46, His 48 and His 120, which are potential copper ligands, are monoprotonated. His 48 and His 120 are protonated on Nδ, whereas the other His residues are protonated on Nɛ. His 43 is biprotonated, as it occurs in the fully metallated dimeric and monomeric form, most likely as a consequence of its interaction with the negative charge of Glu 40, which would increase its pKa. The other expected imidazole NHs probably are not observed due to fast exchange with the solvent (see below). It is worthy to note that the imidazole NH signals of His 48 and His 80 NHs are detected only at 288 K.
Fig. 1.

1H 600 MHz NMR spectra of the histidine ring NH resonances of CuZnM2E133QSOD (A) and of EZnM2E133QSOD (B) in H2O, of EZnM2E133QSOD after 30 min (C), and after 5 d (D) the lyophilized protein has been dissolved in D2O. Signal assignment is as follows: (a) NHɛ2 His 71, (b) NHɛ2 46, (c) NHɛ2 43, (d) NHɛ2 63, (e) NHɛ2 80, (f) NHδ1 43, (g) NHδ1 48, (h) NHδ1 120. In all of the spectra, recorded at 298 K, WATERGATE sequence was used to suppress the water resonance.
The chemical shifts of the ring histidine protons and nitrogens of the wild-type dimeric protein of CuZnM2E133QSOD and CuZnM4SOD are quite similar to those of EZnM2E133QSOD (Table 1), with small differences only for the proton and nitrogen resonances of the histidines involved in the copper binding. In Figure 1 ▶, the spectrum of the present copper-free form (Fig. 1B ▶) is compared with that of the copper-bound form of the same protein (Fig. 1A ▶). This suggests that the copper-binding site has a very similar conformation with and without copper, and that it is preorganized as a result of H-bond interactions (see below), even before the copper is coordinated.
Table 1.
1H and 15N chemical shifts of histidine resonances in the wild-type dimeric protein Cu2Zn2SOD and in the monomeric forms CuZnM2E133QSOD, CuZnM4SOD, and EZnM2E133QSOD at 298 K (pH 5.0) and in water solutiona
| Residue | Cu2Zn2SODb | CuZnM2E133QSODc | CuZnM4SODd | EZnM2E133QSODe | |
| His 43 | Hδ2 | 7.09 | 7.08 | 7.09 | 7.04 |
| Hɛ1 | 8.57 | 8.59 | 8.58 | 8.54 | |
| Hδ1 (Nδ1) | 12.8 (172.4) | 13.0 (173.6) | 12.9 (173.4) | 12.8 (172.9) | |
| Hɛ2 (Nɛ2) | 14.06 (175.4) | 14.1 (176.3) | 14.0 (176.3) | 14.05 (175.1) | |
| His 80 | Hδ2 | 6.79 | 6.85 | 6.85 | 6.76 |
| Hɛ1(Nδ1) | 8.48(f) | 8.49 (201.0) | 8.54(f) | 8.51 (201.7) | |
| Hɛ2 (Nɛ2) | 12.7 (176.8) | 12.8 (177.0) | 12.7 (177.5) | 12.9 (176.4) | |
| His 71 | Hδ2 | 6.75 | 6.80 | 6.76 | 6.76 |
| Hɛ1 (Nδ1) | 7.72(f) | 7.74 (208.6) | 7.71(f) | 7.59 (209.0) | |
| Hɛ2 (Nɛ2) | 15.4 (177.1) | 15.3 (175) | 15.3 (177.7) | 15.0 (176.6) | |
| His 63 | Hδ2 | 5.93 | 5.79 | 5.77 | 6.08 |
| Hɛ1 (Nδ1) | 6.4(f) | 6.47 (210.0) | 6.46(f) | 7.2 (209.0) | |
| Hɛ2 (Nɛ2) | 12.5 (169.6) | 12.6 (171.0) | 12.5 (171.1) | 13.5 (172.9) | |
| His 46 | Hδ2 | 7.07 | 7.08 | 7.00 | 7.23 |
| Hɛ1 (Nδ1) | 6.74(f) | 6.77 (234.6) | 6.76(f) | 8.05 (214.6) | |
| (Nɛ2) | 13.4 (168.9) | 13.4 (178.0) | 13.5 (170.4) | 14.4 (175.7) | |
| His 48 | Hδ2 | 7.09 | 7.01 | f | 6.86 |
| Hɛ1 | 8.35 | 8.57 | 8.35 | 9.45 | |
| Hδ1 (Nδ1) Hɛ2 (Nɛ2) | 12.5 (169.5)(f) | 12.4 (171.0) (215.0) | 12.4 (171.2)(f) | 12.5 (176.9)g(201.7)g | |
| His 120 | Hδ2 | 6.56 | 6.56 | 6.54 | 5.93 |
| Hɛ1 | 8.15 | 8.21 | 8.15 | 9.56 | |
| Hδ1 (Nδ1) (Nɛ2) | 10.5 (164.8)(f) | 10.4 (165.0) (225.0) | 10.4 (165.1)(f) | 10.83 (171.5) (208.9) |
a When copper is present, it is in its reduced state, that is, Cu(1).
b Banci et al. (2002).
c Banci et al. (1998).
d Banci et al. (1999).
e Present work.
f Not assigned.
g Resonances measured at 283 K.
Sequence-specific assignment and structure determination
The 15N and 13C HSQC spectra show well-dispersed resonances indicative of an essentially folded protein. Furthermore, the signal line widths are consistent with a monomeric state of the protein. The spectra are also relatively similar to those observed for the same fully metallated monomeric protein, suggesting that the overall protein fold is maintained.
The backbone resonance assignment, with the exception of the amide nitrogen of Lys 23, Ser 25, and Glu 132, was obtained through a semiautomatic approach (see Materials and Methods section). About 93% of carbon atoms, 95% of nitrogen atoms, and 96% of the protons were assigned.
By analyzing three-dimensional 15N-edited and 13C-edited NOESY spectra and two-dimensional NOESY spectra, 4432 NOE cross-peaks were assigned and transformed into upper-distance limits with the program CALIBA (Güntert et al. 1991). They resulted in 3201 unique upper-distance limits, of which 2467 were meaningful. The number of meaningful NOEs per residue is reported in Figure 2A ▶. The average number of meaningful NOEs per residue is 16. A total of 84 proton pairs were stereospecifically assigned using the program GLOMSA (Güntert et al. 1991). A total of 94 dihedral φ angle constraints were obtained from the analysis of the HNHA spectrum and 76 dihedral ξ angles were obtained from the 15N-edited NOESY spectrum. The constraints used for structure calculations and the obtained stereospecific assignments are reported in the Supplementary material (see www.proteinscience.org.).
Fig. 2.
