

Abstract
FixJ is a two-domain response regulator involved in nitrogen fixation in Sinorhizobium meliloti. Recent X-ray characterization of both the native (unphosphorylated) and the active (phosphorylated) states of the protein identify conformational changes of the β4–α4 loop and the conserved residue Phe101 as the key switches in activation. These structures also allowed investigation of the transition between conformations of this two-component regulatory receiver domain by molecular dynamics simulations. The path for the conformational change was studied with a distance constraint directing the system from one state to the other. The simulations provide evidence for a correlation between the conformation of the β4–α4 loop and the orientation of the residue Phe101. A model presenting the sequence of events during the activation/deactivation process is discussed.
Keywords: Molecular dynamics, response regulator, conformational change, transition pathway, path exploration with distance constraints
The Sinorhizobium meliloti FixJ protein is at the top of the regulation cascade of N2 fixation. It belongs to the family of response regulator proteins that function within two-component signal transduction pathways. The sensor kinase FixL autophosphorylates under low concentrations of oxygen. The phosphoryl group is then transferred to the N-terminal receiver domain of FixJ (FixJN) at the aspartate residue at position 54, leading to activation of FixJ. The phosphorylation induced conformational change of the response regulator is responsible for the modulation of its biological function, a mechanism of activation widely shared among two-component systems (Cho et al. 2001).
To date, several native (unphosphorylated) receiver domains have been characterized structurally (Stock et al. 1989; Volz and Matsumara 1991; Stock et al. 1993; Volkman et al. 1995; Baikalov et al. 1996; Madhusudan et al. 1997; Djordjevic et al. 1998; Zapf et al. 1998; Gouet et al. 1999; Solà et al. 1999; Lewis et al. 2000). They all consist of a central five-stranded β-sheet surrounded by five α-helices (Fig. 1 ▶). The short lifetime of the phosphoaspartate has hampered structural characterization of phosphorylated response regulators. However, three-dimensional structures for four of them have been reported recently. FixJN and Spo0A receiver domains were characterized by X-ray crystallography as isolated phosphorylated species (Birck et al. 1999; Lewis et al. 1999). The NtrC receiver domain was characterized by NMR using in situ phosphorylation with low-molecular-weight phosphodonors (Kern et al. 1999). The CheY protein was characterized by X-ray crystallography and by NMR spectroscopy as analogs (BeF3 or phosphono derivatives) of the activated state (Cho et al. 2000; Halkides et al. 2000; Lee et al. 2001). Comparison of the backbone coordinates for the active and inactive states revealed that the largest changes are generally observed in the β4–α4 loop.
Fig. 1.

General view of the FixJ receiver domain. This structure corresponds to the JN structure to which a phosphoryl group has been added in silico (JN[+P]). Helices α1 to α5 are colored from green to blue, the central β sheet is orange, and the β4–α4 loop is red. Important residues are shown: Pas54, phosphorylated aspartate (phosphate group added in silico), Thr82 at the N-terminal end of the β4–α4 loop and Phe101 in the outside orientation. This figure was prepared using Bobscript (Esnouf 1997) and Raster 3D (Merritt and Bacon 1997).
In most receiver domains, the residue equivalent to position 101 in FixJ is conserved as either a phenylalanine or a tyrosine. This conserved aromatic residue, found in strand β5, is exposed to the solvent in the native X-ray structures, whereas in the activated state it is buried within a cavity created by a repositioning of helix α4. It has been shown that Phe101 plays a major role in the switch from the inactive to the active state of FixJ (Schumacher 1999). A mechanism for the activation of the FixJ receiver domain has been proposed by the comparison of the unphosphorylated and phosphorylated crystal structures (Birck et al. 1999). In this model, the conformational change is initiated, after phosphorylation, by the conserved Thr82 residue forming a hydrogen bond with the phosphoryl group. This movement of Thr82 induces a rearrangement of the β4–α4 loop that extends to helix α4, creating a cavity between the helix and the adjacent strand β5 . As a consequence, the conserved aromatic residue Phe101 rotates into the newly created cavity where its conformation is stabilized by hydrophobic interactions. In addition to the conformational change, it has been shown that phosphorylation of the FixJ receiver domain induces its dimerization (Da Re et al. 1999).
The transition pathway between the two states of FixJ cannot be assessed directly using biochemical experiments. Therefore, we carried out molecular dynamics (MD) simulations in combination with path exploration by distance constraints (PEDC) (Guilbert et al. 1995). This technique allows investigation of the mechanism of the conformational change between the unphosphorylated (FixJN) and phosphorylated (FixJN∼P) states of the FixJ receiver domain. Analysis of the trajectories revealed a correlation between the conformation of the β4–α4 loop and the Phe101 flip during the activation of the FixJ receiver domain.
