

Abstract
The flavoenzyme dihydroorotate dehydrogenase A from Lactococcus lactis is a homodimeric protein of 311 residues/subunit, and the two active sites are positioned at a distance from the dimer interface. To promote formation of the monomeric form of the enzyme, we changed the residues involved in formation of two salt bridges formed between the residues Glu206 of the one polypeptide and Lys296 of the other polypeptide. The mutant enzymes formed inactive precipitates when cells were grown at 37°C, but remained soluble and active when cells were grown at 25°C. The salt bridges were not needed for activity, because the mutant enzymes in which one of the residues was converted to an alanine (E206A or K296A) retained almost full activity. The mutant enzymes in which the charge of one of the residues of the salt bridge was inverted (i.e., E206K or K296E) were severely impaired. The double mutant E206K-K296E, which has the possibility of forming salt bridges in the opposite orientation of the wild type, was fully active in concentrated solutions, but dissociated into inactive monomers upon dilution. The KD for the dimer to monomer dissociation reaction was 12 μM, and dimer formation was favored by the product, orotate, or by high ionic strength, indicating that the hydrophobic interactions are important for the subunit contacts. Wild-type dihydroorotate dehydrogenase A was similarly found to dissociate into inactive monomers, but with a KD for dissociation equal to 0.12 μM. These results imply that the dimeric state is necessary for activity of the enzyme.
Keywords: Nucleotide synthesis, oligomerization, quaternary structure, dissociation kinetics, oxido-reductase, flavin
Dihydroorotate dehydrogenase (DHOD) catalyzes the oxidation of dihydroorotate (DHO) to orotate (OA), which is the fourth step in de novo biosynthesis of pyrimidine nucleotides. The DHODs represent a divergent family of enzymes (Björnberg et al. 1997) and have been divided into two major classes, Classes 1 and 2. The Class 1 enzymes are soluble proteins that use small soluble electron acceptors; the Class 2 enzymes are attached to membranes and use respiratory quinones as electron acceptors (Jensen and Björnberg 1998).
Two types of Class 1 DHODs, 1A and 1B, have been defined. The Class 1A DHODs are found in Lactococcus lactis (Nielsen et al. 1996b), Enterococcus faecalis (Marcinkeviciene et al. 2000), anaerobic yeast (Nagy et al. 1992; Jordan et al. 2000), and some protozoan parasites (Pascal et al. 1983; Gao et al. 1999). They are dimeric proteins that use fumarate, quinones, and O2 as electron acceptors (Rowland et al. 1997a; Björnberg et al. 1999, 2001). The DHODs of Class 1B are prevalent in Gram-positive bacteria such as L. lactis (Nielsen et al. 1996a), E. faecalis (Marcinkeviciene et al. 1999), Bacillus subtilis (Kahler et al. 1999), and Clostridium oroticum (Argyrou et al. 2000). They are heterotetrameric proteins (PyrDb2 PyrK2) which utilize a subunit belonging to the ferredoxin reductase family (PyrK) to channel the electrons from the oxidation of DHO to NAD+ (Nielsen et al. 1996a; Rowland et al. 2000).
We have investigated dihydroorotate dehydrogenase A (DHODA) from L. lactis as a representative for the Class 1A DHODs. This enzyme contains one molecule of FMN bound to each subunit (Nielsen et al. 1996b), and the crystal structure of the protein is known (Rowland et al. 1997b; 1998). Each subunit folds into an (α/β)8-barrel structure, and the active site is formed by several loops that protrude from the top of the barrel. When bound in the active site, the substrate dihydroorotate is stacked with the isoalloxazine ring of FMN in an orientation favorable for hydride-transfer from the C6 position of DHO to the N5-position of FMN (Rowland et al. 1998; Fraaije and Mattevi 2000; Palfey et al. 2001). The active site base, Cys130, initiates the reaction by abstracting the 5-proS proton from DHO (Björnberg et al. 1997, 2001), to facilitate hydride transfer to the flavin. The pKa of the 5-proS proton of DHO is 20–21 (Argyrou and Washabaugh 1999). However, many tight hydrogen bonds between the substrate and totally conserved asparagine side chains of the enzyme, notably Asn132 and Asn193, which interact with the O4 position of DHO, may serve to increase the acidity of the 5-proS proton in the enzyme-substrate complex (Fraaije and Mattevi 2000; Björnberg et al. 2001). Furthermore, the reaction may be facilitated by the interaction of Lys43 with the flavin, which stabilizes the negative charge that develops on the isoalloxazine ring upon hydride transfer (Björnberg et al. 1997; Jiang et al. 2000).