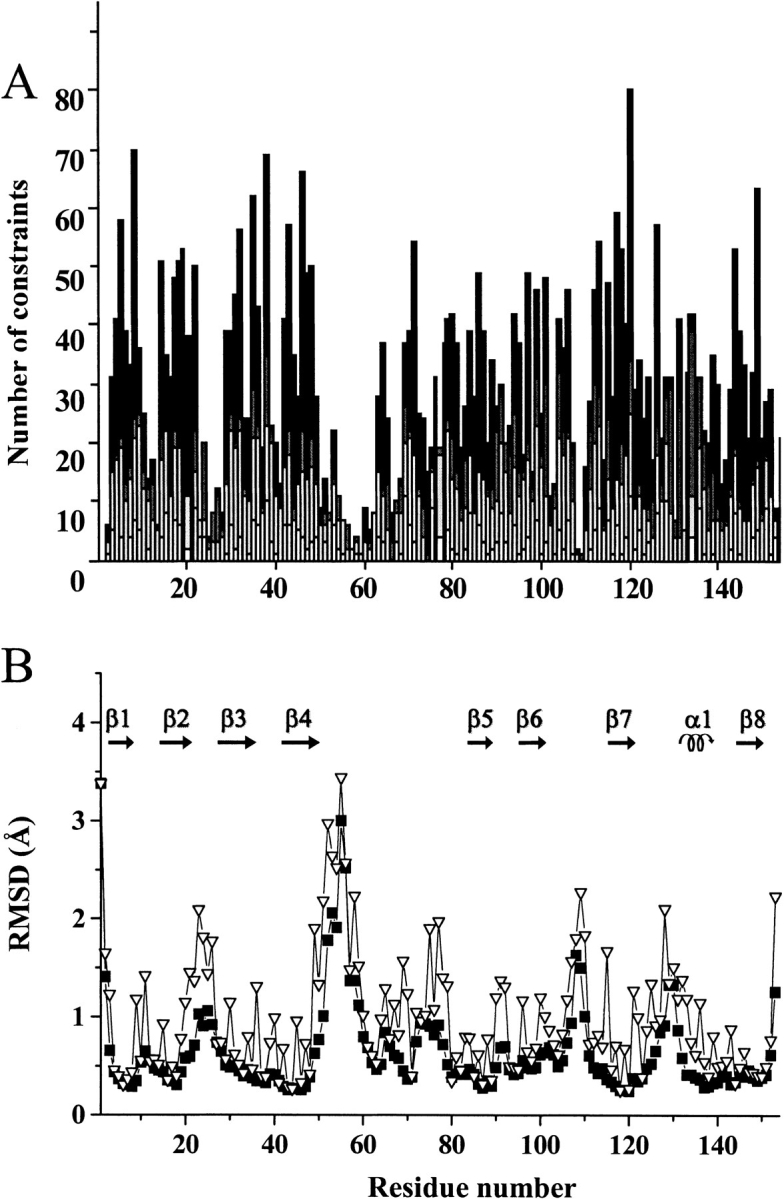
(A) Meaningful NOEs per residue for EZnM2E133QSOD. White, gray, and black bars indicate intraresidue, sequential, and medium/long range connectivities, respectively; (B) RMSD per residue to the mean structure of EZnM2E133QSOD for the backbone (▪) and all heavy atoms () of the REM structure family of 35 conformers.
The 35 conformers constituting the final DYANA family have an average target function of 1.46 ± 0.16 Å2 and an average RMSD to the mean structure (for residues 3–150) of 0.77 ± 0.08 Å for the backbone and 1.22 ± 0.10 Å for the heavy atoms. Each conformer of the family was then subjected to further refinement with the program AMBER. The family has a total NMR constraint penalty of 83.7 ± 8.4 kJ mole−1, which corresponds to a target function of 0.62 ± 0.05 Å2. The average RMSD values for the family are 0.76 ± 0.09 Å for the backbone and 1.22 ± 0.13 Å for the heavy atoms. Statistics of the structure analysis are reported in Table 2. According to the program PROCHECK, secondary structure elements were found for: 2–8 (β1), 15–22 (β2), 29–36 (β3), 41–48 (β4), 82–88 (β5), 94–100 (β6), 114–120 (β7), 130–136 (α1) and 141–148 (β8). These secondary structure elements, connected by loops, produce the typical SOD Greek key, formed by an eight-stranded β-barrel. The completeness of experimentally observed NOE restraints is also shown in Table 2.
Table 2.
Statistical analysis of the final REM family and the mean structure of EZnM2E133QSOD
| RSM violations per experimental distance constraint (Å)b | REMa (35 structures) | 〈REM〉a (mean) |
| Intraresidue (454) | 0.0507 ± 0.0740 | 0.0218 |
| Sequential (670) | 0.0162 ± 0.0012 | 0.0162 |
| Medium range (271)c | 0.0119 ± 0.0027 | 0.0164 |
| Long range (1072) | 0.0869 ± 0.0013 | 0.0136 |
| Total (2467) | 0.0733 ± 0.0009 | 0.0163 |
| RSM violations per experimental dihedral angle constraints (deg)b | ||
| φ (94) | 0.60 ± 0.28 | 0.46 |
| ξ (76) | 2.61 ± 0.91 | 2.16 |
| Average number of violations per structure | ||
| Intraresidue | 12.3 ± 2.8 | 15 |
| Sequential | 18.2 ± 2.5 | 18 |
| Medium range | 4.5 ± 1.6 | 9 |
| Long range | 20.6 ± 3.5 | 16 |
| Total | 55.7 ± 5.2 | 58 |
| Phi | 2.4 ± 1.2 | 2 |
| psi | 5.8 ± 1.7 | 7 |
| Average no. of NOE violations larger than 0.3 Å | 0 | 0 |
| Average no. of NOE violations between 0.1 Å and 0.3 Å | 17.4 ± 2.4 | 22 |
| Structural analysisd | ||
| % of residues in most favorable regions | 65.1 | 67.5 |
| % of residues in allowed regions | 30.3 | 27.5 |
| % of residues in generously allowed regions | 4.0 | 5.0 |
| % of residues in disallowed regions | 0.6 | 0.0 |
| Experimental restraints analysise | ||
| % completeness of experimentally observed NOE up to 4 Å cut-off distance | 63% | 58% |
| % completeness of experimentally observed NOE up to 5 Å cut-off distance | 38% | 42% |
a REM indicates the energy minimized family of 35 structures; ≤REM> is the energy minimized mean structure obtained from the coordinates of the individual REM structures.
b The number of experimental constraints for each class is reported in parenthesis.
c Medium range distance constraints are those between residues (i,i + 2), (i,i + 3), (i,i + 4) and (i,i + 5).
d As it results from the Ramachandran plot analysis.
e As it results from the AQUA analysis. Laskowski et al. (1996, 1998).
In Figure 2B ▶, the RMSD values per residue to the mean structure for the EZnM2E133QSOD structure, shown in Figure 3 ▶, are reported. The secondary structure is well defined and the β-strands are characterized by a lower disorder than the connecting loops. The average RMSD values for the segments involved in the β-barrel are 0.38 ± 0.07 Å and 0.81 ± 0.13 Å for the backbone and all heavy atoms, respectively. On the contrary, the most disordered regions are those involving residues 22–27 (located in loop II), residues 52–61 (located in loop IV), and residues 107–110 (located in loop VI). This behavior is very similar to that observed for CuZnM2E133QSOD (Koradi et al. 1996). These high RMSD values are due to the paucity of NOEs (Fig. 2 ▶) as a consequence of the location of these segments in external loops projected toward the solvent. Again, as observed for the other two solved monomeric structures, the largest disorder is observed for even-numbered loops. Loops IV and VI partially constitute the interface between the two subunits in the holo protein.
Fig. 3.

Solution structure of EZnM2E133QSOD, showing the backbone atoms represented as a tube with a radius proportional to the backbone RMSD value of each residue. The Zn(II) ion is also shown. The figure was generated with the program MOLMOL 37.
Deuterium-exchange analysis
The amide protons of EZnM2E133QSOD can be grouped into three categories according to their behavior with respect to exchange with the bulk solvent in D2O solution. They were classified according to the time needed for the signal to disappear when the lyophilized protein is dissolved in D2O, fast exchange (<10 min), intermediate exchange (10 min to 10 h), and slow exchange (>10 h). Their mapping on the protein is shown in Figure 4 ▶. The fast exchanging residues (white in Fig. 4 ▶) are located on the surface of the molecule. Residues located in the electrostatic channel, such as 132 and 133, also experience a fast exchange with the bulk solvent. Slow exchange (black in Fig. 4 ▶) is observed for amide protons of residues 3, 6, 8, 18–22, 29–36, 38, 42–48, 82–89, 95–100, 114–120, 144–145, and 147–150 (they are still present after 5 d). These slow-exchanging amide protons are essentially located in the β-strands, and some of them are involved in hydrogen bonds. The backbone amide proton of the residues in metal sites do experience slow exchange too, because their signal is intensly constant up to 5 d, whereas their ring NH resonances do exchange with the solvent. By comparing the 1H 1D NMR spectra for the histidine ring NH resonances in water (Fig. 1B ▶) with those recorded at different times after the protein is dissolved in D2O (Fig. 1C,D ▶), it appears that the signals are completely exchanged already after 30 min.