Results
Using free (unconstrained) MD simulations up to 1 nsec we did not observe a conformational change corresponding to the transition between the inactive (unphosphorylated) and active (phosphorylated) states of FixJN (data not shown). This can be explained by the fact that such conformational changes usually take place on the micro- to millisecond time scale. We therefore used the PEDC procedure that consists of the addition of a constraint term to the potential energy to direct the system from one initial state towards a target structure (see Materials and Methods).
The conformational change between the two states of FixJN was investigated for both the phosphorylation and dephosphorylation pathways. Movie presentations of representative trajectories can be found at the URL http://www.toulouse.inra.fr/lbmrpm/dk/Fixj_index.html.
Phosphorylation pathway
To study the conformational change between the native and active conformations of FixJN, a phosphoryl group was added in silico to the Asp54 residue of the unphosphorylated form to give JN[+P] (see Materials and Methods). Two independent trajectories of 1200 psec each were generated as follows: The in silico phosphorylated structure (JN[+P]) was minimized to reduce conformational stress, heated to 300 K (30 psec), equilibrated (200 psec), and subjected to free molecular dynamics (200 psec). At the end of the free molecular dynamics, the potential energy reached a plateau (Fig. 2A ▶). The PEDC procedure was then applied using constraints on residues 3 to 122 to direct the structure obtained at the end of the free MD (JNI[+P]) to a target structure (JNT∼P) corresponding to the modified crystallographic structure of phosphorylated FixJN (see Materials and Methods). It is important to point out that JNI[+P] was derived from the native state of FixJN, whereas JNT∼P was derived from the active state. The mass-weighted root-mean-square (mrms) difference between JNI[+P] and JNT∼P was 3.7 Å (Fig. 2B ▶) and 3.4 Å for the first and second trajectories, respectively. Although two independent trajectories were analyzed, the results of only one are shown in the figures for clarity.
Fig. 2.

Molecular dynamics of the activation pathway before the PEDC constraint was applied: heating (30 psec), equilibration (200 psec), and free MD (200 psec) steps. (A) Potential energy in kcal•mole−1; (B) mrms distance in Å with regard to the phosphorylated target structure (JNT∼P).
The progress of the conformational transition was monitored by the mrms of the system to the final structure. As expected, the mrms decreased gradually during the time course of the simulation under PEDC constraints.
Description of the conformational change
A significant structural change between FixJN and FixJN∼P centers on the β4–α4 loop, which changes from a β-turn to an extended conformation (Birck et al. 1999). This rearrangement was monitored by measuring the distance between Cα atoms of the two extreme residues of the β4–α4 loop, that is, Thr82 and Val87 (Fig. 3A ▶), and by following the variation of pseudodihedral angle formed by Cα atoms of four consecutive amino acid residues within the loop (Fig. 3B ▶). Another functionally important change is the conformation of residue Phe101, which rotates into a hydrophobic cavity in FixJN∼P (Birck et al. 1999). To characterize its orientation we measured its χ1 dihedral angle along the trajectories. The flip between the out and in conformations was observed to occur after the rearrangement of the β4–α4 loop (Fig. 3C ▶). Finally, monitoring the distance between the γ-hydroxyl group of Thr82 and the O2 atom of the Asp54 phosphoryl group showed that these two groups come close to each other, allowing the formation of a hydrogen bond at the end of the transition (Fig. 3D ▶). The common features between the two independent trajectories allowed us to define the key events during the phosphorylation pathway. The conserved sequence of events could be summarized as follows: (1) rearrangement of the β4–α4 loop (Figs. 3A, 3B ▶); (2) followed by rotation of the side chain of residue Phe101 within the cavity (Fig. 3C ▶); finally, (3) followed by formation of a hydrogen bond between the hydroxyl group of the Thr82 residue and the O2 of the Asp54 phosphoryl group (Fig. 3D ▶). To illustrate this sequence of events, snapshots of key intermediate structures are shown in Figure 4 ▶.
Fig. 3.

Distance and angle analyses showing the sequence of events during the phosphorylation pathway. The PEDC constraint was applied either on residues 3–122 (A–D) or on residues 82–86 (E–H). (A) and (E): distance between the Cα atoms of Thr82 and Val87 (rearrangement of the β4–α4 loop); (B) and (F): pseudodihedral angle (Psdie) formed by Cα atoms of residues 83 to 86 (rearrangement of the β4–α4 loop); (C) and (G): χ1 dihedral angle of residue Phe101 (rotation of Phe101); (D) and (H): distance in Å between the hydrogen atom of the hydroxyl group of Thr82 and the O2 of the phosphoryl group (formation of a hydrogen bond). Transitions discussed in the text are indicated by black triangles.
