

Abstract
Previous studies have shown that reduced hen egg white lysozyme refolds and oxidizes according to a linear model, in which the number of disulfide bonds increases sequentially. In this study, we describe the kinetics of native tertiary structure formation during the oxidative-renaturation of reduced hen egg white lysozyme, as monitored using an immunochemical pulsed-labeling method based on enzyme-linked immunosorbent assay (ELISA) in conjuction with two monoclonal antibodies (mAb). Each of these antibodies recognizes a separate face of the native lysozyme surface and, more importantly, each epitope is composed of discontinuous regions of the polypeptide chain. Renaturation kinetics were studied under the same refolding conditions as previous investigations of the kinetics of the regain of far-UV CD, fluorescence, enzymatic activity, and disulfide bonds. Comparison of our results with the results from those studies showed that the immunoreactivity (i.e., the native fold) of the α-domain appeared in intermediates containing two SS bonds only (C6–C127 and C30–C115), while the immunoreactivity of the β-domain appeared together with the formation of the third SS bond (C64–C80). Thus, the α-domain folds before the β-domain during the oxidative folding of reduced lysozyme.
Keywords: Reduced lysozyme, disulfide, folding kinetics, ELISA, monoclonal antibody
Hen egg white lysozyme (HEWL) is a convenient model for protein-folding studies. It is a small (129 amino acid) globular enzyme that catalyzes the hydrolysis of polysaccharide chains in bacterial cell walls. It contains eight cysteines that form disulfide bonds between residues C6–C127, C30–C115, C64–C80, and C76–C94; in its oxidized form, it represents one of the most extensively characterized refolding systems available for study. The structure of the native protein comprises two domains, α and β, and was first solved using X-ray crystallography by Blake et al. in 1965. The α domain contains four α-helices and a 310-helix; the β-domain consists of a second 310-helix as well as two antiparallel β-strands and an irregular loop.
Previous studies using stopped-flow technology have revealed that the refolding of oxidized unfolded lysozyme proceeds through several intermediates and is complete in about 1 sec (Radford et al. 1992). However, the folding of oxidized lysozyme is not the same as that which occurs in vivo, where folding begins with cysteines reduced; thus, our interest in the refolding of reduced lysozyme. Whereas the steps in the refolding of oxidized lysozyme occur on a millisecond time scale, those in the refolding of reduced lysozyme occur over a period of minutes or hours. Previous studies by Roux et al. (1999) proposed that the refolding of reduced/denatured lysozyme is a sequential process involving three intermediate stages, I0, I1, and I2, and the native structure. The first intermediate, I0, contains one disulfide bond and has no enzymatic activity; lack of secondary structure is marked by the absence of change in far-UV ellipticity at this stage. I1 has two disulfide bonds and no activity but shows 20% of the native CD signal at 222 nm. I2 has three disulfides and shows 100% of the native CD signal and 80% of native activity. Rate constants for the formation of I0, I1, I2, and the fully refolded enzyme, N, were found to be 0.148 min−1, 0.065 min−1, 0.032 min−1, and 0.005 min−1, respectively, under experimental conditions that were optimal for the renaturation yield but not for the renaturation rate (25°C, very low redox catalyst concentration, and 0.5 M guanidinium chloride). However, these studies revealed nothing about the homogeneity of each intermediate population, in particular whether the observed secondary structure and disulfide bonds undergo further rearrangement after their appearance. Nor do they indicate the stage at which a native-like tertiary fold of the polypeptide chain is formed.
To determine the intermediate stage at which local tertiary structures exist in their native arrangement, we chose to investigate the kinetics of appearance of immunoreactivity toward monoclonal antibodies (mAb) that recognize epitopes dependent on native structure. Two such antibodies have been well characterized: D1.3 and HyHel-5. Binding fragments of each monoclonal antibody have been crystallized in complex with HEWL (Amit et al. 1986; Sheriff et al. 1987). As shown in Figure 1 ▶, the antibody epitopes are nonoverlapping, occupying separate domains; D1.3 binds the α-helical domain, while HyHel-5 binds the β-domain on the opposite surface of the enzyme. Most importantly, both epitopes are discontinuous, occupying nonadjacent portions of the amino acid chain. Therefore, for the lysozyme to be immunoreactive, residues in disparate regions of the polypeptide chain must be brought into proximity and specifically arranged according to native conformation. Using these antibodies in conjunction with a quantitative enzyme-linked immunosorbant assay (ELISA) will enable us to follow the kinetics of the appearance of local native three-dimensional structure elements in reduced/denatured lysozyme.
Fig. 1.
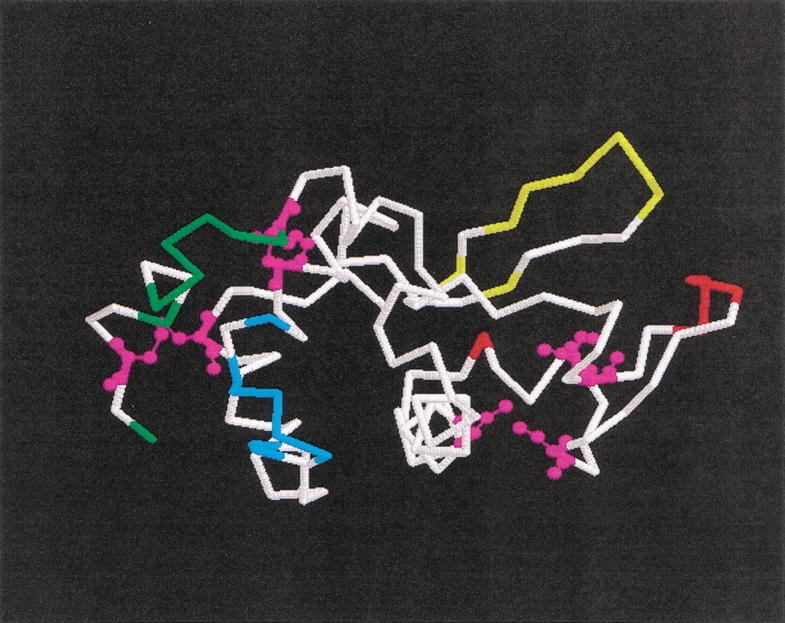
Location of the epitopes in the structure of oxidized hen egg white lysozyme. The D1.3 epitope (in the α-domain, left) is depicted in green (upwards: residues 129, 124, 121–116) and cyan (downwards: residues 27, 25–21, 19–18). The HyHel-5 epitope (in the β-domain, right) is shown in red (right to left: residues 70–67, 84) and yellow (from left side of upper strand: 41, 43–49, 53). Differing colors within each epitope signify distinct, relatively continuous, segments of the polypeptide chain. The cysteinyl residues involved in native disulfide bonds (from left to right on the figure: C6–C127, C30–C115, C76–C94, and C64–C80) are shown in magenta using ball-and-stick representation.