The two subunits of DHODA are related by a noncrystallographic twofold axis. Twenty-eight amino acid residues from each subunit form the dimer interface through hydrophobic and hydrophilic interactions. There are two intersubunit salt bridges formed between E206 of one subunit and K296 of the other subunit (Fig. 1 ▶). E206 is part of a small 310-helix, and K296 is placed at the end of helix 8. These two residues are completely conserved within the Class 1A DHODs. A rather hydrophobic cavity (109 Å3) is formed in dimer interface around the center of the twofold axis and contains two water molecules (Rowland et al. 1997b).
Fig. 1.
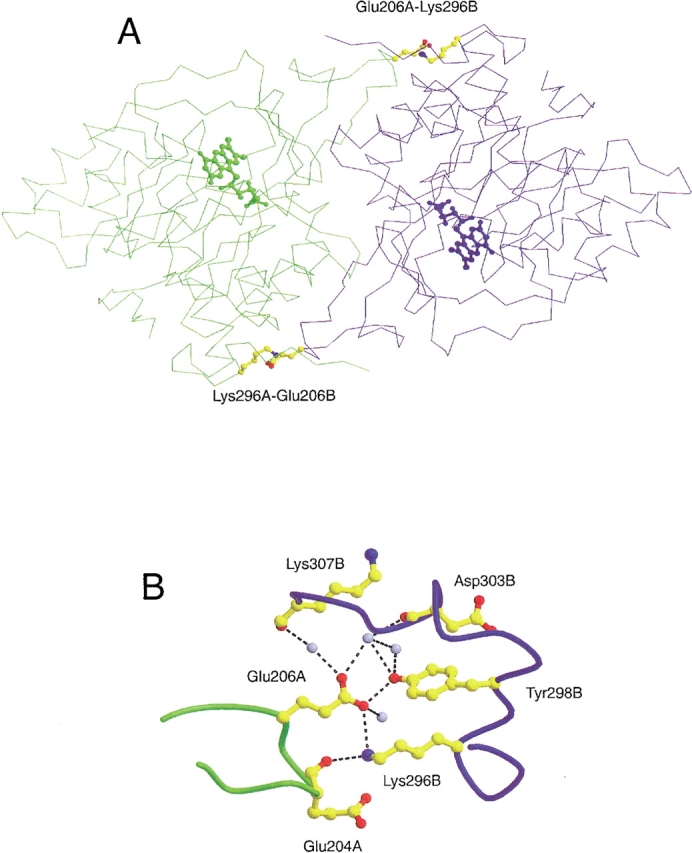
Position and environment of the E206-K296 salt bridges in the subunit interface of DHODA. (A) The two subunits are presented in different colors ("A", green, and "B", blue). The residues involved in formation of the intersubunit salt bridges, Glu206 and Lys296, are shown as balls and sticks in CPK colors, and the position of the active sites are indicated by their FMN cofactors. (B) The amino acid residues are represented as balls and sticks in CPK colors, and the colors of the peptide backbones have the same meaning as above. The light blue spheres represent water molecules, and the broken lines show hydrogen bonds with distances shorter than 3.4 Å. "A" and "B" specify the particular subunits as above. The distances from the carboxylate oxygen atom of Glu206 to the Nɛ of Lys296 and to the OH oxygen of Tyr298 are 3.1 Å and 2.6 Å, respectively. The programs Molscript (Kraulis 1991) and Raster 3D (Merritt and Bacon 1997) were used to prepare the figure, which is based on the coordinates from a recent structure determination of DHODA to 1.7 Å resolution (S. Nørager, S. Jensen, O. Björnberg, M. Oddosen, L. Leggio, K. Jensen, S. Larsen, in prep. pdb code number 1JUE.).