Fig. 4.
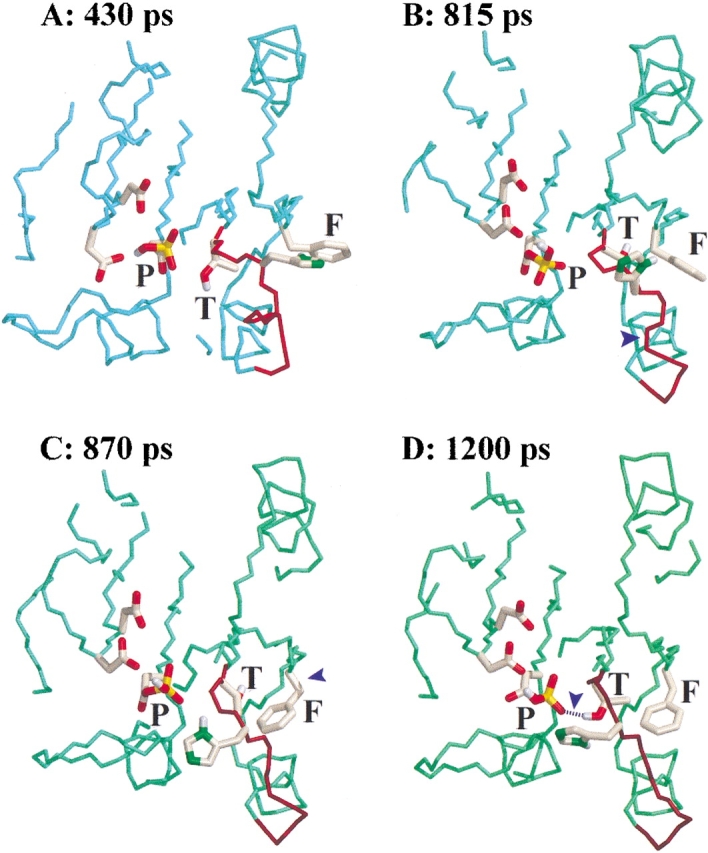
Views of four intermediate structures during the activation process. The PEDC constraint was applied to residues 3 to 122. The three residues of interest (P: phosphorylated Asp54, T: Thr82, and F: Phe101) as well as neighboring residues are highlighted. (A) Starting structure of the MD under constraints corresponding to the last structure of the free MD run (430 psec); (B) structure showing the rearrangement of the β4–α4 loop (815 psec); (C) structure after the rotation of residue Phe101 into the hydrophobic cavity (870 psec); (D) last structure of the simulation (1200 psec in which the hydrogen bond between Thr82 and the phosphate group is highlighted). Key changes are indicated with blue arrowheads.
Rotation of residue Phe101
Residue Phe101 is observed in two orientations in the X-ray structures of the native and the phosphorylated FixJ receiver domains (Birck et al. 1999). In the native structure the side chain of Phe101 adopts the gauche+ conformation (χ1 ≈ +60°) and is exposed to the solvent, whereas in the phosphorylated structure Phe101 is in the trans conformation (χ1 ≈ 180°), and is buried in a hydrophobic pocket. During the time course of the dynamics, a third conformation was observed: the gauche− conformer (χ1 ≈ −60°) (Fig. 3C ▶). In this conformation, that has already been described in the X-ray structures of PhoB and CheY (Zhu et al. 1996, 1997; Solà et al. 1999), the aromatic side chain is not completely buried inside the cavity but is less exposed to the solvent than in the gauche+ conformation.
Importance of the β4–α4 loop
To investigate the possible role of the β4–α4 loop in inducing the rotation of Phe101, we generated two new independent trajectories in which the PEDC constraint was applied solely to the loop (residues 82 to 86) to induce the transition between the β-turn and the extended conformations. The rest of the molecule was allowed to evolve freely. The loop clearly underwent the expected rearrangement during the simulation (Figs. 3E, 3F ▶), and this conformational change was rapidly followed by rotation of the side chain of residue Phe101 (Fig. 3G ▶). This indicates that the movement of residue Phe101 is dependent on the rearrangement of the loop because the flip was observed without any constraint on Phe101. As with the simulations in which the PEDC procedure was applied to residues 3 to 122, the hydrogen bond between Thr82 and the phosphate was formed stably after rotation of residue Phe101, indicating that formation of this hydrogen bond is not a prerequisite for the conformational change (Fig. 3H ▶).