Results
The quantitative ELISA we used requires that the concentration of antibody bound to the plate should be proportional to the concentration of antibody in solution (Friguet et al. 1985). We first verified that this requirement was satisfied when the ELISA was performed in the presence of the same residual GuHCl concentration as in the actual immunochemical assay, using antibody concentrations ranging from 1.0 × 10−10 M to 1.6 × 10−8 M mAb. The resulting absorbances were plotted against the antibody concentration. The relationship between mAb and plate-bound lysozyme was linear up to 3 × 10−9 M D1.3 and 2 × 10−9 M HyHel-5. Based on these results, the concentration of antibody used during competition ELISA—2.0 × 10−9 M for D1.3, and 1.5 × 10−9 M for HyHel-5—was selected from well within the upper limits of linearity. The concentration of antigen originally selected was that which resulted in approximately 70–80% recognition of the antigen after incubation with the chosen amount of antibody (4 μg/mL for incubations with D1.3, and 1.33 μg/mL for incubations with HyHel-5). Percent recognition in the competition ELISA was defined as
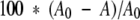 |
(1) |
where A0 and A represent the absorbances at 405 nm of the antibody in the absence of antigen, and of the absorbance of the antibody/antigen mixtures, respectively. Early results with D1.3 displayed considerable scattering for refolding times exceeding 2 h (data not shown), presumably because the antibody in solution was too close to saturation. To increase precision, lysozyme samples refolded/oxidized for more than 2 h were therefore diluted to 1.33 μg/mL in the mixture with mAb D1.3. As later discussed, the concentration of antigen always far exceeded that of the antibody, a condition essential for a simple quantitative interpretation of the ELISA data (Friguet et al. 1985). Lastly, controls demonstrated that neither antibody recognized lysozyme in its reduced/denatured form.
mAb-Lysozyme equilibrium dissociation constants in the presence of 0.5 M GuHCl
Determinations of the equilibrium dissociation constants were performed multiple times and the data treated according to Klotz (1953). Figure 2 ▶ shows results typically obtained during these experiments. Under these conditions, D1.3 was found to have an average dissociation constant of 84 ± 30 nM, compared with literature values (in the absence of GuHCl) of 3.5 nM determined either by fluorescence quenching (Foote and Winter 1992) or by microcalorimetry (Tello et al. 1993) and of 2.3 nM determined by the same competition ELISA method as that used here (Tello et al. 1990). An average value of 13.9 ± 1.8 nM was obtained for HyHel-5, markedly higher than literature values ranging between 6.7 and 25 pM (Lavoie et al. 1990; Xavier et al., 1998) obtained in the absence of GuHCl by PCFIA and fluorescence anisotropy, respectively.
Fig. 2.

Klotz plots of the binding of native lysozyme to mAb D1.3 and mAb HyHel-5. Binding of native oxidized lysozyme at various concentrations to the antibodies was measured by solution phase competition ELISA at room temperature (Friguet et al. 1985). The concentration of mAb D1.3 was 2.0 × 10−9 M, and that of mAb HyHel-5 was 1.5 × 10−9 M. The reciprocal of the fraction recognition [defined as (Ao − A)/Ao, where Ao is the absorbance at 405 nm in the absence of free antigen, and A is the absorbance in the presence of antigen] is shown as a function of the reciprocal of the antigen concentration. The straight lines result from fitting by linear regression which, in these examples, provided dissociation constants of 95.5 nM for D1.3 (crossed squares) and 13.3 nM for HyHel-5 (filled triangle).
Immunopulsed labeling of refolding HEWL
Using the procedure and timing as detailed in Materials and Methods, aliquots of unfolded-reduced HEWL were diluted in renaturation buffer, incubated at 20°C for the required refolding time, and rapidly centrifuged. Samples were diluted twofold with the desired antibody, incubated for 5 min, transferred to an ELISA plate, and the enzyme-linked immunosorbent assay brought to completion. The average absorbance readings resulting from triplicate measurements for each sample were used to determine the concentration of free antigenic lysozyme at "τ," the time of addition of antibody to refolding samples, using the following formula derived from the Klotz equation:
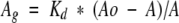 |
(2) |
where Ao represents the average absorbance in the presence of mAb but absence of antigen; A represents the absorbance of the mAb/refolded antigen sample; and Kd the equilibrium dissociation constant determined above. To render results independent of the different antigen concentrations and any fluctuations in precision of dilutions, buffer pH, and other assay conditions, an internal standard of native lysozyme at a known concentration was incubated alongside the refolding samples and included on each ELISA plate. The concentration of refolded antigenic lysozyme was expressed as a fraction of the concentration of native lysozyme, calculated from the internal control in the same manner.
Refolding time corrections
To determine the concentration of immunoreactive antigen as a function of the effective renaturation time, it was necessary to take into account the fact that the antigen continued to fold both during preincubation of the antigen/antibody mixture in solution and during incubation on the plate.
To understand the influence of the continued refolding on our results, each antibody was preincubated with native lysozyme for varying lengths of time (20 sec, 1, 2, 3, 4, 5, and 6 min) before it was transferred to the ELISA plate. We found that the amount of free antibody in solution did not vary significantly over this preincubation time range, indicating that the antibody–antigen association equilibrium was reached nearly instantaneously. Therefore, any antigenic molecules that formed during the preincubation period (i.e., after the time of addition of the mAb, defined as time τ) influenced the measurements. To compensate for this effect, the 5-min preincubation time was added to τ in the calculation of the refolding time.
- Refolding also continued during the 5-min incubation of the antibody/antigen mixture on the plates. Because it was impossible to quantify the amount of antigenic lysozyme that appeared during this time, an estimation was made according to the following rationale: (a) the amount of mAb immobilized, in the absence of free antigen, on the ELISA plate by binding to the coated lysozyme was analyzed as a function of incubation time on the plate and found to be linear, as the amount of immobilized antibody increased proportionally with increasing incubation times. Therefore, immobilized antibody, B, was proportional to a constant k and to time (t). Under our experimental conditions, the concentration of immobilized mAb, B, also varied directly with the concentration of unbound (i.e., not bound to antigen at equilibrium in the preincubation mixture) mAb, NSτ, present at time τ in solution. The amount of immobilized antibody may therefore be expressed as
The rate of antibody binding at time τ, the time at which the antibody was introduced, is equal to the time derivative of the above equation: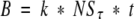
(3)
(b) However, due to the increase in antigenic lysozyme resulting from continued folding during the 5-min incubation, the concentration of unbound antibody decreased with time. Using a linear interpolation with a factor of α, then the amount of unbound antibody present after t minutes of incubation in the well may be written: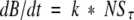
(4)
where θ represents the time of introduction of the antigen/antibody mixture in the well (i.e., τ + 5). (c) Equation 4 may now be expressed as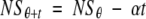
(5)
(d) Integration of Equation 6 from time θ to θ + 5 yields the amount of antibody bound during the 5-min incubation on the ELISA plate: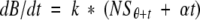
(6)
where (NSθ+t + NSθ)/2 may be interpreted as the average of the unbound antibody concentrations present at the start and end of the incubation in the well. Within the precision of the linear interpolation, this average is equal to the amount of unbound antibody present at time θ + 2.5 min, so the data was shifted an additional 2.5 min toward longer incubation times to take into account the further folding that occurs in the well. Thus, a total of 7.5 min (5 for the preincubation in solution and 2.5 to compensate for the incubation on the plate) was added to each refolding time.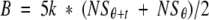
(7)
Kinetics of renaturation
The amount of immunoreactive lysozyme in each aliquot was calculated according to Equation 1 and plotted against refolding time (including the 7.5-min adjustment made for refolding during the preincubation in solution and incubation on the plate). The results are shown in Figure 3 ▶. As expected, refolding was reflected by an overall increase in the concentration of immunoreactive lysozyme for both D1.3 and HyHEL-5. The formation of both antigenic determinants displayed an initial lag time followed by a fast phase, although due to the 12.5-min dead time of the experiment, the exact "shape" of the lag remains in question. The lack of data prior to 12.5 min reflects the limitations imposed by centrifugation and preparation of the refolding aliquots immediately prior to the antigen/antibody mixtures, as well as the previously discussed adjustment to the calculation of refolding times. In both sets of experiments, the concentration of free immunoreactive lysozyme reached a maximum of approximately 60% of the concentration of native lysozyme included in the internal control. However, given that mAb D1.3 had a native lysozyme recognition level of approximately 75%, the actual refolding yield may have been nearer to 80%. Likewise, the mAb HyHel-5 recognized approximately 85% of native lysozyme, so the actual refolding yield of the HyHel-5 epitope may have been closer to 70%. As a control, refolding aliquots were assayed for activity according to the methods of Roux et al. (1997); we found that they displayed about 75% of the activity level of native lysozyme incubated under the same conditions, in fair agreement with the yields in immunoreactivity obtained from the two mAbs. Comparing the kinetics of regain of antigenicity of the two epitopes revealed that the refolding of the D1.3 antigenic determinant occurred approximately 2.5 times faster than that of the HyHel-5 epitope. Attempts to fit the kinetics of regain of immunoreactivity to a single exponential resulted in negative values when the fit curve was extrapolated to time zero, indicating that the kinetics exhibit a lag and therefore should not be fit using a single exponential. Unfortunately, mAb D1.3 data taken from refolding samples with shorter incubation times displayed considerable scattering; this may be due in part to the fact that each time point was obtained from a different refolding sample, reflecting difficulties in performing quantitatively reproducible renaturation experiments with low concentrations of immunoreactive antigen.