Fig. 4.

Deuterium-exchange analysis for the amide protons of EZnM2E133QSOD. The color code is as follows: white for fast exchanging (<10 min), light gray for intermediate exchanging (10 min to 10 h), and black for slow exchanging protons (>10 h). Proline residues or residues that could not be accurately quantified due to spectral overlap are in gray.
Mobility measurements
The 15N backbone R1 and R2 relaxation rates and 1H–15N NOEs were measured on EZnM2E133QSOD at 600 MHz. Average values of R1 and R2 with a spin echo delay (τCPMG) of 450 μs are 1.43 ± 0.09 s−1 and 12.0 ± 0.5 s−1, respectively. The average 1H–15N NOE value is 0.79 ± 0.10. The values are close to those found at the same magnetic field for the CuZnM2E133QSOD form (Banci et al. 2000), that is, 1.36 ± 0.29 s−1 and 13.4 ± 0.9 s−1 for R1 and R2, respectively, and 0.81 ± 0.10 for NOE. The differences observed for R1, R2, and 1H–15N NOE, between CuZnM2E133QSOD and EZnM2E133QSOD, are reported in Figure A of the Supplementary Material (see www.proteinscience.org).
The presence of conformational exchange processes occurring in the ms-μs time range were further analyzed by R2 measurements as a function of the τCPMG length (see Material and Methods section).
The R1 and R2 rates in EZnM2E133QSOD are almost homogenous along the entire protein, with the exception of a few residues (Table 3) that show either R2 values higher than the average and/or a dependence of R2 with the τCPMG length. This behavior suggests the presence of exchange processes involving NH groups. These residues are located mainly in the β3 strand, at the subunit interface ( which encompasses residues 49–55, 113–115, and 148–153 ), and in the helix α1. R2 experiments with variable τCPMG length were recorded also for histidine ring NH nitrogens. These measurements indicate a conformational exchange process in the ms-μs time scale for NH protons NHɛ2 of residues 43 and 63, and NHδ1 of residue 120. For the cases in which the R2 rates are higher than the average value, but constant with τCPMG, the exchange rate is faster than the reciprocal of the shortest τCPMG value.
Table 3.
Backbone amide nitrogens and histidine ring NH nitrogens involved in conformational exchange processes in EZnM2E133QSODa and CuZnE133QM2SODb
| τex (μs)c | τex (μs)c | ||||
| Backbone NH | EZnSOD | CuZnSOD | Backbone NH | EZnSOD | CuZnSOD |
| 3 | 114 ± 29 | — | 127 | 128 ± 35 | <15d |
| 19 | 156 ± 60 | — | 130 | 291 ± 18 | — |
| 35 | 155 ± 9 | — | 134 | 155 ± 32 | — |
| 39 | 156 ± 18 | — | 135 | <100d | — |
| 55 | 155 ± 11 | — | 145 | <100d | — |
| 56 | <100d | 102 ± 15 | 146 | <100d | 92 ± 13 |
| 61 | 261 ± 10 | 86 ± 9 | 147 | <100d | 71 ± 18 |
| 108 | 156 ± 20 | — | 148 | 115 ± 35 | — |
| 115 | <100d | 40 ± 8 | 149 | <100d | — |
| 118 | <100d | 104 ± 7 | |||
a Measured from the dependence of the 15N CPMG R2 relaxation rates on τCPMG length at 298 K.
b Measured from the dependence of the R2 on the spin lock power in 15N R1p measurements.
c Correlation time for the exchange process, τex.
d A lower limit can be provided only on the basis of the maximum experimental effective magnetic field.
In Table 3, the residues that experience conformational exchange processes in CuZnM2E133QSOD are also reported for comparison. It can be seen that only a few residues involved in exchange processes in the copper-free form are also mobile in the copper-bound form. For CuZnM2E133QSOD, the mobile residues are all located at the dimer interface. No residues in the metal binding region nor in the active channel show exchange processes.
The Model-Free parameters were determined for each backbone NH and for the histidine ring NHs that were resolved in the two-dimensional maps. The experimental data, similar to the copper-bound form (Banci et al. 2000) can be fitted using an isotropic model for the overall protein tumbling as it results from the F-test of the Model-Free analysis. The optimized value of Diso is (1.95 ± 0.01) 107 s−1, which compares with the (1.80 ± 0.01) 107 s−1 value found for the CuZnM2E133QSOD (Banci et al. 2000). The Model-Free calculations provide a τm value of 8.4 ± 0.3 nsec and a mean S2 of 0.87 ± 0.03. The corresponding parameters for CuZnM2E133QSOD are 9.10 ± 0.1 nsec and 0.89 ± 0.10, respectively. The results of the Model-Free analysis, in terms of S2 values for CuZnM2E133QSOD and EZnM2E133QSOD, are compared in Figure 5 ▶. Both forms are characterized by a larger amplitude of internal motions (i.e., lower values of S2) within the subunit interface as compared with the rest of the protein. Furthermore, some residues of loops IV and VI, of helix α1 and of β-sheet consisting of β2, β3, and β6 strands, show lower S2 values for EZnM2E133QSOD than in the fully metallated form (Fig. 5 ▶). Significantly, the copper ligands ( residues 46 and 120 ) and the zinc ligands ( residues 83 and 63 ) are localized within these regions; this behavior points out an increased mobility in the subnanosecond time range for backbone metal ligands in the copper-free form with respect to the copper-bound form.
Fig. 5.
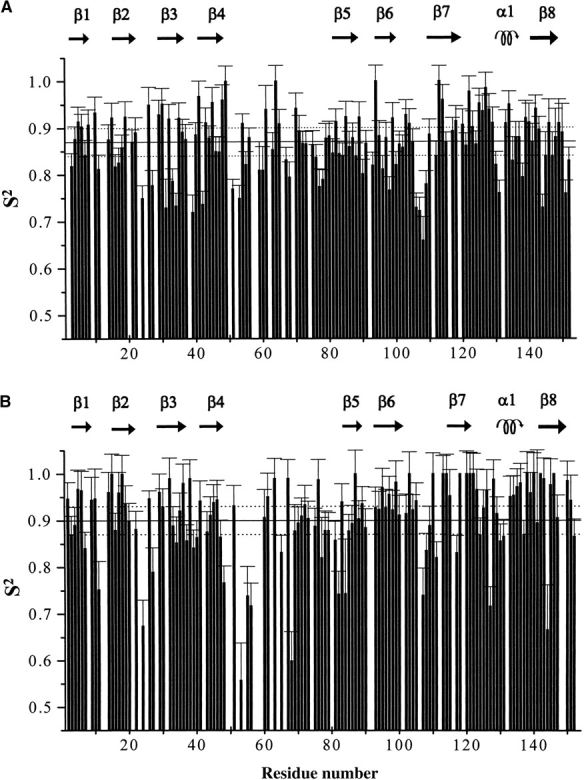
S2 values of (A) EZnM2E133QSOD and (B) CuZnM2E133QSOD.
Overall, for the exchange processes occurring in the ms-μs time range, the copper-free protein behaves like the fully metallated protein, with the exception of the region around the active site channel, which displays increased mobility in the former protein. Also, in the subnanosecond time scale, EZnM2E133QSOD shows features very similar to CuZnM2E133QSOD. However, the residues in the metal-binding site and near the active-site channel show an increased mobility in the absence of copper within this time range.