Dephosphorylation pathway
To study the dephosphorylation pathway, the phosphoryl group was removed in silico from the X-ray structure of phosphorylated FixJN (see Materials and Methods). Two independent trajectories of 1200 psec each were generated as follows: The in silico dephosphorylated structure (JNP[−P]) was minimized, heated to 300 K, equilibrated, and subjected to free molecular dynamics. The PEDC method was then applied to direct the last structure of the free MD (JNI[−P]) to a target structure (JNT) derived from the crystallographic structure of FixJN (see Materials and Methods). Thus, (JNI[−P]) was derived from the active structure, whereas (JNT) was obtained from the native structure. The mrms difference between JNI[−P] and JNT was 2.7 Å and 2.85 Å for the first and second trajectories, respectively. The trajectories were analyzed as described for the phosphorylation pathway. In both trajectories, the rotation of residue Phe101 (Fig. 5A ▶) was observed prior to the rearrangement of the β4–α4 loop (Fig. 5B ▶). The latter was immediately followed by the movement of residue Thr82, leading to the disruption of the hydrogen bond between the hydroxyl group of Thr82 and the carboxylate group of Asp54 (Fig. 5C ▶).
Fig. 5.

Distance and angle analyses showing the sequence of events during the dephosphorylation pathway. The PEDC constraint was applied either on residues 3–122 (A–C) or residues 82–86 (D–F). (A) and (D): χ1 dihedral angle of residue Phe101 (rotation of Phe101); (B) and (E): distance between the Cα atoms of Thr82 and Val87 (rearrangement of the β4–α4 loop); (C) and (F): distance between the hydroxyl group of Thr82 and the CG atom of residue Asp54 (disruption of hydrogen bond). The conformational changes discussed in the text are indicated with black triangles.
The PEDC procedure was also applied specifically to residues 82 to 86 to study the importance of the β4–α4 loop. The rotation of residue Phe101 from an inward to an outward orientation was observed without any constraint on this residue (Fig. 5D ▶), which indicates a correlation between the conformation of the β4–α4 loop (Fig. 5E ▶) and that of residue Phe101. The movement of the loop is concomitant with the disruption of the hydrogen bond between Thr82 and Asp54 (Fig. 5F ▶).
Discussion
We have performed molecular dynamics simulations to study the activation/deactivation process of the FixJ response regulator. Our goal was to provide further insight into the mechanism of activation of the receiver domain. Given that the time scale accessible to free MD is not compatible with the time scale of the conformational change, we used the PEDC method to direct the system from one state to the other. This was done by applying a constraint in two different ways: either on almost the whole protein (residues 3 to 122) or only on the β4–α4 loop (residues 82 to 86). Similar results were obtained in both procedures, showing that the observed process is inherent to the system rather than to the application of specific restraints.
Analysis of the different simulations revealed a correlation between the conformation of the β4–α4 loop and the orientation of the conserved residue Phe101. In the dephosphorylation process the rotation of the aromatic residue was observed prior to the rearrangement of the loop, whereas in the phosphorylation process the flip occurred after the loop conformational change. A model can be proposed, based on the simulations for the activation/deactivation of the FixJ receiver domain (Fig. 6 ▶). This model supports the hypothesis that the β4–α4 loop acts as a switch, as proposed previously (Birck et al. 1999; Simonovic and Volz 2001). In the activation process, the rearrangement of the β4–α4 loop is a prerequisite for the rotation of residue Phe101. The trans-conformation of the loop induces the displacement of helix α4 that leads to the creation of a cavity. This hydrophobic pocket is then filled by the side chain of residue Phe101. The active conformation is finally stabilised by the formation of a hydrogen bond between Thr82 and the phosphate group. In the deactivation process, the rotation of residue Phe101 occurs prior to the rearrangement of the loop and the release of the hydrogen bond between the conserved Asp54 and Thr82. In this scheme Phe101 has to rotate before Thr82, and the β4–α4 loop can move and close the cavity.
Fig. 6.

Proposed model for the activation/deactivation of the FixJ receiver domain. Upon phosphorylation, the β4–α4 loop undergoes a conformational change from a β-turn to an extended conformation (A). This event is followed by the rotation of Phe101 (B). Finally, the loop is stabilized by the formation of a hydrogen bond between Thr82 and the phosphate group (C). After dephosphorylation, residue Phe101 can rotate to the outside (D) and the β4–α4 loop adopts a β-turn conformation that disrupts the hydrogen bond between Thr82 and Asp54 (E).