Fig. 3.

Kinetics of immunoreactivity regain during lysozyme oxidative folding. The fraction of lysozyme immunoreactive towards mAbs D1.3 (crossed squares) and HyHel-5 (filled triangles), expressed in fraction of immunoreactive native lysozyme at the same total protein concentration, was determined by immunochemical pulsed-labeling (see text), and is plotted as a function of the renaturation time. The time indicated in abscissa was corrected (as indicated in the Results section) for the folding that occurs during centrifugation, preincubation in solution, and incubation on the ELISA plate. The curves correspond to the simulations of the appearance of 2SS (short dashed line) and 3SS (long dashed line) intermediates obtained from the rate constant determined by Roux et al. (1999), and to the kinetics of recovery of native ellipticity at 222 nm (continuous line) obtained from the data of Roux et al. (1997). When several measurements were performed with mAb D1.3 at the same renaturation time in distinct experiments, the data point represented corresponds to the average value, and the error bars indicate the extreme values, obtained for that incubation time.
The data obtained were compared with the appearance of 2SS and 3SS intermediate species (Fig. 3 ▶), as simulated using the rate constants determined by Roux et al. (1999). Curves were constructed by simulating a linear three-step model, assuming that intermediates prior to the species in question made no contribution to the signal. Conversely, subsequent species were assumed to contribute 100%, because the lysozyme–antibody interaction continues in 3SS and 4SS species. We found that the D1.3 antigenic determinant appeared after the formation of refolding intermediates with two disulfide bonds, but prior to the formation of a third disulfide bond. D1.3 immunoreactivity was not observed to appear either prior to the appearance of the second disulfide or after the third. Early formation of the HyHel-5 epitope was remarkably coincident with the simulated appearance of intermediates with three or more disulfide bonds.
The regain of native ellipticity at 222 nm was also plotted in Figure 3 ▶. To normalize the kinetics to the same scale as the immunoreactivity, we used the rate constants and intermediate dichroic signals values obtained by Roux et al. (1997), which resulted from an excellent fitting to the experimental curve, and normalized the plateau to the final concentration of antigenic lysozyme. The simulated recovery of native ellipticity was observed to occur approximately midway between the formation of the 2SS and 3SS refolding intermediates, and appeared to present a much more accurate "fit" to the D1.3 data than the appearance of either the 2SS or 3SS species.
Immunoreactivity of 2SS variants
Because the kinetics of appearance of immunoreactivity to mAb D1.3 preceded the formation of species with 3SS bonds, it was concluded that some species with 2SS bonds should be recognized by that antibody. Recent results (Tachibana et al. 2001; Bai and Peng 2001) indicated that the α-domain may exist as an independent folding domain at equilibrium, displaying both native-like secondary and tertiary structure. Moreover, Noda et al. (2002) reported that a 2SS variant with native disulfides C6–C127 and C30–C115 displayed native-like secondary and tertiary structure in the α-domain. Because the epitope recognized by mAb D1.3 is carried by the α-domain of lysozyme, these results suggested that formation of at least one of the native SS bonds within the α-domain (C6–C127 and/or C30–C115) might be involved in the immunoreactivity to mAb D1.3. To identify the disulfides involved in the appearance of immunoreactivity, the immunoreactivity of 2SS lysozyme variants carrying either the C6–C127 or the C30–C115 disulfide bond or both was investigated. The immunoreactivities of the five 2SS variants containing at least one native disulfide in the α-domain were therefore studied, using mAb D1.3 as a probe. Their affinities for mAb D1.3 were determined using the ELISA method (Friguet et al. 1985). The dissociation constant obtained for the variant with disulfides C6–C127 and C30–C115 was 170 ± 53 nM, compared to 84 ± 30 nM (see above) for native lysozyme. Because of the very small amounts of variant protein available, its concentration after solubilization could not be measured directly and was estimated only approximately. We therefore consider the affinities of the wild-type and variant lysozymes to be very similar. Conversely, the four 2SS variants with only one intra α-domain SS bond failed to give rise to any detectable specific binding to mAb D1.3, up to concentrations of 300 nM. For comparison, native lysozyme at concentrations about 100-fold lower gave rise to a clear, unambiguous binding signal. These results indicated that the native epitope recognized by this antibody was not present on these variants. Similar studies with mAb HyHel-5 failed to reveal any significant specific interaction between that antibody and any of the 2SS variants investigated.
Discussion
The immunochemical pulsed-labeling method used in this study enabled us to monitor the kinetics of regain of antigenicity during the oxidative renaturation of reduced hen lysozyme. The regain of antigenicity could provide valuable structural information only because monoclonal antibodies endowed with the following properties were available: (a) they had to be "conformation dependent," that is, to bind strongly to the native protein but not to interact with the denatured polypeptide chain; (b) they had to recognize epitopes located far from each other on the protein surface, and hence, "probe" distinct regions of the protein surface; (c) each epitope had to be made of several residues carried by regions of the polypeptide chain that are distant in the sequence, and hence should be immunoreactive only if these regions of the polypeptide chain adopt a fold and relative positioning that closely resemble those present in the native protein. In the case of mAbs D1.3 and HyHEL-5, the first property was verified by the lack of reactivity of reduced lysozyme (see Results) and the two latter were ascertained by examination of Figure 1 ▶, which summarizes the relevant results of X-ray crystallographic studies of the two antigen–antibody complexes. Thus, that the two epitopes regain their antigenicities with affinities equivalent to those of the native enzyme warrants that the two cognate regions have regained their native-like fold. Because of the specificity of the two mAbs we used, and of the high sensitivity of ELISA methods, valuable information could be obtained on the kinetics of local 3D structure recovery under conditions where no other structural method was applicable. Indeed, neither X-ray crystallography nor NMR could be applied to a rapidly evolving, heterogeneous, and highly diluted mixture like that present during lysozyme oxidative folding.