Comparison with the existing structures
The solution structure of EZnM2E133QSOD is very similar to CuZnM2E133QSOD, (Fig. 6 ▶), with a RMSD between the backbone atoms of the two proteins of 1.66 Å. All secondary structure elements are conserved in terms of number and length. When the RMSD per residue between the two structures is compared with the sum of the RMSD of the structures (which measures their precision), we can see that some regions experience meaningful differences, that is, outside of the uncertainty of the structures of the two protein forms. They are located in loop I, loop IV, and in the α-helix.
Fig. 6.

Comparison between EZnM2E133QSOD and CuZnM2E133QSOD structures. Backbone of EZnM2E133QSOD (dark gray) and CuZnM2E133QSOD (light gray). The zinc ions are white and the copper ion is dark gray. (Inset) Metal-binding sites, including metal-coordinating amino acid residues.
In loop IV, the residues with the largest differences are 49–54 and 65–69. Loop IV is involved in the subunit interactions in the dimeric protein and contains copper ligands and residues forming the active-site channel. Among residues at the dimer interface, Thr 54 and Leu 67 show a sizable change in conformation; in the copper-free form they point toward the solution, whereas they are buried in the copper-bound form.
The active-site channel, formed by residues of loop IV on one side and by loop VII and α-helix on the other, is characterized by several charged residues which produce an optimal electrostatic field to attract, drive, and increase the diffusion rates of the substrate to the copper site. The active-site channel of the present monomer (magenta) compared with the copper-bound form (green), is shown in Figure 7A ▶. The most significant differences in the active-site channel are observed for the side chains of Glu 132, Gln 133, Lys 136, Thr 137, and Arg 143. In the copper-depleted protein, the side chain of Lys 136 has moved toward the protein surface. Nζ of Lys 136 is ∼17 Å away from the copper location. In CuZnM2E133QSOD, this distance is significantly smaller (11.5 Å). The α-helix backbone, with the side chain of Glu 132, shows a displacement that determines a more open cavity. Glu 132 Oɛ is 23 Å from the Cγ of Thr 58, which is located on the opposite side of the channel entrance. The same distance in the copper-bound form is 19 Å. Cζ of Arg 143 and the Cγ of Thr 137, which form a bottleneck in the channel close to the active site, are 10.2 Å apart in the copper-depleted protein, whereas this distance is ∼7.4 Å in the monomeric copper-bound form and 5.7 Å in the dimeric form 9; (Banci et al. 1998, 2002; Ferraroni et al. 1999). The lack of copper ion clearly produces a significant increase in width of the active site channel (see Fig. 7B,C ▶).
Fig. 7.

(A) Comparison between active-site channel in EZnM2E133QSOD (magenta) and in CuZnM2E133QSOD (green). (B) Surface of CuZnM2E133QSOD and (C) EZnM2E133QSOD, showing atoms within 5 Å from the copper ion (magenta), between 5 and 10 Å (orange) and farther than 15 Å (yellow).
Still, the loops forming the channel in which superoxide diffuses maintain the optimal conformation for generating the suitable electrostatic field. The conformation of the loops is maintained by several H-bonds, which form a highly conserved network in the human CuZnSOD structures solved to date, either in solution or in the crystal (Parge et al. 1992; Hart et al. 1998; Banci et al. 1997, 1999, 2002). Also, in the present case, the H-bonds of the electrostatic loop are conserved. They include the hydrogen bonds between backbone NH of Lys 136 and CO of Glu 132, backbone NH of Gly 138 and CO of Gln 133, and backbone NH of Thr 137 and CO of Gln 133.
The guanidinium group of Arg 143 does not form the H-bonds with the CO groups of Cys 57 and Thr 58, which are present in the dimeric forms of SOD and keep Arg 143 close to the copper ion. The lack of these H-bonds is a feature of monomeric forms independent of their metallation condition (Parge et al. 1992; Banci et al. 1999). In the disulfide bridge, Cys 146 has the same position in all of the human structures, both monomeric and dimeric (Parge et al. 1992; Banci et al. 1998, 1999, 2002; Hart et al. 1998; Ferraroni et al. 1999), whereas Cys 57 has a large variability in conformation.
As already discussed, the metal ligands have a conformation, in the present copper-free form, very close to that found in the copper-bound form (Fig. 6 ▶, inset). Although this seems somehow expected for the zinc ligands, this finding for the copper ligands is quite relevant and indicates that the copper-binding site is preorganized and well ordered in the absence of the copper ion also. Its histidine ligands are involved in a series of H-bonds, which stabilize the structure of the copper site. This network of H-bonds is quite conserved with respect to the holoproteins. H-bonds involving the main and side chains of the metal ligands are summarized and compared with the copper-bound form in Table 4.
Table 4.
Backbone and side chain hydrogen bonds involving metal binding residues in EZnM2E133QSOD and CuZnE133QM2SOD
| Donor | Acceptor | EZnSOD | CuZnSOD | ||||
| 71 | His | NH | 135 | Thr | O | x | x |
| 80 | His | NH | 83 | Asp | +OD1 | x | |
| 43 | His | Hδ1 | 120 | His | O | x | x |
| 120 | His | Hδ1 | 141 | Gly | O | x | x |
| 48 | His | Hδ1 | 61 | Gly | O | x | x |
| 48 | His | NH | 116 | Thr | O | x | x |
Discussion
In this work, the solution structure of the copper-free form of M2E133QSOD, with a good degree of resolution, is determined. The protein is well folded and the elements of secondary structure are the same as in the copper-bound protein. The copper site is well ordered and the metal-binding residues are in the proper orientation to bind the copper ion, suggesting that the zinc ion has a determinant structural effect in preorganizing both metal sites, even in absence of copper.
On the other hand, relaxation measurements reveal an increase in mobility for some metal-binding residues, indicating that copper binding decreases the dynamics of the coordinated histidines.
The copper domain is largely solvent exposed in the absence of copper as shown by the deuterium-exchange analysis of histidine protons attached to the potential copper donors, whereas the zinc site is still buried inside of the protein. The location of the latter, which results in being sheltered by the α-helix, is consistent with its main structural role.
The active-site channel shows a sizable increase in its width with respect to the copper, zinc monomeric protein as a result of a different orientation of the side chain of some residues. Furthermore, residues of this region experience an increased mobility also.
The wider and more mobile channel, together with the conformational exchange process shown by the copper-site histidine rings and their solvent exposure, can significantly contribute to the transfer of the copper ion from its chaperone, the CCS protein (Culotta et al. 1997).
The CCS protein is composed of three domains, of which domain III is proposed to carry out insertion of the copper ion into copper-free SOD (Culotta et al. 1997; Schmidt et al. 1999), whereas domain I is functionally relevant only in extremely low concentrations of copper (Schmidt et al. 1999), and domain II is essential for the specificity of CCS interaction with SOD. Although the structures of both SOD and CCS are available (Banci et al. 1998, 1999, 2002; Lamb et al. 1999; Hall et al. 2000), the details of copper chaperone (CCS) (Culotta et al. 1997) and target protein (SOD) recognition and interaction for metal ion transfer are not yet understood.
Two possible models of CCS–SOD interaction have been proposed. One involves the interaction between the two proteins (CCS and SOD), both in the dimeric form (Hall et al. 2000), the other suggests the formation of a heterodimer between one subunit of each protein (Rae et al. 2001). For the latter model, the X-ray structure has been solved recently (Lamb et al. 2001) for a yeast CCS–SOD complex, in which one copper ligand in SOD has been mutated to produce a copper-free form. In this complex, domain II of CCS interacts with SOD through hydrophobic conserved residues and through four strong interprotein hydrogen bonds. These interactions are similar to those found in both structures at their dimeric interface (Lamb et al. 2001).