It is not known whether different receiver domains share a similar molecular switch mechanism. However, in most receiver domains, the residue equivalent to position 82 in FixJ is conserved as either a threonine or a serine, and position 101 is usually conserved as either a phenylalanine or a tyrosine. Biochemical studies have suggested that these two residues are involved in the activation of the receiver domain (Zhu et al. 1996, 1997; Appleby and Bourret 1998). In the case of FixJ, the importance of Phe101 for activation was demonstrated by the functional analysis of a Phe101 to alanine mutation (Schumacher 1999). Comparisons of receiver domain structures in the unphosphorylated and activated states show that the largest structural changes usually occur in the β4–α4 loop region. Furthermore, the orientation of the aromatic residue at position 101 (FixJ numbering) correlates with the signaling state of receiver domains in the case of CheY (Cho et al. 2000; Halkides et al. 2000), FixJ (Birck et al. 1999), and Spo0A (Lewis et al. 1999). This residue is exposed to the outside of the protein in the native state, whereas it is buried in a hydrophobic cavity in the active state (Birck et al. 1999; Lewis et al. 1999; Cho et al. 2000; Lee et al. 2001). This results in the reshaping of the active α4–β4 face of the receiver domain, thereby altering its interactions with the other domains or proteins. In the case of FixJ, this new shape is required for the formation of the FixJ dimerization interface (Birck et al. 1999).
In most response regulators that have been characterized in the active state by X-ray crystallography, the residue corresponding to Thr82 forms a hydrogen bond with the phosphoryl group. In our simulations of the activation process, formation of the hydrogen bond between the Thr82 residue and the phosphoryl group was always observed after the rotation of residue Phe101. Therefore, the formation of this hydrogen bond appears not to be a prerequisite for activation. This result is compatible with the population shift model that has been postulated for other receiver domains (Simonovic and Volz 2001; Volkman et al. 2001). In this model, the role of the phosphoryl group is not to activate the conformational change but rather to stabilize the loop in its active conformation.
In conclusion, we propose that the correlation between the conformation of the β4–α4 loop and the rotation of Phe101 is the key to the coupling between Phe101 and the phosphorylation site that underlies two-component signal transduction. In this model, the switch of the aromatic residue will depend on the flexibility of the β4–α4 loop, which appears indeed to be widely shared by two-component receiver domains (Djordjevic and Stock 1998; Feher and Cavanagh 1999; Jiang et al. 1999; Kern et al. 1999; Lewis et al. 1999; Cho et al. 2000). The active extended conformation of the β4–α4 loop is then further stabilized by hydrogen bonding of Thr82 in the phosphorylated form, thus providing the framework for a general mechanistic hypothesis for two-component signal transduction.
Materials and methods
Program used
All molecular dynamics simulations were performed with the CHARMM package (Brooks et al. 1983) in which the PEDC method was implemented, using a Silicon Graphics IRIX 6.5, Indigo R10000 workstation. VMD and Insight were used for visualization.
Preparation of the structures
Seven X-ray structures have been characterized for the native (unphosphorylated) receiver domain (Gouet et al. 1999). The structure obtained at a resolution of 2.0 Å (PDB ID: 1dck) was used in this work. Two molecules were present in the asymmetric unit for this structure; only coordinates corresponding to the B chain were used for the purpose of this study. Because Val50 presented two orientations in the initial structure (1dck), the orientation found in all other FixJN structures was used. The side-chain coordinates of Lys73 were not resolved for 1dck, and were added after superposition with another FixJN structure (1dbw). The modified structure was named JN.
The activated (phosphorylated) receiver domain was resolved at 2.3 Å (1d5w). Three molecules were present in the asymmetric unit (Birck et al. 1999), and the C protomer as defined in the original paper was chosen as representative. The unresolved side chain of Arg56 was added after superposition with the A protomer. Moreover, missing C-terminal residues (123 to 125) were added after superposition with a FixJN structure (1dck). The modified structure was named JN∼P.
Water molecules present in the crystals as well as SO42− anions and PEG were not taken into account for the simulations. All polar hydrogen atoms were included explicitly in the calculation, whereas the others were treated with a united atom model. Coordinates of polar hydrogen atoms were determined using the hbuild algorithm in CHARMM.
Energy minimization
All calculations were performed on the protein in vacuum, with the solvent being approximated using a linear distance-dependent dielectric constant. Electrostatic interactions were switched to zero for interactions beyond 10 Å. The energy of structures JN and JN∼P was minimized by 700 steps of conjugate gradient minimization. Harmonic constraints were applied to nonhydrogen atoms to allow smooth minimization. Harmonic forces were decreased every 100 steps from 250 to 0 kcal/mole•Å2.