Some constraints inherent to the immunochemical pulsed-labeling method we used will now be discussed. An obvious drawback of the procedure we used is its relatively long dead time. One could have envisaged bypassing this problem by blocking the refolding process, either through changing the buffer (pH, . . .) or by blocking the unreacted cysteines with a specific reagent like N-ethylmaleimide. We did not do so in order not to perturb the conformation and/or immunoreactivity of the folding intermediates. Indeed, folding intermediates are often quite unstable, and their conformation (and hence their immunoreactivity) might easily be perturbed by changes in the environment. Moreover, because cysteines are present in the vicinity of the two epitopes investigated, we feared that introducing a chemical reagent on such cysteines might affect the interaction between the antibodies and the blocked intermediate, thus masking the appearance of folded epitopes. Hence, our choice to perform all the immunochemical pulsed-labeling and assay procedure in the refolding buffer and in the absence of a blocking agent. The rather long dead time in our procedure results from the need to centrifuge the mixture after the renaturation, to preincubate the antibody/antigen mixture in solution, and to incubate it in the ELISA plate. Because reduced hen lysozyme has a strong tendency to form aggregates at early stages of renaturation even at low protein concentrations (Goldberg et al. 1991), and because we observed that these aggregates adhere strongly to the ELISA plate, resulting in an irreproducible background signal, these aggregates had to be removed by centrifugation which, when including introduction of the samples in the rotor, centrifugation and removal of the samples, resulted in a delay of about 3 min. With proteins less prone to aggregation, or less "sticky," this centrifugation step might be omitted to minimize the dead time. The second important component of the dead time was the time of preincubation of the antibody with the antigen in solution. We arbitrarily chose an incubation time of 5 min. On the basis of available data on the kinetics of association of lysozyme with mAbs (Braden et al. 1995; Xavier and Willson 1998) this ensured that association would be completed at the end of the preincubation. Retrospectively, this incubation could have been shortened considerably because, within the precision of the ELISA measurement, no difference was detected between samples preincubated for periods ranging between 30 sec and 5 min. Reducing this preincubation period to 30 sec would have reduced by half the dead time of the experiment. The last component of the dead time stemmed from the incubation of the antigen/antibody mixture in the ELISA well. Although this incubation could have been somehow shortened, the signal of "bound" antibody would have been reduced and, hence, the sensitivity and precision of the measurement diminished. Thus, even under favorable conditions where no centrifugation would be needed and the preincubation time would be reduced to 30 sec, the dead time would still be about 3 min. We consider this as the lower limit of the dead time of such immunochemical pulsed-labeling ELISA techniques, which therefore can be used only for studying rather slow processes. In view of the rather low rate we observed for the immunoreactivity regain during lysozyme oxidative folding, we did not attempt to reach this lower limit.
Another difficulty of the immunochemical pulsed-labeling method resides in the quantification of the immunoreactive species. Indeed, this quantification relies on the value of the affinity of the antibody for the antigenic protein. In early studies using antibodies as conformational probes for protein folding, it was widely accepted that the antigen is either nonreactive, or has the same affinity for the antibody as the native protein (Sachs et al. 1972; Furie et al. 1975). However, detailed studies with monoclonal antibodies indicated that this is not always the case (Friguet et al. 1986; Goldberg et al. 1999), and that intermediate affinities can be observed for some forms of the antigenic protein. Because we used the affinities of the monoclonal antibodies for native lysozyme in our calculations, the validity of our estimates of the immunoreactive intermediate concentrations must be discussed. Let us assume that there exists a partly folded intermediate endowed with an affinity Kint for the antibody lower than that, Knat, of the native protein. According to Equation 2 (see Results section), if one uses Knat instead of Kint in the calculation of the immunoreactive species calculations, the absorbance decrease (I0 − A) produced in the ELISA by the low-affinity complex would result in an underestimate of the partly folded species concentration by a factor Knat/Kint. Thus, the contribution of low affinity intermediates to the estimate of the concentration of immunoreactive species corresponds to only about Kint/Knat times the concentration of the same amount of species with the native affinity. Thus, any intermediate with an affinity below about 1/10th that of native lysozyme will be barely detectable in the assay used. Because no immunoreactivity whatsoever was detected at the 1 disulfide stage, because the 2SS variant with the 6–127 and 30–115 disulfide bonds had an affinity for mAb D1.3 indistinguishable from that of native lysozyme, because the other 2SS variants tested showed no detectable interaction with mAb D1.3, and because the kinetics of immunoreactivity regain could be fitted with a sequential model in which the entire amplitude change occurred during a unique phase, we can safely conclude that, within the precision of our analysis, intermediates with low affinity did not significantly affect our quantitative analysis. Although perhaps present during lysozyme oxidative folding, such intermediates did not accumulate in any detectable amount. Therefore, the quantitative analysis leading to the data of Figure 3 ▶ should represent a good approximation of the amount of species with native-like epitopes.
Effect of GuHCl on equilibrium dissociation constants
The presence of GuHCl considerably increased the equilibrium dissociation constants for interactions between lysozyme and mAbs D1.3 and HyHel-5. These variations may be attributed to the ionic and/or chaotropic effects of guanidine on the antibody/antigen binding. In this case, the increase in equilibrium dissociation was advantageous in that it contributed to the precision of the ELISA analyses by permitting the concentration of antibody to be raised despite the experimental requirements of the ELISA competition method. One requirement is to maintain the antigen concentrations in large excess compared to the antibody to allow for the quantitative analysis of the ELISA while the other is to maintain the antigen concentration not much higher than the equilibrium dissociation constant, to prevent antibody saturation (Friguet et al. 1985).
Implications for oxidative refolding of lysozyme
Past studies on the refolding of lysozyme with its disulfide bonds intact indicated that the tertiary structure of the α-domain forms before that of the β-domain (Radford et al. 1992), and it has been proposed that the partially folded intermediate on the slow-folding pathway has a nativelike α-domain but an unfolded β-domain (Wildegger and Kiefhaber 1997). Results presented here show that the D1.3 epitope in the α-domain refolded prior to the formation of the HyHel-5 epitope in the β-domain. This indicates that, as in the refolding of oxidized HEWL, native-like secondary and tertiary structure is achieved first in the α-domain and then in the β-domain during oxidative refolding of reduced HEWL. Thus, while occurring on radically different time scales, the refolding of reduced and of oxidized HEWL seem to obey common patterns.
Although it has been previously shown that 2SS intermediates occur well prior to 3SS intermediates, little characterization of the 2SS intermediates had been attempted until very recently. It is therefore of interest to note that, as indicated in Figure 3 ▶, the D1.3 antigenic determinant appeared between the development of species with two or more disulfide bonds and the formation of species with three or more disulfide bonds, thus demonstrating a lag time between the formation of the second SS bond and the formation of the native surface structure in the α-domain. This lag suggests that the "I1" intermediate, as defined by Roux et al. (1999), is a heterogeneous population that actually comprises several conformers, some without sufficient native-like structure to form the antigenic determinant, and others in which the polypeptide chain acquires at least the local secondary and tertiary configurations necessary for epitope formation. Strikingly, the recovery of native ellipticity at 222 nm, indicative of secondary structure formation, also fell between the formation of the second and third disulfide intermediates. From the similarities of the rate constants obtained for CD and disulfide bond recoveries, Roux et al. (1999) concluded that native ellipticity was recovered with the formation of the third disulfide intermediate. The more careful analysis achieved by the simulations reported here clearly indicate that the recovery of native ellipticity occurs prior to the formation of a third disulfide. Furthermore, the kinetics observed for the recovery of native ellipticity appeared to fit well with the kinetics of appearance of the D1.3 epitope—which itself includes elements of α-helix secondary structure.