On the basis of the latter hypothesis (Rae et al. 2001) and within the frame of the characterization of the mechanism of copper transfer and insertion in SOD, the present solution structure of copper-free human superoxide dismutase is therefore quite meaningful as a model for the protein form that interacts with its copper chaperone CCS. Interestingly, the structural and dynamical changes between the copper-free and the copper-bound form are observed in loop IV and loop VII, containing the copper ligands and residues forming the active-site channel and being involved in the subunit interactions in the dimeric protein. All of these changes could likely facilitate the interaction of copper-free SOD with CCS and the insertion of copper.
Materials and methods
Sample isolation and preparation
The expression of the monomeric form of human SOD and its purification are reported elsewhere (Getzoff et al. 1992). Two 2.2-mM samples of 15N-enriched and 15N-13C-enriched proteins were prepared in 20 mM phosphate buffer (pH 5). The copper-free derivative was prepared as reported in Beem et al. (1974) and McCord and Fridovich (1969).
NMR measurements and structure calculation
The NMR spectra were acquired on Avance 800, 700, and 600 Bruker spectrometers operating at proton nominal frequencies of 800.13, 700.13,and 600.13 MHz, respectively. All of the triple resonance (TXI 5-mm) probes used were equipped with Pulsed Field Gradients along the z-axis.
The backbone sequential assignments were performed using through-bond heteronuclear correlation experiments; experimental details are summarized in Table 5. For triple-resonance experiments, quadrature detection in the indirect dimensions was performed in the Echo/Antiecho-TPPI mode (Marion and Wüthrich 1983).
Table 5.
Acquisition parameters for NMR experiments performed on EZnM2E133QSOD
| Dimension of acquired data (nucleus) | Spectral width (ppm) | ||||||
| Experiments | t1 | t2 | t3 | F1 | F2 | F3 | na |
| [1H-1H]-NOESYb | 102 (1H) | 1024 (1H) | 25 | 25 | 64 | ||
| 1H-15N-HSQCb | 256 (15N) | 1024 (1H) | 38 | 7 | 16 | ||
| CBCA(CO)NHb | 144 (13C) | 70 (15N) | 66 | 38 | 15 | 16 | 63 |
| DBCANHb | 144 (13C) | 70 (15N) | 1024 (1H) | 66 | 38 | 15 | 16 |
| HNCOb | 144 (13C) | 70 (15N) | 1024 (1H) | 66 | 38 | 15 | 16 |
| HN(CA)COb | 144 (13C) | 70 (15N) | 1024 (1H) | 66 | 38 | 15 | 16 |
| 13CHCCH-TOCSYc | 208 (1H) | 144 (13C) | 1024 (1H) | 15 | 74 | 15 | 16 |
| CC(CO)NH-TOCSYc | 128 (15C) | 64 (15N) | 1024 (1H) | 80 | 38 | 15 | 16 |
| 15C-edited [1H-1H]-NOESYb | 256 (1H) | 128 (13C) | 1024 (1H) | 10 | 80 | 10 | 16 |
| 15N-edited [1H-1H]-NOESYb | 320 (1H) | 64 (15N) | 1024 (1H) | 10 | 40 | 7 | 16 |
| HNHAb | 144 (1H) | 128 (15N) | 1024 (1H) | 15 | 48 | 15 | 32 |
a Number of acquired scans.
b Data acquired on a 700-MHz spectrometer.
c Data acquired on an 800-MHz spectrometer.
Two two-dimensional NOESY experiments (80 msec mixing time) (Wider et al. 1984) were carried out to identify connectivities involving histidines of the binding sites at 298 and 288 K, respectively. A 1H–15N HSQC experiment (Bodenhausen et al. 1980) with a spectral width of 13 ppm centered at 12 ppm for the proton resonances and of 36 ppm centered at 175 ppm for the nitrogen resonances was recorded to assign the 15N resonances of histidine rings.
Distance constraints for structure determination were obtained from a 15N-edited and a 13C-edited three-dimensional NOESY–HSQC experiment (Wider et al. 1989) (see Table 5) with a mixing time of 80 msec and from two-dimensional NOESY. HCCH–TOCSY (Kay et al. 1993) and CC(CO)HN–TOCSY (Gardner et al. 1996) experiments allowed the assignment of the proton and carbon side-chain resonances. 3JHNHα-coupling constants were determined by the HNHA experiment (Vuister et al. 1993) (Table 5). Histidine coordination mode was determined by 1H–15N heteronuclear experiments at 298 and 283 K, by detecting the 2J 1H–15N coupling between the imidazole nitrogen and nonexchangeable imidazole protons (Bertini et al. 1994).
The backbone assignment was facilitated by using the automatic assignment program GARANT (Bartels et al. 1997) run on a Linux cluster processor. Input consisted of three peak-picked three-dimensional spectra CBCANH, CBCACONH, and HNCO (Kay et al. 1990) without an input structure. The program assigned 70% of the backbone resonances. The backbone assignment was then completed manually. Stereospecific assignments of diastereotopic protons were obtained using the program GLOMSA (Güntert et al. 1991).
Relaxation experiments were collected on a Bruker Avance 600 spectrometer. The 15N backbone longitudinal relaxation rates, R1, were measured as described previously (Kay et al. 1992) using 12 different delays (T) in the pulse sequence ranging from 5 to 2250 msec. The 15N backbone transverse relaxation rates, R2, were measured with the CPMG sequence as described elsewhere (Kay et al. 1992) using a refocusing delay (τCPMG) of 450 μs. R2 rates were also measured at six different τCPMG lengths from 450 to 1150 μs, to monitor their dependence on weak effective fields (Kay et al. 1992; Peng and Wagner 1994). Relaxation delays varied from 7 to 290 msec, the exact values depending on τCPMG. Heteronuclear 1H–15N NOEs were measured using reported methodologies (Grzesick and Bax 1993).
The exchangeability of the backbone amide hydrogens with solvent protons was investigated through a series of 1H–15N HSQC experiments performed along 5 d on the EZnM2E133QSOD protein previously frozen-dry and then dissolved in D2O.
For all of the experiments, quadrature detection in the indirect dimensions was obtained in the TPPI mode Marion and Wüthrich (1983), and water suppression was achieved through WATERGATE sequence (Piotto et al. 1992), except for the heteronuclear 1H–15N NOE experiments in which flip back pulses were used. The two-dimensional spectra for measuring R1, R2, and 1H–15N NOEs two-dimensional spectra consisted of 2K data points in the acquisition dimension and of 256 experiments in the indirect dimension; 8 scans were collected for each experiment.
All of the spectra were analyzed with the XEASY program (Eccles et al. 1991). Integration of cross-peaks in the R1, R2, and 1H–15N NOEs measurements was performed by using the standard routine of the XWINNMR program.
The NOESY cross-peak volumes were converted into upper-distance limits through the program CALIBA (Güntert et al. 1991), in which the calibration curves were adjusted iteratively during the structure calculations. For the protons of the histidine ligands, an independent calibration was used. The elements of secondary structure were determined on the basis of the chemical shift index (CSI) (Wishart and Sykes 1994), of the 3JHNHα-coupling constants and of the backbone NOEs. The CSI analysis provided the dihedral φ and ξ angles according to the nature of the secondary structural elements (Gagné et al. 1994). Backbone dihedral φ angles were also derived from 3JHNHα-coupling constants through the Karplus equation (Vuister et al. 1993). Backbone dihedral ξ angles for residue (i-1) were also determined from the ratio of the intensity of the dαN(i-1,i) and dNα(i,i) NOEs, present on the 15N (i) plane of residue (i) in the 15N NOESY–HSQC.