Molecular dynamics
Molecular dynamics (MD) simulations were performed using the Shake algorithm (Ryckaert et al. 1977). The integration step was 1 fsec. A typical trajectory was prepared as follows: The minimized structures were heated gradually to 300 K (30 psec), equilibrated (200 psec), subjected to free MD (200 psec), and finally the PEDC procedure was applied (770 psec). The final structure of the free MD was considered as the initial structure in the PEDC procedure.
PEDC procedure
The PEDC procedure (Path Exploration with Distance Constraints) (Guilbert et al. 1995) involves the introduction of an additional constraint term (Vdist) to the potential energy, to force the system towards a given target structure. This potential has recently been described in detail (Mouawad et al. 2002). The distance constraint corresponds to a bi-harmonic potential with a flat-bottomed well given by the general formula:
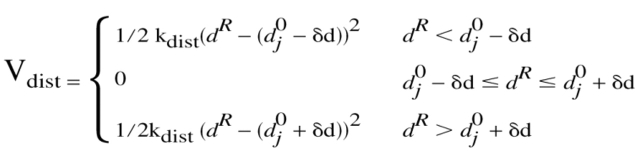 |
where kdist is the force constant, dR is the mrms between the constrained residues of the instantaneous structure and the target, d0j − δd and d0j + δd are the mrms interval that the system must reach, and j is an index corresponding to a discrete displacement. The width of the flat-bottomed well (2δd) was chosen to correspond to the average mrms fluctuation of JN (or JN∼P) at 300 K (0.8 Å). As the mrms is averaged over the whole protein (or several residues), imposing a given value of mrms (d0j) between the structure that moves and the target structure will induce conformational changes in the protein that cost the least energy while satisfying the constraint.
Each displacement lasted 55 psec, and was divided into two phases: in the first 5 psec, the kdist value was gradually increased from 200 to 106 kcal/mole•Å2 to move the system into the desired interval (driving phase). In the last 50 psec, the Vdist constraint was used with the kdist value equal to 106 kcal/mole•Å2 (confinement phase). During this latter phase, the system was not subjected to excessive forces because the structure broadly met its target and Vdist is zero within the broad well.
The PEDC procedure was applied either to residues 3–122 to study the overall conformational change (N- and C-terminal residues were not constrained because of their high flexibility) or to residues 82–86 to induce selectively the rearrangement of the β4–α4 loop. Two independent trajectories were generated for each transition. Thus, eight trajectories were generated in total to study both phosphorylation and dephosphorylation pathways.
Phosphorylation pathway
Initial structure
To study the structural transition upon phosphorylation, a phosphoryl group was added to the JN structure (1dck modified as previously described). The modified structure was named JN[+P]. Charges for the phosphoryl group were calculated with the MOPAC program (assuming a total charge of −1), and its geometrical properties were taken from the 5′-phosphate-methyl-tetrahydrofuran (T5PH) defined in CHARMM force field. The in silico phosphorylated structure (JN[+P]) was minimized, progressively heated to 300 K, equilibrated and subjected to free MD. The final structure of the free MD was considered as the initial structure (JNI[+P]) for the PEDC procedure.
Target structure
The JN∼P structure prepared as mentioned above was minimized and heated to 300 K. The final structure was used as the target (JNT∼P) in the PEDC procedure.
Dephosphorylation pathway
Initial structure
To study the dephosphorylation pathway, the phosphate group was removed in silico from the JN∼P structure. The structure was then minimized, heated, equilibrated, and subjected to free MD. The final structure of the free MD was considered as the initial structure (JNI[−P]) for the PEDC procedure.
Target structure
The JN structure was minimized and heated at 300 K. The final structure was used as the target (JNT) in the PEDC procedure.
Acknowledgments
We thank Dr. Robert M. Esnouf for helpful discussions and a critical reading of the manuscript. This work was supported, in part, by the ‘Action Informatique Mathématique Physique pour la Génomique’ (IMPG) of the French Ministry for Research.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
MD, molecular dynamics
PEDC, path exploration with distance constraints
mrms, mass-weighed root-mean-square
FixJN, nonphosphorylated, N terminal domain of FixJ
FixJN∼P, phosphorylated, N terminal domain of FixJ
JN, FixJN structure modified to allow MD studies
JN∼P, FixJN∼P structure modified to allow MD studies
JNP[−P], in silico dephosphorylated JN∼P
JNI[−P]
initial structure in the PEDC dephosphorylation pathway
JNT, target structure in the PEDC dephosphorylation pathway
JN[+P], in silico phosphorylated JN
JNI[+P], initial structure in the PEDC phosphorylation pathway
JNT∼P, target structure in the PEDC phosphorylation pathway
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0218802.