There are several possible explanations for the delay between the formation of a second disulfide bond and the appearance of the D1.3 epitope. Because it has been demonstrated that the formation of native-like secondary and tertiary structures in lysozyme occurs rapidly once native disulfide bonds have been formed (Goldberg and Guillou 1994; Guez et al. 2002), it is unlikely that the lag is due merely to the gradual formation and isomerization of native structures within the 2SS species. More likely, the lag would result from disulfide exchange within the 2SS population. Thus, the 2SS intermediate as defined by Roux et al. (1999) may initially contain at least one non-native disulfide bond, because research by Tachibana et al. (2001) demonstrated that lysozyme variants containing one or more non-native disulfide bonds show little evidence of secondary structure, and no evidence of tertiary structure. Alternatively, the initial 2SS intermediate population may contain native disulfides that fail to induce or stabilize native-like structure within the α-domain. In contrast, some 2SS species with native disulfides that could accumulate at a later stage through disulfide exchange may have a native-like α-domain. This is illustrated by the 2SS variant with the C6–C127 and C30–C115 disulfides, which exhibits a strong near-UV CD and contains a native-like α-domain (Tachibana et al. 2001), indicating that the presence of these disulfides suffices to promote the formation of the native structure in the α-domain. This is confirmed by our observation that this variant has an affinity for mAb D1.3 similar to that of native wild-type lysozyme. The failure reported here of mAb D1.3 to react with the four variants containing either the C6–C127/C64–C80, or the C30–C115/C64–C80, or the C6–C127/C76–C94, or the C30–C115/C76–C94 pairs of disulfides, lead us to conclude that the D1.3 epitope carried by the α-domain, results from the formation of the C6–C127 and C30–C115 disulfides. Thus, as this particular pair of "structure-determining" native bonds within the α-domain prevails over non-native, or native but nonstructure-determining bonds, native spatial configurations and interactions are fostered among amino acid residues, the D1.3 epitope is formed, and mAb binding occurs. Such a model obeys what Welker et al. (2001b) referred to as a quasi-stochastic folding mechanism, in which disulfide bonds form first, thereby engendering the environments necessary for the secondary and tertiary structures to arise. Neither the proximity of cysteines in the α-domain (residues 6, 30, 115, and 127 with native bonds existing as C6–C127 and C30–C115), nor the exchange-permissive conditions under which this experiment was performed (Roux et al. 1999; near-neutral pH, redox mixture), would preclude this hypothesis. Further support, albeit highly speculative, might come from the data reported by Roux et al. (1997), who found a 10% difference in amplitude at the 2SS intermediate stage between the CD signal corresponding to native-like structure at 222 nm and the signal corresponding to the contribution of disulfide bonds at 228 nm, compared to the signal of oxidized/refolded lysozyme. If not due to margins of error, this difference may be indicative of an initially non-native arrangement of disulfide bonds.
Another hypothesis that might account for the apparent lag between the formation of the second SS bond and that of the D1.3 epitope assumes that only native bonds are formed during lysozyme oxidative folding, but that these bonds are formed independently of each other, in a nonsequential order and with different rates of formation. If D1.3 were to bind only after the formation of two specific disulfide bridges, then the two possible orders in which these bridges could form would give rise to two different phases in the kinetics of antigenicity regain. Moreover, while the beginning of both phases would essentially correspond to the oxidation of a 1SS into a 2SS intermediate, the second specific disulfide would also, at later times, be formed inside species already containing two SS bonds, one involved in the antigenicity and the other uninvolved, thus yielding 3SS species. Intuitively, these combined effects might well explain why the kinetics of appearance of the D1.3 epitope would be intermediate between those of appearance of 2SS and 3SS species. However, when this situation was simulated using Dynafit software (data not shown) the resulting fit was reasonable, but the simulated rate constants for the formation of species with two, three, and four disulfides varied by three to five orders of magnitude from the constants published by Roux et al. (1999). Therefore, it is more likely that the lag between the formation of a second disulfide bond and the appearance of D1.3 antigenicity is due to disulfide bond rearrangement.
Unlike the refolding of the D1.3 antigenic domain, the appearance of the HyHel-5 epitope in the β-sheet domain coincides with the simulated appearance of the third disulfide bond. Because our experiments concerning refolding during the incubation period, as well as the available HyHel-5 association rate constants (Xavier and Willson 1998) suggested that HyHel-5/lysozyme association may be considered nearly instantaneous, it is highly plausible that the residues involved in antigenicity attain their native conformation with the formation of the third disulfide bond. Evidence from studies by Noda et al. (2002) demonstrated that although a native-like β-hairpin does in fact exist in the β-domain of the 2SS intermediate containing bonds C6–C127 and C30–C115, most of the β-domain is disorganized. This fits with our observations that HyHel-5 was not observed to bind well prior to the formation of the third disulfide, and confirms that this third disulfide is crucial to the formation of the HyHel-5 epitope. There are two disulfides in close proximity to the HyHel-5 epitope, C64–C80 and C76–C94, both of which could be involved in the appearance of the immunoreactivity towards HyHel-5. However, the 3SS intermediate recognized by this antibody clearly corresponds to the 3SS intermediate that accumulates during the oxidative refolding of HEWL and was shown to be that lacking the C76–C94 disulfide bond (des-[76–94]) (van den Berg et al. 1999). Furthermore, analysis using circular dichroism studies and NMR spectroscopy (van den Berg et al. 1999) revealed that des-[76–94] contains highly native-like tertiary structure in the α-domain, and, to a more limited extent, in the β-domain as well—specifically, the partial formation of the antiparallel β-sheet. Several of the residues in this sheet, including Thr43, Asn44, Arg45, Asn46, and Tyr53, have also been identified as participating in the HyHel-5 antigenic determinant (Smith-Gill et al. 1982). Taken together, these observations clearly indicate that the appearance of the immunoreactivity to HyHel-5 depends on the formation of disulfide bond between cysteines 64 and 80. Because this disulfide bond is inside the β-domain and close to the HyHel-5 epitope, it could have been inferred that formation of this disulfide might suffice to fold the β-domain and structure the epitope. However, our failure to detect any interaction between HyHel-5 and the two 2SS variants containing this disulfide (and either the C6–C127 or the C30–C115 disulfide) indicate that formation of the C64–C80 disulfide is not sufficient in itself to ensure the proper folding of the β-domain. Rather, it appears that proper folding of the α-domain, and presumably interactions between the two domains, are needed for the proper folding and/or stabilization of the β-domain.
The 75% activity levels observed in controls of incubated lysozyme were consistent with the maximum refolding yield of approximately 80% observed by Roux et al. (1997), and the 70–80% antigen refolding yield achieved in the D1.3 experiment. In contrast, the 60–70% maximum seen in HyHel-5 experiments was slightly lower than expected. Roux et al. (1999) observed the presence of a trapped 3SS species at the end of the renaturation process; this population consisted of incompletely or misfolded molecules, and accounted for approximately 20–30% of the initial lysozyme concentration. Thus, the lower final regain of imunoreactivity observed with mAb HyHel-5 may originate from a lack of reactivity of this population of misfolded 3SS species.