Structure calculations were performed using DYANA (Güntert et al. 1997). A total of 400 random conformers were annealed in 18,000 steps using NOE and dihedral angles constraints. The zinc ion was included in the structural calculations and energy minimization by adding a new residue in the amino acid sequence following a procedure already reported by us (Banci et al. 1998). The presence of the disulfide bridge between Cys 57 and Cys 146 was checked through the analysis of the 13C shifts of the Cβ of the cysteines. Hydrogen-bond constraints were introduced at later steps of structure calculations for backbone amide protons that were within hydrogen-bond distance and have the correct orientation with respect to hydrogen-bond acceptors in structures calculated without inclusion of these constraints (for Supplementary material, see www.proteinscience.org). The distance of the NH proton and the nitrogen with the oxygen atom acceptor were constrained in the 1.8–2.4 Å and in the 2.6–3.3 Å intervals by inclusion of the corresponding upper and lower distance limits.
The final family consists of 35 conformers with the lowest target function. REM was then applied within the molecular mechanics and dynamic module SANDER of the AMBER package (Pearlman et al. 1997) on each member of the DYANA family. The value of NOE and torsion-angle potentials was calculated with force constants of 134 kJ mole−1 Å−2 and 209 kJ mole−1 rad−2, respectively.
The program CORMA (Borgias et al. 1989), which is based on relaxation matrix calculations, was used to back calculate the NOESY cross-peaks from the calculated structure to check the consistency of the analysis. The quality of the structure was evaluated through Ramachandran plots using the programs PROCHECK-NMR (Laskowski et al. 1996). Experimental restraints were analyzed using the program AQUA (Laskowski et al. 1996, 1998).
The average minimized structure of EZnM2E133QSOD is available at the protein Data Bank, PDB entry 1KMG and RCSB ID RCSB015109.
Relaxation data analysis
R 1 and R2 relaxation rates were determined by fitting the cross-peak volumes measured as a function of the delay (T) in the suitable pulse sequences, to a single exponential decay by using the Levenburg-Marquardt algorithm (Marquardt 1963). The errors on the rates were evaluated with a Monte Carlo approach (Palmer III et al. 1991).
The experimental relaxation rates (R1, 1H–15N NOE and R2 with τCPMG of 450 μs) were analyzed with the Model-Free 4.0 program (Mandel et al. 1995), within the Lipari-Szabo approach (Lipari and Szabo 1982). According to this analysis (Mandel et al. 1995), the rates are expressed in terms of spectral density functions J(ω), which depend on the overall rotational correlation time τm, on the order parameter S2, and on the correlation time for internal motions, which can be considered as arising from two components, one describing faster (τf) and one slower (τs) motions (collectively called τe), but always faster than τm.
By use of the three-dimensional structure of EZnM2E133QSOD, determined in the present research, the rotational diffusion tensor can be estimated from the R2/R1 ratio. Once the best model for the molecular motions is selected on the basis of an F-statistical test (Mandel et al. 1995) of the Model-Free analysis, the overall τm, the ratio D∥/D⊥, and the internal motion parameters for each spin are optimized by fitting the experimental relaxation parameters R1, R2, and NOE to their equations.
The presence of exchange contributions in R2 was evaluated by measuring the R2 dependence on the τCPMG length in the CPMG R2 experiments.
Acknowledgments
This work was supported by European Community (Contract Number HPRI-CT-1999-00009 and QLG2-CT-1999-01003), by Italian CNR (Progetto Finalizzato Biotecnologie 99.00286.PF49 and 99.00950.CT03), and by MIUR-ex 40%.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
CCS, copper chaperone for SOD
SOD, superoxide dismutase
M2SOD, Phe50Glu, Gly51Glu superoxide dismutase
M2E133QSOD, Phe50Glu, Gly51Glu, Glu133Gln superoxide dismutase
M4SOD, Phe50Glu,Gly51Glu, Val148Lys, Ile151Lys superoxide dismutase
EZnM2E133QSOD, empty, zinc M2E133QSOD
TPPI, time-proportional phase incrementation
NOESY, nuclear Overhauser effect spectroscopy
HSQC, heteronuclear single quantum coherence
TOCSY, total correlation spectroscopy
CPMG, Carr-Purcell-Meiboom-Gill
WATERGATE, water suppression by gradient-tailored excitation
NOE, Nuclear Overhauser effect
CSI, chemical shift index
REM, restrained energy minimization
R1, longitudinal relaxation rate
R2, transverse relaxation rate
RMSD, root-mean-square deviation
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0210802.
References
- Arnesano, F., Banci, L., Bertini, I., Huffman, D.L., and O'Halloran ,T.V. 2001. Solution structure of the Cu(I) and Apo forms of the yeast metallochaperone, Atx1. Biochemistry 40 1528–1539. [DOI] [PubMed] [Google Scholar]
- Banci, L., Bertini, I., Chium, C.Y., Mullenbach, G.T., and Viezzoli, M.S. 1995. Synthesis and characterization of a monomeric mutein of Cu/Zn superoxide dismutase with partially reconstituted enzymatic activity. Eur. J. Biochem. 234 855–860. [DOI] [PubMed] [Google Scholar]
- Banci, L., Bertini, I., Viezzoli, M.S., Argese, E., Orsega, E., Choi, Y.C., Mullenbach, G.T. 1997. Tuning the activity of Cu,Zn superoxide dismutase through site directed mutagenesis: A relatively active monomeric species. J. Biol. Inorg. Chem. 2 295–301. [Google Scholar]
- Banci, L., Benedetto, M., Bertini, I., Del Conte, R., Piccioli, M., and Viezzoli, M.S. 1998. Solution structure of reduced monomeric Q133M2 copper, zinc superoxide dismutase. Why is SOD a dimeric enzyme? Biochemistry 37 11780–11791. [DOI] [PubMed] [Google Scholar]
- Banci, L., Bertini, I., Del Conte, R., Mangani, S., Viezzoli, M.S., and Fadin, R. 1999. The solution structure of a monomeric reduced form of human copper, zinc superoxide dismutase bearing the same charge as the native protein. J. Biol. Inorg. Chem. 4 795–803. [DOI] [PubMed] [Google Scholar]
- Banci, L., Bertini, I., Cramaro, F., Del Conte, R., Rosato, A., and Viezzoli, M.S. 2000. Backbone dynamics of human Cu, Zn superoxide dismustase and of its monomeric F50/EG51E/E133Q mutant: The influence of dimerization on mobility and function. Biochemistry 39 9108–9118. [DOI] [PubMed] [Google Scholar]
- Banci, L., Bertini, I., Cramaro, F., Del Conte, R., and Viezzoli, M.S. 2002. The solution structure of reduced dimeric copper zinc SOD: The structural effects of dimerization. Eur. J.Biochem. 269 1905–1915. [DOI] [PubMed] [Google Scholar]
- Bartels, C., Güntert P., Billeter, M., and Wüthrich, K. 1997. GARANT - A general algorithm for resonance assignment of multidimensional nuclear magnetic resonance spectra. J. Comp. Chem. 18 139–149. [Google Scholar]
- Beem, K.M., Rich, W.E., and Rajagopalan, K.V. 1974. Total reconstitution of copper-zinc superoxide dismutase. J. Biol. Chem. 249 7298–7305. [PubMed] [Google Scholar]
- Bertini, I., Piccioli, M., Viezzoli, M.S., Chiu, C.Y., and Mullenbach, G.T. 1994. A spectroscopic characterization of a monomeric analog of copper-zinc superoxide dismutase. Eur. J. Biophys. 23 167–176. [DOI] [PubMed] [Google Scholar]
- Bertini, I., Mangani, S., and Viezzoli, M.S. 1998. Structure and properties of copper/zinc superoxide dismutases. In: Advanced inorganic chemistry. (ed. A.G. Sykes), pp. 127–250. Academic Press, San Diego, CA.