References
- Appleby, J.L. and Bourret, R.B. 1998. Proposed signal transduction role for conserved CheY residue Thr87, a member of the response regulator active-site quintet. J. Bacteriol. 180 3563–3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baikalov, I., Schroder, I., Kaczor-Grzeskowiak, M., Grzeskowiak, K., Gunsalus, R.P., and Dickerson, R.E. 1996. Structure of the Escherichia coli response regulator NarL. Biochemistry 35 11053–11061. [DOI] [PubMed] [Google Scholar]
- Birck, C., Mourey, L., Gouet, P., Fabry, B., Schumacher, J., Rousseau, P., Kahn, D., and Samama, J.P. 1999. Conformational changes induced by phosphorylation of the FixJ receiver domain. Struct. Fold. Des. 7 1505–1515. [DOI] [PubMed] [Google Scholar]
- Brooks, B., Bruccoleri, R.E., Olafson, B.D., States, D.J., Swaminathan, S., and Karplus, M. 1983. Charmm: A program for macromolecular energy minimization and dynamics calculations. J. Comput. Chem. 4 187–217. [Google Scholar]
- Cho, H.S., Lee, S.Y., Yan, D., Pan, X., Parkinson, J.S., Kustu, S., Wemmer, D.E., and Pelton, J.G. 2000. NMR structure of activated CheY. J. Mol. Biol. 297 543–551. [DOI] [PubMed] [Google Scholar]
- Cho, H.S., Pelton, J.G., Yan, D., Kustu, S., and Wemmer, D.E. 2001. Phosphoaspartates in bacterial signal transduction. Curr. Opin. Struct. Biol. 11 679–684. [DOI] [PubMed] [Google Scholar]
- Da Re, S., Schumacher, J., Rousseau, P., Fourment, J., Ebel, C., and Kahn, D. 1999. Phosphorylation-induced dimerisation of the FixJ receiver domain. Mol. Microbiol. 34 504–511. [DOI] [PubMed] [Google Scholar]
- Djordjevic, S. and Stock, A.M. 1998. Structural analysis of bacterial chemotaxis proteins: Components of a dynamic signalling system. J. Struct. Biol. 124 189–200. [DOI] [PubMed] [Google Scholar]
- Djordjevic, S., Goudreau, P.N., Xu, Q., Stock, A.M., and West, A.H. 1998. Structural basis for methylesterase CheB regulation by a phosphorylation-activated domain. Proc. Natl. Acad. Sci. 95 1381–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esnouf, R.M. 1997. An extensively modified version of Molscript that includes greatly enhanced coloring capabilities. J. Mol. Graph. 15 133–138. [DOI] [PubMed] [Google Scholar]
- Feher, V.A. and Cavanagh, J. 1999. Millisecond-timescale motions contribute to the function of the bacterial response regulator protein Spo0F. Nature 400 289–293. [DOI] [PubMed] [Google Scholar]
- Gouet, P., Fabry, B., Guillet, V., Birck, C., Mourey, L., Kahn, D., and Samama, J.P. 1999. Structural transitions in the FixJ receiver domain. Struct. Fold. Des. 7 1517–1526. [DOI] [PubMed] [Google Scholar]
- Guilbert, C., Perahia, D., and Mouawad, L. 1995. A method to explore transition paths in macromolecules. Applications to hemoglobin and phosphoglycerate kinase. Comput. Phys. Commun. 91 263–274. [Google Scholar]
- Halkides, C.J., McEvoy, M.M., Casper, E., Matsumura, P., Volz, K., and Dahlquist, F.W. 2000. The 1.9 Å resolution crystal structure of phosphono-CheY, an analogue of the active form of the response regulator, CheY. Biochemistry 39 5280–5286. [DOI] [PubMed] [Google Scholar]
- Jiang, M., Tzeng, Y.L., Feher, V.A., Perego, M., and Hoch, J.A. 1999. Alanine mutants of the Spo0F response regulator modifying specificity for sensor kinases in sporulation initiation. Mol. Microbiol. 33 389–395. [DOI] [PubMed] [Google Scholar]
- Kern, D., Volkman, B.F., Luginbuhl, P., Nohaile, M.J., Kustu, S., and Wemmer, D.E. 1999. Structure of a transiently phosphorylated switch in bacterial signal transduction. Nature 402 894–898. [DOI] [PubMed] [Google Scholar]
- Lee, S.Y., Cho, H.S., Pelton, J.G., Yan, D., Berry, E.A., and Wemmer, D.E. 2001. Crystal structure of activated CheY. Comparison with other activated receiver domains. J. Biol. Chem. 276 16425–16431. [DOI] [PubMed] [Google Scholar]