In conclusion, using as a conformational probe the immunoreactivity of HEWL toward two monoclonal antibodies recognizing discontinuous epitopes on the α- and β-domains, the following dominant pathway can be proposed for the oxidative refolding of hen lysozyme. Populations of intermediates with one, two, and three disulfide bonds accumulate sequentially prior to the formation of the native enzyme (Roux et al. 1999). Species with one disulfide form rapidly, but fail to exhibit stable secondary and tertiary structures. Next, heterogeneous 2SS species form and isomerize to yield molecules containing two native disulfide bonds (disulfides C6–C127 and C30–C115) and native-like secondary and tertiary structures in the α-domain, although the β-domain remains poorly organized. The C64–C80 disulfide bond appears next, together with the native-like tertiary structure in the β-domain, either by direct oxidation of cysteines 64 and 80 or indirectly through disulfide exchange involving a non-native or the C76–C94 transient disulfide. Finally, formation of the last disulfide bond (C76–C94) locks together and stabilizes the two folded domains to provide the native protein.
It is important to point out that the proposed pathway is expected to account for the folding of a majority of, but probably not all, the lysozyme molecules. It is likely that some molecules follow different pathways. Indeed, the refolding conditions used throughout this study were initially chosen on the basis of two requirements: produce high recoveries of native lysozyme by avoiding aggregation, and allow far UV CD measurements to be done by keeping the far UV absorbance reasonably low (Roux et al. 1997). As indicated in a previous report (Roux et al. 1999) these conditions result in the oxidation rate being low, thus providing time for rapid intramolecular disulfide reshuffling to occur and preventing the accumulation of the least stable intermediates within the 1SS, 2SS, and 3SS populations. Also, the presence of 0.5 M GuHCl in the renaturation buffer may be sufficient to affect marginally stable intermediates, and hence modify the folding pathway significantly. These are presumably the reasons why some intermediates previously identified by other groups (Anderson and Wetlaufer, 1976; van den Berg et al. 1999), although likely to be transiently formed, do not appear as significantly populated species under the conditions used in the present study. Furthermore, the possibility that molecules would fold via the 3SS intermediates lacking the C64–C80 disulfide is not ruled out by our experiments. However, several pieces of evidence indicate that the 3SS species that accumulates under our experimental conditions is essentially that lacking the C76–C94 disulfide. Indeed, its far UV ellipticity and specific activity (Roux et al. 1999) are identical to those reported for the pure, 3SS species lacking the C76–C94 bond (van den Berg et al. 1999). Moreover, the kinetics of oxidation of the 3SS into the 4SS species can be well fit with a monophasic model (Roux et al. 1999). This would not be the case if significant amounts of the des[64–80] species were present, because this 3SS intermediate was reported to oxidize to the 4SS native molecule much more rapidly than the des[76–94] species (van den Berg et al. 1999). These considerations rule out that the des[64–80] species might be a major component of the 3SS intermediate that accumulates under our experimental conditions.
According to our results, the model proposed by Welker et al. (2001a) to account for the folding of a one-domain protein might be extended to lysozyme, a two-domain protein. Indeed, it fits well the proposed lysozyme pathway, in which folding of the α-domain occurs first and coincides with the accumulation of the C6–C127 and C30–C115 disulfides. This "disulfide secure species" (according to the terminology of Welker et al. 2001a) would protect the formed disulfide bonds against reshuffling and allow the formation of the two last bonds by oxidation and rapid exchange among the remaining four cysteines. Thus, the independent early folding of the α-domain would enhance the regeneration and make the des[64–80] and des[76–94] pathways more populated than the two other des pathways. A similar mechanism might apply inside the β-domain, where the formation of the C64–C80 disulfide bond would lead to the 3SS disulfide secure des[76–94] species accounting for its prevalence over the des[64–80] as a 3SS folding intermediate.
In conclusion, despite their limited resolution as conformational probes, monoclonal antibodies have provided valuable structural informations on a variety of lysozyme folding intermediates and clarified the main oxidative-folding pathway leading to native HEWL under a defined set of renaturation conditions.
Materials and methods/experimental procedures
Materials
Reagents were purchased from Merck unless stated otherwise. Native HEWL (muramidase) and oxidized glutathione (GSSG), were purchased from Boehringer Mannheim; immunoconjugate Ig-alkaline phosphatase directed against mouse immunogloblins was purchased from BioSYS; DTT, from Research Organics Inc.; Guanidine HCl, from ICN Biomedicals Inc; Tris base ("Trizma base") was purchased from Sigma, as was alkaline phosphatase substrate, pNPP, in the form of a Fast Kit.
Preparation of reduced/denatured lysozyme, and subsequent renaturation, was performed as described by Roux et al. (1999). Protein concentrations were determined spectrophotometrically using ɛ280 values of 2.63 cm2/mg and 2.37 cm2/mg for native and reduced/denatured lysozyme, respectively.
Monoclonal antibodies
Anti-lysozyme D1.3 was a gift from Ginette Boulot and Roberto Poljak. Anti-lysozyme HyHel-5 was prepared as described in Xavier et al. (1997).
2SS Lysozyme variants
The preparation of 2SS variant (C6–C127, C30–C115) has been described (Tachibana et al. 2001). The gene for 2SS variant (C6–C127, C64–C80) was constructed by recombining a 216 bp Csp45I-MluI fragment derived from the 3SS variant (C76A, C94A) gene (Tachibana et al. 1994), with a 2837-bp Csp45I- MluI fragment derived from a plasmid in which the 3SS variant (C30A,C115A) gene was incorporated between the HindIII and BamHI sites of a pUC18 vector. The former fragment spans the polypeptide region from E35 to N106 with replacements of C76A and C94A, and the latter fragment contained the gene portion for K1 to F34 as well as that for A107 to L129 with replacements of C30A and C115A. The genes for the 2SS variants (C30–C115, C64–C80), (C6–C127, C76–C94), and (C30–C115, C76–C94) were constructed as described above for the (C6–C127, C64–C80) variant using, respectively, the 3SS variant (C76A, C94A) and (C6S, C127A) genes, (C64A, C80A) and (C30A,C115A) genes, and (C64A, C80A) and (C6S, C127A) genes. Nucleotide sequences of the variant genes were confirmed with dideoxy sequencing. Direct expression of the gene and purification of the polypeptide from inclusion bodies as reduced form were carried out as described previously. The oxidative folding reactions for the 2SS variants (C6–C127, C64–C80), (C30–C115, C64–C80), (C6–C127, C76–C94), and (C30–C115, C76–C94) were carried out as described for 2SS variant (C6–C127, C30–C115) in the presence of 30% glycerol, except that the reaction temperature and time were 20°C for 3 h for (C6–C127, C64–C80), and 10°C for 6 h for the other three variants. The reoxidized materials were purified with RPHPLC and freeze dried. The disulfide linkage patterns in the purified 2SS variants were analyzed as described previously, and confirmed to be native. The freeze-dried proteins were dissolved and incubated for 30 min in 6 M GuHCl (pH 5.2) at concentratrions ranging between 0.1 and 0.25 mg/mL. The samples were then diluted to 8–10 μg/mL in Refolding/ELISA buffer (see below) devoid of DTT and GSSG, allowed to refold at 20°C for 30 min, and used immediately for ELISA tests.