- Bodenhausen, G. and Ruben, D.J. 1980. Natural abundance nitrogen-15 NMR by enhanced heteronuclear spectroscopy. Chem. Phys. Lett. 69 185–188. [Google Scholar]
- Borgias, B., Thomas, P.D., and James, T.L. 1989. COmplete Relaxation Matrix Analysis (CORMA). University of California, San Francisco, CA.
- Culotta, V.C., Klomp, L.W, Strain, J., Casareno, R.L., Krems, B., and Gitlin, J.D. 1997. The copper chaperone for superoxide dismutase. J. Biol. Chem. 272 23469–23472. [DOI] [PubMed] [Google Scholar]
- Eccles, C., Güntert, P., Billeter, M, and Wüthrich, K. 1991. Efficient analysis of protein 2D NMR spectra using the software package EASY. J. Biomol. NMR 1 111–130. [DOI] [PubMed] [Google Scholar]
- Ferraroni, M., Rypniewski, W., Wilson, K.S., Viezzoli, M.S., Banci, L., and Mangani, S. 1999. The crystal structure of the monomeric human SOD mutant F50/G51E/E133Q at atomic resolution. The enzyme mechanism revisited. J. Mol. Biol. 288 413–426. [DOI] [PubMed] [Google Scholar]
- Fisher, C.L., Cabelli, D.E., Tainer, J.A., Hallewell, R.A., and Getzoff, E.D. 1994. The role of Arginine 143 in the electrostatic and mechanism of Cu,Zn superoxide dismutase: Computational and experimental evaluation of site-directed mutants. Proteins Struct. Funct. Genet. 19 24–34. [DOI] [PubMed] [Google Scholar]
- Gagné, R.R., Tsuda, S., Li, M.X., Chandra, M., Smillie, L.B., and Sykes, B.D. 1994. Quantification of the calcium-induced secondary structural changes in the regulatory domain of troponin-C. Protein Sci. 3 1961–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner, K.H., Konrat, R., Rosen, M.K., and Kay, L.E. 1996. An (H)C(CO)NH-TOCSY pulse scheme for sequential assignment of protonated methyl groups in otherwise deuterated 15N, 13C-labeled proteins. J. Biomol. NMR 8 351–356. [DOI] [PubMed] [Google Scholar]
- Getzoff, E.D., Cabelli, D.E., Fisher, C.L., Parge, H.E., Viezzoli, M.S., Banci, L., and Hallewell, R.A. 1992. Faster superoxide dismutase mutants designed by enhancing electrostatic guidance. Nature 358 347–351. [DOI] [PubMed] [Google Scholar]
- Goto, J.J., Zhu, H., Sanchez, R.J., Gralla, E.B., and Valentine, J.S. 2000. Loss of in vitro metal ion binding specificity in mutant copper-zinc superoxide dismutase associated with familial amyotrophic lateral sclerosis. J. Biol. Chem. 141007–1014. [DOI] [PubMed] [Google Scholar]
- Graden, J.A., Ellerby, L.M., Roe, J.A., and Valentine, J.S. 1994. Role of the bridging histidyl imidazolate ligand in yeast copper-zinc superoxide dismutase. Characterization of the His63Ala mutant. J. Am. Chem. Soc. 116 9743–9744. [Google Scholar]
- Grzesiek, S. and Bax, A. 1993. The importance of not saturating H2O in protein NMR. Application to sensitivity enhancement and NOE measurements. J. Am. Chem. Soc. 115 12593–12594. [Google Scholar]
- Güntert, P., Braun, W., and Wüthrich, K. 1991. Efficient computation of three-dimensional protein structures in solution from Nuclear Magnetic Resonance data using the program DIANA and the supporting programs CALIBA, HABAS and GLOMSA. J. Mol. Biol. 217 517–530. [DOI] [PubMed] [Google Scholar]
- Güntert, P., Mumenthaler, C., and Wüthrich, K. 1997. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol. 273 283–298. [DOI] [PubMed] [Google Scholar]
- Hall, L.T., Sanchez, R.J., Holloway, S.P., Zhu, H., Stine, J.E., Lyons, T.J., Demeler, B., Schirf, V., Hansen, J.C., Nerissian, A.M., et al. 2000. X-ray crystallographic and analytical ultracentrifugation analyses of truncated and full-length yeast copper chaperones for SOD (LYS7): A dimer-dimer model of LYS7-SOD association and copper delivery. Biochemistry 39 3611–3623. [DOI] [PubMed] [Google Scholar]
- Hart, J.P., Liu, H., Pellegrini, M., Nersissian, A.M., Gralla, E.B., Valentine, J.S., and Eisenberg, D. 1998. Subunit asymmetry in the three-dimensional structure of a human CuZnSOD mutant found in familial amyotrophic lateral sclerosis. Protein Sci. 7 545–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huffman, D.L. and O'Halloran, T.V. 2001. Function, structure, and mechanism of intracellular copper trafficking proteins. Annu. Rev. Biochem. 70 677–701. [DOI] [PubMed] [Google Scholar]
- Kay, L.E., Ikura, M., Tschudin, R., and Bax, A. 1990. Three-dimensional triple-resonance NMR spectroscopy of isotopically enriched proteins. J. Magn. Reson. 89 496–514. [DOI] [PubMed] [Google Scholar]
- Kay, L.E., Nicholson, L.K., Delaglio, F., Bax, A., and Torchia, D.A. 1992. Pulse sequences for removal of the effects of cross correlation between dipolar and chemical-shift anisotropy relaxation mechanisms on the measurement of heteronuclear T1 and T2 values in proteins. J. Magn. Reson. 97 359–375. [Google Scholar]
- Kay, L.E., Xu, G.Y., Singer, A.U., Muhandiram, D.R., and Forman-Kay, J.D. 1993. A gradient-enhanced HCCH-TOCSY experiment for recording side-chains 1H and 13C correlations in H2O samples of proteins. J. Magn. Reson. Ser. B 101 333–337. [Google Scholar]
- Koradi, R., Billeter, M., and Wüthrich, K. 1996. MOLMOL: A program for display and analysis of macromolecular structure. J. Mol. Graphics 14 51–55. [DOI] [PubMed] [Google Scholar]
- Lamb, A.L., Wernimont, A.K., Pufahl, R.A., Culotta, V.C., O'Halloran, T.V., and Rosenzweig, A.C. 1999. Crystal structure of the copper chaperone for superoxide dismutase. Nature Struct. Biol. 6 724–729. [DOI] [PubMed] [Google Scholar]
- Lamb, A.L., Torres, A.S., O'Halloran, T.V., and Rosenzweig, A.C. 2000. Heterodimer formation between superoxide dismutase and its copper chaperone. Biochemistry 39 14720–14727. [DOI] [PubMed] [Google Scholar]
- ———. 2001. Heterodimeric structure of superoxide dismutase in complex with its metallochaperone. Nat. Struct. Biol. 8 751–755. [DOI] [PubMed] [Google Scholar]
- Laskowski, R.A., Rullmann, J.A.C., MacArthur, M.W., Kaptein, R., and Thornton, J.M. 1996. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8 477–486. [DOI] [PubMed] [Google Scholar]
- Laskowski, R.A., MacArthur, M.W., and Thornton, J.M. 1998. Validation of protein models derived from experiment. Curr. Opin. Struct. Biol. 8 631–639. [DOI] [PubMed] [Google Scholar]
- Lipari, G. and Szabo, A. 1982. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity. J. Am. Chem. Soc. 104 4546–4559. [Google Scholar]