- Lewis, R.J., Brannigan, J.A., Turkenburg, J.P., Muchova, K., Barak, I., and Wilkinson, A.J. 1999. Phosphorylated aspartate in the structure of a response regulator protein. J. Mol. Biol. 294 9–15. [DOI] [PubMed] [Google Scholar]
- Lewis, R.J., Muchova, K., Brannigan, J.A., Barak, I., Leonard, G., and Wilkinson, A.J. 2000. Domain swapping in the sporulation response regulator Spo0A. J. Mol. Biol. 297 757–770. [DOI] [PubMed] [Google Scholar]
- Madhusudan, M., Zapf, J., Hoch, J.A., Whiteley, J.M., Xuong, N.H., and Varughese, K.I. 1997. A response regulatory protein with the site of phosphorylation blocked by an arginine interaction: Crystal structure of Spo0F from bacillus subtilis. Biochemistry 36 12739–12745. [DOI] [PubMed] [Google Scholar]
- Merritt, E.A. and Bacon, D.J. 1997. Raster3D photorealistic molecular graphics. Methods Enzymol. 277 505–524. [DOI] [PubMed] [Google Scholar]
- Mouawad, L., Perahia, D., Robert, C.H., and Guilbert, C. 2002. New insights into the allosteric mechanism of human hemoglobin from molecular dynamics simulations. Biophys. J. 82 3224–3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryckaert, J.P., Ciccotti, G., and Berendsen, H.J.C. 1977. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comp. Phys. 23 327–341. [Google Scholar]
- Schumacher, J. 1999. The phosphorylation induced activating mechanism of the response regulator FixJ. PhD thesis, University of Cologne, Cologne, Germany.
- Simonovic, M. and Volz, K. 2001. A distinct meta-active conformation in the 1.1 Å resolution structure of wild-type, apo-CheY. J. Biol. Chem. 276 28637–28640. [DOI] [PubMed] [Google Scholar]
- Solà, M., Gomis-Ruth, F.X., Serrano, L., Gonzalez, A., and Coll, M. 1999. Three-dimensional crystal structure of the transcription factor PhoB receiver domain. J. Mol. Biol. 285 675–687. [DOI] [PubMed] [Google Scholar]
- Stock, A.M., Mottonen, J.M., Stock, J.B., and Schutt, C.E. 1989. Three-dimensional structure of CheY, the response regulator of bacterial chemotaxis. Nature 337 745–749. [DOI] [PubMed] [Google Scholar]
- Stock, A.M., Martinez-Hackert, E., Rasmussen, B.F., West, A.H., Stock, J.B., Ringe, D., and Petsko, G.A. 1993. Structure of the Mg(2+)-bound form of CheY and mechanism of phosphoryl transfer in bacterial chemotaxis. Biochemistry 32 13375–13380. [DOI] [PubMed] [Google Scholar]
- Volkman, B.F., Nohaile, M.J., Amy N.K., Kustu, S., and Wemmer, D.E. 1995. Three-dimensional solution structure of the N-terminal receiver domain of NtrC. Biochemistry 34 1413–1424. [DOI] [PubMed] [Google Scholar]
- Volkman, B.F., Lipson, D., Wemmer, D.E., and Kern, D. 2001. Two-state allosteric behavior in a single-domain signaling protein. Science 291 2429–2433. [DOI] [PubMed] [Google Scholar]
- Volz, K. and Matsumura, P. 1991. Crystal structure of Escherichia coli CheY refined at 1.7-Å resolution. J. Biol. Chem. 266 15511–15519. [DOI] [PubMed] [Google Scholar]
- Zapf, J., Madhusudan, M., Grimshaw, C.E., Hoch, J.A., Varughese, K.I., and Whiteley, J.M. 1998. A source of response regulator autophosphatase activity: The critical role of a residue adjacent to the Spo0F autophosphorylation active site. Biochemistry 37 7725–7732. [DOI] [PubMed] [Google Scholar]
- Zhu, X., Amsler, C.D., Volz, K., and Matsumura, P. 1996. Tyrosine 106 of CheY plays an important role in chemotaxis signal transduction in Escherichia coli. J. Bacteriol. 178 4208–4215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, X., Rebello, J., Matsumura, P., and Volz, K. 1997. Crystal structures of CheY mutants Y106W and T87I/Y106W. CheY activation correlates with movement of residue 106. J. Biol. Chem. 272 5000–5006. [DOI] [PubMed] [Google Scholar]