ELISA
All ELISAs were performed in triplicate as detailed in Friguet et al. (1989). Refolding/ELISA buffer contained 0.1 M TrisHCl, 1 mM EDTA, 0.5 M GuHCl, 20 μM DTT, and 60 μM oxidized GSSG, pH 8.2. Plates were coated with 100 μL per well of 5 μg/mL native lysozyme in PBS, sealed with adhesive film, and incubated overnight at 4°C.
Affinity measurements
The equilibrium dissociation constants of mAb/lysozyme complexes were determined in the presence of GuHCl by the method of Friguet et al. (1985) for both mAbs. Native lysozyme, at concentrations ranging from 4 to 0.1 μg/mL, was preincubated for 5 min with a constant amount of antibody (2.0 × 10−9 M for D1.3, and 1.5 × 10−9 M for HyHel-5). Aliquots of each lysozyme/mAb complex solution were then transfered on a lysozyme-coated ELISA plate and incubated for 5 min, and the amount of antibody that remained unsaturated at each antigen concentration was determined using a classical indirect ELISA. The same procedure was used for the 2SS variants, except for DTT and GSSG, which were omitted from all buffers. Data was treated according to Klotz (1953): the average absorbance at each antigen concentration was divided by the difference between that value and the absorbance of the internal control. When this value was plotted against the inverse of the lysozyme concentration, the slope of the resulting line indicated the dissociation constant of lysozyme with either D1.3 or HyHel-5.
Kinetics
The appearance of lysozyme immunoreactivity during oxidative refolding was monitored using quantitative competition ELISA, based on methods previously described by Friguet et al. (1984). All ELISA were performed at ambient temperature. (a) Coating: plates were coated with 100 μL per well of 5 μg native lysozyme as described above. (b) Reduced/denatured lysozyme was dissolved in 6 M GuHCl and 0.1 M acetic acid, pH 2.5, and prepared to 10 mg/mL. Oxidized lysozyme was dissolved in PBS and prepared to the same concentration. (c) Oxidative refolding was initiated by diluting 10 μL samples of reduced lysozyme with 1 mL of freshly prepared refolding buffer at varying times (0, 2, 4, and 6 h, for example), using strong vortex agitation to minimize aggregation. A control of native lysozyme at the same concentration was prepared alongside the first refolding sample. All samples were incubated at 25°C and withdrawn from the water bath at a given end-time, yielding incubations of varying lengths. (In the previous example, withdrawing all samples at 6 h would yield incubation times of 6, 4, 2, and 0 h, respectively.) Refolding samples were centrifuged for 1 min at 11.5 K rpm to reduce aggregate and diluted to twice the concentration required for the subsequent competition. Indirect ELISA was used to measure the concentration of antigenic lysozyme present in each sample in the following manner:
At a chosen time τ = "0," an aliquot of monoclonal antibody at twice the final concentration was added to an equal volume of each aliquot of refolded lysozyme. These mixtures of mAb and refolding lysozyme were then preincubated for 5 min at room temperature, a length of time sufficient for binding equilibrium to be established. Aliquots withdrawn from samples with longer refolding incubations contained a higher concentration of refolded antigenic lysozyme, and therefore a decreased amount of unbound antibody in solution. The concentration of free antibody in each mixture was measured by transferring the lysozyme/mAb mixtures to a lysozyme-coated ELISA plate and incubating them in the wells for 5 min at 25°C, after which an ELISA measurement was performed according to Friguet et al. (1985). This incubation time was kept short to prevent the dissociation of preexisting refolded lysozyme–mAb complexes, as dissociation would lead to a higher apparent concentration of unbound mAb in solution and yield incorrect measurements.
Plates were developed using alkaline phosphate immunoconjugate and pNPP substrate (Sigma Fast Kit). The absorbance of each well was measured at 405 nm using a Multiskan MS (Life Sciences International, Cergy Pontoise), and the concentration of refolded antigenic lysozyme was calculated from the absorbance as indicated in Results.
Analysis of kinetics
Simulations of the appearance of SS intermediates were performed using the rate constants reported by Roux et al. (1999) for the formation of folding intermediates (k0 = 0.148 min−1, k1 = 0.065 min−1, and k2 = 0.032 min−1) and Personal VisSim software (Visual Solutions Inc.). The regain of native ellipticity at 222 nm was simulated using the same software and the rate constants reported by Roux et al. (1997): k1 = 0.056 ± 0.068 min−1, and k2 = 0.058 ± 0.073 min−1, which well reflected the experimental data.
Acknowledgments
The authors are very grateful to Dr. Robert Poljak and Mrs. Ginette Boulot for providing the antibody D1.3 used in this study. The contribution of Mr. H. Sawano in the construction of variant lysozyme genes is gratefully acknowledged. Research in the Protein Folding and Modeling Unit was supported by funds from the Institut Pasteur and the Centre National de Reherche Scientifique (CNRS-URA2185). Research at Kobe University was partly supported by a Grant-in-Aid for Scientific Research (13558082) from the Ministry of Education, Culture, Sports, Science and Technology of Japan. Research at the University of Houston was partly supported by a grant from the R.A. Welch Foundation. N.M.J. received a grant from the BRAVO program of the Univeristy of Arizona, which was supported by NIH Grant TW00036.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
CD, circular dichroism
DTT, reduced dithiothreitol
EDTA, ethylene diamine tetracetic acid sodium salt
ELISA, enzyme-linked immunosorbent assay
GSSG, oxidized glutathione
GuHCl, guanidinium chloride
HEWL, hen egg white lysozyme
Ig, immunoglobulin
mAb, monoclonal antibody
PBS, phosphate-buffered saline
pNPP, alkaline phosphatase substrate
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0221802.
References
- Amit, A.G., Mariuzza, R.A., Phillips, S.E.V., and Poljak, R.J. 1986. Three dimensional structure of an antigen–antibody complex at 6Å resolution. Annu. Rev. Biophys. Biophys. Chem. 16 139–159. [DOI] [PubMed] [Google Scholar]
- Anderson, W.L. and Wetlaufer, D.B. 1976. The folding pathway of reduced lysozyme. J. Biol. Chem. 251 3147–3153. [PubMed] [Google Scholar]
- Bai, P. and Peng, Z. 2001. Cooperative folding of the isolated α-domain of hen egg-white lysozyme. J. Mol. Biol. 314 321–329. [DOI] [PubMed] [Google Scholar]
- Blake, C.C.F., Koenig, D.F., Mair, G.A., North, A.C.T., Phillips, D.C., and Sarma, V.R. 1965. Structure of hen egg white lysozyme. A three-dimensional Fourier analysis at 2 Å resolution. Nature 206 757–761. [DOI] [PubMed] [Google Scholar]
- Braden, B.C., Cauerhff, A., Dall’Acqua, W., Fields, B.A., Goldbaum, F.A., Malchiodi, E.L., Mariuzza, R.A., Poljak, R.J., Schwarz, F.P., Ysern, X., and Bhat, T.N. 1995. Structure and thermodynamics of antigen recognition by antibodies. Ann. N.Y. Acad. Sci. 764 315–327. [DOI] [PubMed] [Google Scholar]
- Foote, J. and Winter, G. 1992. Antibody framework residues affecting the conformation of the hypervariable loops. J. Mol. Biol. 224 487–499. [DOI] [PubMed]
- Friguet, B., Chaffotte, A.F., Djavadi-Ohaniance, L., and Goldberg, M.E. 1985. Measurements of the true affinity constant in solution of antigen–antibody complexes by enzyme-linked immunosorbent assay. J. Immunol. Methods 77 305–319. [DOI] [PubMed] [Google Scholar]
- Friguet, B., Djavadi-Ohaniance, L., and Goldberg, M.E. 1984. Some monoclonal antibodies raised with a native protein bind preferentially to the denatured antigen. Mol. Immunol. 21 673–677. [DOI] [PubMed] [Google Scholar]
- ———. 1986. Conformational changes induced by domain assembly within the β2 subunit of Escherichia coli tryptophan synthase analyzed with monoclonal antibodies. Eur. J. Biochem. 160 593–597. [DOI] [PubMed] [Google Scholar]
- ———. 1989. Immunochemial analysis of protein conformation. In Protein structure: a practical approach (ed. T. Creighton), pp. 287–310. IRL Press, Oxford.