- Lippard, S.J., Burger, A.R., Ugurbil, K., Pantoliano, M.W., and Valentine, J.S. 1977. Nuclear magnetic resonance and chemical modification studies of bovine erythrocyte superoxide dismutase: Evidence for zinc-promoted organization of the active site structure. Biochemistry 16 1136–1141. [DOI] [PubMed] [Google Scholar]
- Lyons, T.J., Gralla, E.B., and Valentine, J.S. 1999. Biological chemistry of copper-zinc superoxide dismutase and its link to amyotrophic lateral sclerosis. Metal Ions Biol. Syst. 36 125–177. [PubMed] [Google Scholar]
- Lyons, T.J., Nerissian, A., Huang, H., Yeom, H., Nishida, C.R., Graden, J.A., Grolla, E.B., and Valentine, J.S. 2000. The metal binding properties of the zinc site of yeast copper-zinc superoxide dismutase: implications for amyotrophic lateral sclerosis. J. Biol. Inorg. Chem. 5 189–203. [DOI] [PubMed] [Google Scholar]
- Mandel, M.A., Akke, M., and Palmer III, A.G., 1995. Backbone dynamics of Escherichia coli ribonuclease HI: correlations with structure and function in an active enzyme. J. Mol. Biol. 246 144–163. [DOI] [PubMed] [Google Scholar]
- Marion, D. and Wüthrich, K. 1983. Application of phase sensitive correlated spectroscopy (COSY) for measurements of proton-proton spin-spin coupling constants in proteins. Biochem. Biophys. Res. Commun. 113 967–974. [DOI] [PubMed] [Google Scholar]
- Marquardt, D.W. 1963. An algorithm for least-squares estimation of nonlinear parameters. J. Soc. Ind. Appl. Math. 11 431–441. [Google Scholar]
- McCord, J.M. and Fridovich, I. 1969. Superoxide dismutase. Enzymic function for erythrocuprein. J. Biol. Chem. 244 6049–6055. [PubMed] [Google Scholar]
- O'Halloran, T.V. and Culotta, V.C. 2000. Metallochaperones: An intracellular shuttle service for metal ions. J. Biol. Chem. 275 25057–25060. [DOI] [PubMed] [Google Scholar]
- Palmer III, A.G., Rance, M., and Wright, P.E. 1991. Intramolecular motions of a zinc finger DNA-binding domain Xfin characterized by proton-detected natural abundance 13C heteronuclear NMR spectroscopy. J. Am. Chem. Soc. 113 4371–4380. [Google Scholar]
- Pantoliano, M.W., Valentine, J.S., Burger, A.R., and Lippard, S.J. 1982. A pH-dependent superoxide dismutase activity for zinc-free bovine erythrocuprein. Reexamination of the role of zinc in the holoprotein. J. Inorg. Biol. Chem. 17 325–341. [DOI] [PubMed] [Google Scholar]
- Parge, H.E., Getzoff, E.D., Scandella, C.S., Hallewell, R.A., and Tainer, J.A. 1986. Crystallographic characterization of recombinant human CuZn superoxide dismutase. J. Biol. Chem. 261 16215–16218. [PubMed] [Google Scholar]
- Parge, H.E., Hallewell, R.A., and Tainer, J.A. 1992. Atomic structures of wild-type and thermostable mutant recombinant human Cu,Zn superoxide dismutase. Proc. Natl. Acad. Sci. 89 6109–6114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearlman, D.A., Case, D.A., Caldwell, J.W., Singh, U.C., Weiner, P.K., Seibel, G.L., Cheatham III, T.E., Ross, W.S., Simmerling, C.L., Darden, T.A., et al. 1997. AMBER 5.0. University of California, San Francisco, CA.
- Peng, J.W. and Wagner, G. 1994. Investigation of protein motions via relaxation measurements. Methods Enzymol. 239 563–596. [DOI] [PubMed] [Google Scholar]
- Piotto, M., Saudek, V., and Sklenar, V. 1992. Gradient-tailored excitation for single quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR 2 661–666. [DOI] [PubMed] [Google Scholar]
- Rabizadeh, S., Gralla, E.B., Borchelt, D.R., Gwinn, R., Valentine, J.S., Sisodia, S., Wong, P., Lee, M., Hahn, H., and Bredesen, D.E. 1995. Mutations associated with amyotrophic lateral sclerosis convert superoxide dismutase from an antiapoptotic gene to a proapoptotic gene: Studies in yeast and neural cells. Proc. Natl. Acad. Sci. 92 3024–3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rae, T., Schmidt, P.J., Pufahl, R.A., Culotta, V.C., and O'Halloran, T.V. 1999. Undetectable intracellular free copper: The requirement of a copper chaperone for superoxide dismutase. Science 284 805–808. [DOI] [PubMed] [Google Scholar]
- Rae, T.D., Torres, A.S., Pufahl, R.A., and O'Halloran, T.V. 2001. Mechanism of Cu,Zn-superoxide dismutase activation by the human metallochaperone hCCS. J. Biol. Chem. 276 5166–5176. [DOI] [PubMed] [Google Scholar]
- Rosenzweig, A.C. and O'Halloran, T.V. 2000. Structure and chemistry of the copper chaperone proteins. Curr. Opin. Chem. Biol. 4 140–147. [DOI] [PubMed] [Google Scholar]
- Rosenzweig, A.C., Huffman, D.L., Hou, M.Y., Wernimont, A.K., Pufahl, R.A., and O'Halloran, T.V. 1999. Crystal structure of the Atx1 metallochaperone protein at 1.02 Å resolution. Structure Fold Des. 7 605–617. [DOI] [PubMed] [Google Scholar]
- Schmidt, P.J., Rae, T.D., Pufahl, R.A., Hamma, T., Strain, J., O'Halloran, T.V., and Culotta, V.C. 1999. Multiple protein domains contribute to the action of the copper chaperone for superoxide dismutase. J. Biol. Chem. 274 23719–23725. [DOI] [PubMed] [Google Scholar]
- Schmidt, P.J., Kunst, C., and Culotta, V.C. 2000. Copper activation of superoxide dismutase 1 (SOD1) in vivo. Role for protein-protein interactions with the copper chaperone for SOD1. J. Biol. Chem. 275 33771–33776. [DOI] [PubMed] [Google Scholar]
- Torres, A.S., Petri, V., Rae, T.D., and O'Halloran, T.V. 2001. Copper-stabilized heterodimer of the yCCS metallochaperone and its target superoxide dismutase. J. Biol. Chem. 276 38410–38426. [DOI] [PubMed] [Google Scholar]
- Valentine, J.S. and Gralla, E.B. 1997. Delivering copper inside yeast and human cells. Science 278 817–818. [DOI] [PubMed] [Google Scholar]
- Vuister, G.W. and Bax, A. 1993. Quantitative J correlation: A new approach for measuring homonuclear three-bond J(HNHα) coupling constants in 15N enriched proteins. J. Am. Chem. Soc. 115 7772–7777. [Google Scholar]
- Wider, G., Macura, S., Kumar, A., Ernst, R.R., and Wüthrich, K. 1984. Homonuclear two-dimensional 1H NMR of proteins. Experimental procedures. J. Magn. Reson. 56 207–234. [Google Scholar]
- Wider, G., Neri, D., Otting, G., and Wüthrich, K. 1989. A heteronuclear three-dimensional NMR experiment for measurements of small heteronuclear coupling constants in biological macromolecules. J. Magn. Reson. 85 426–431. [Google Scholar]
- Wishart, D. and Sykes, B.D. 1994. The 13C chemical shift index: A simple method for the identification of protein secondary structure using 13C chemical shift data. J. Biomol. NMR 4 171–180. [DOI] [PubMed] [Google Scholar]