- Furie, B., Schechter, A.N., Sachs, D.H., and Anfinsen, C.B. 1975. An immunological approach to the conformational equilibrium of staphylococcal nuclease. J. Mol. Biol. 92 497–506. [DOI] [PubMed] [Google Scholar]
- Goldberg, M.E. and Guillou, Y. 1994. Native disulfide bonds greatly accelerate secondary structure formation in the folding of lysozyme. Protein Sci. 3 883–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg, M.E., Rudolph, R., and Jaenicke, R. 1991. A kinetic study of the competition between renaturation and aggregation during the refolding of denatured-reduced egg white lysosyme. Biochemistry 30 2790–2797. [DOI] [PubMed] [Google Scholar]
- Goldberg, M.E., Schaeffer, F., Guillou, Y., and Djavadi-Ohaniance, L. 1999. Pseudo-native motifs in the noncovalent heme-apocytochrome c complex: Evidence from antibody binding studies by ELISA and microcalorimetry. J. Biol. Chem. 274 16052–16061. [DOI] [PubMed] [Google Scholar]
- Guez, V., Roux, P., Navon, A., and Goldberg, M.E. 2002. Role of individual disulfide bonds in hen lysozyme early folding steps. Protein Sci. 11 1136–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klotz, I.M. 1953. Protein interactions. In The proteins (eds. H. Neurath and K. Bailey), pp. 727–806, vol. 1. Academic Press, New York.
- LaVoie, T.B., Kam-Morgan, L.N.W., Mallett, C.P., Schilling, J.W., Prager, E.M., Wilson, A.C., and Smith-Gill, S.J. 1990. Analysis of antibody-protein interactions utilizing site-directed mutagenesis and a new evolutionary variant of lysozyme. In Use of X-ray crystallography in the design of antiviral agents (eds. W.G. Laver and G.M. Air), pp. 213–232. Academic Press, San Diego, CA.
- Noda, Y., Yokota, A., Horii, D., Tominaga, T., Tankiska, Y., Tachibana, H., and Segawa, S.I. 2002. NMR structural study of two-disulfide variant of hen lysozyme: 2SS[6–127, 30–115]—A disulfide intermediate with a partly unfolded structure. Biochemistry 41 2130–2139. [DOI] [PubMed] [Google Scholar]
- Radford, S.E., Dobson, C.M., and Evans, P.A. 1992. The folding of hen lysozyme involves partially structured intermediates and multiple pathways. Nature 358 302–307. [DOI] [PubMed] [Google Scholar]
- Roux, P., Delepierre, M., Goldberg, M.E., and Chaffotte, A.F. 1997. Kinetics of secondary structure recovery during the refolding of reduced hen egg white lysozyme. J. Biol. Chem. 272 24843–24849. [DOI] [PubMed] [Google Scholar]
- Roux, P., Ruoppolo, M., Chaffotte, A.F., and Goldberg, M.E. 1999. Comparison of the kinetics of S-S bond, secondary structure, and active site formation during refolding of reduced denatured hen egg white lysozyme. Protein Sci. 8 2751–2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs, D.H., Schechter, A.N., Eastlake, A., and Anfinsen, C.B. 1972. An immunologic approach to the conformational equilibria of polypeptides. Proc. Natl. Acad. Sci. 69 3790–3794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheriff, S., Silverton, E.W., Padlan, E.A., Cohen, G.H., Smith-Gill, S.J., Finzel, B.C., and Davies, D.R. 1987. Three-dimensional structure of an antibody–antigen complex. Proc. Natl. Acad. Sci. 84 8075–8079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith-Gill, S.J., Wilson, A.C., Potter, M., Prager, R.J., Feldman, R.J., and Mainhart, C.R. 1982. Mapping the antigenic epitope for a monoclonal antibody against lysozyme. J. Immunol. 128 314–322. [PubMed] [Google Scholar]
- Tachibana, H., Ohta, Y., Sawano, H., Koumoto, Y., and Segawa, S. 1994. Relationship between the optimal temperature for oxidative refolding and the thermal stability of refolded state of hen lysozyme three-disulfide derivatives. Biochemistry 3315008–15016. [DOI] [PubMed] [Google Scholar]
- Tachibana, H., Oka, T., and Akasaka, K. 2001. Native-like tertiary structure formation in the α-domain of a hen lysozyme two-disulfide variant. J. Mol. Biol. 314 311–320. [DOI] [PubMed] [Google Scholar]
- Tello, D., Spinelli, S., Souchon, H., Saul, F.A., Riottot, M.M., Mariuzza, R.A., Lascombe, M.B., Houdusse, A., Eisele, J.L., and Fischmann, T. 1990. Three-dimensional structure and antigen binding specificity of antibodies. Biochimie 72 507–512. [DOI] [PubMed] [Google Scholar]
- Tello, D., Goldbaum, F.A., Mariuzza, R.A., Ysern, X., Schwarz, F.P., and Poljak, R.J. 1993. Three-dimensional structure and thermodynamics of antigen binding by anti-lysozyme antibodies. Biochem. Soc. Transact. 21 943–946. [DOI] [PubMed] [Google Scholar]
- van den Berg, B., Chung, E.W., Robinson, C.V., and Dobson, C.M. 1999. Characterisation of the dominant oxidative folding intermediate of hen lysozyme. J. Mol. Biol. 290 781–796. [DOI] [PubMed] [Google Scholar]
- Welker, E., Narayan, M., Wedemeyer, W.J., and Scheraga, H.A. 2001a. Structural determinants of oxidative folding in proteins. Proc. Natl. Acad. Sci. 98 2312–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welker, E., Wedemeyer, W.J., Narayan, M., and Scheraga, H.A. 2001b. Coupling of conformational folding and disulfide-bond reactions in oxidative folding of proteins. Biochemistry 40 9059–9064. [DOI] [PubMed] [Google Scholar]
- Wildegger, G. and Kiefhaber, T. 1997. Three-state model for lysozyme folding: Triangular folding mechanism with an energeticaly trapped intermediate. J. Mol. Biol. 270 294–304. [DOI] [PubMed] [Google Scholar]
- Xavier, K.A. and Willson, R.C. 1998. Association and dissociation kinetics of anti-hen egg lysozyme monoclonal antibodies HyHEL-5 and HyHEL-10. Biophys. J. 74 2036–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xavier, K.A., Shick, K.A., Smith-Gill, S.J., and Willson, R.C. 1997. Involvement of water molecules in the association of monoclonal antibody HyHEL-5 with bobwhite quail lysozyme. Biophys. J. 73 2116–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]