Because the membrane-bound Class 2 DHODs are monomeric proteins (Liu et al. 2000; Nørager et al. 2002), we were interested in learning whether the subunits of the dimeric DHODA might have activity in a monomeric state. To make this investigation, we changed the nature of the amino acid residues involved in salt-bridge formation to destabilize the dimer and promote dissociation into monomers. We found that although the salt bridges were not necessary for activity, perturbation of the salt bridges destabilized the dimer and resulted in dissociation into inactive monomers upon dilution.
Results
Mutations introduced in DHODA
The subunits of the dimeric DHODA are held together in part by two salt bridges formed between the Glu206 residue of one subunit and the Lys296 residue of the other subunit (Fig. 1 ▶). To test the importance of these salt bridges for activity, we changed the residues involved in forming the salt bridges to either alanine or the amino acid of opposite charge, that is, lysine to glutamate or glutamate to lysine. The following amino acid replacements were constructed: E206A, E206K, K296A, K296E, and also the double replacement mutant E206K-K296E, in which the polarity of the salt bridge was inverted. The genes encoding these mutations contained on the inducible multicopy vector pUHE23-2 were introduced into the E. coli strain SØ6645, which lacks the chromosomal pyrD gene and requires an exogenous source of pyrimidine due to lack of dihydroorotate dehydrogenase (Nielsen et al. 1996b).
Complementation and analysis in crude extracts
Bacterial strains carrying plasmids with mutated as well as wild-type versions of pyrDa were spread as single colonies on agar plates in the absence of uracil (without induction). When plates were incubated at 37°C, only the wild-type pyrDa gene gave rise to colony formation; none of the mutant plasmids showed any complementation. The response was more differentiated at 25°C, as the single mutant E206A relieved the pyrimidine requirement of SØ6645 as well as the wild-type gene on pFN1, and the single mutant K296A and the double mutant E206K-K296E gave rise to fair growth in the absence of uracil. The two single mutants, E206K and K296E, complemented the pyrD deletion of the host extremely poorly if at all (Table 1).
Table 1.
Complementation of the uracil requirement of strain SØ6645(ΔpyrD) by plasmids harboring wild-type and mutant pyrD genes and specific enzyme activities in extracts
| Plasmid with mutation | Growth 37°C minus uracila | Growth 25°C minus uracila | Growth plus uracila | Spec. act. (U/mg)c |
| pFN1 (wt) | ++ | ++ | +++ | 0.84 |
| pE206A | − | ++ | +++ | 0.78 |
| pE206K | − | (−)b | +++ | 0.14 |
| pK296A | − | + | +++ | 0.51 |
| pK296E | − | (−)b | +++ | 0.14 |
| pK206E-E296K | − | + | +++ | 0.63 |
a Growth was monitored after ∼48 h at 25°C and after 24 h at 37°C in the absence of induction. In the presence of uracil, the colonies had approximately the same size after the two incubation periods. All plasmids seemed to complement the growth when the cells were induced, but there was a strong growth inhibition also in the presence of uracil.
b Denotes very tiny, shadow-like colonies.
c The cells were grown at 25°C with induction overnight. Specific enzymatic activity in noncentrifuged extracts measured by the DCIP assay. When analyzed by electrophoresis in SDS gel, the DHODA protein bands appeared equally strong in the different extracts. The background enzyme activity in extracts of the strain transformed with the vector plasmid pUHE23-2 was zero due to the deletion of the chromosomal pyrD gene of strain SØ6645.
When the transformed cells were grown at 37°C in liquid culture and disrupted by ultrasonic treatment, all of the mutant enzymes precipitated together with cell debris during low-speed centrifugation as revealed by SDS-PAGE of resuspended pellets and supernatants, and no enzymatic activity could be detected in any of the sonicated extracts except for the wild type. However, the mutant proteins proved to be soluble and active to a variable extent when the cells were grown at 25°C. The enzymatic activity of the E206A, K296A, and E206K-K296E mutant enzymes were similar to the activity of the wild-type enzyme, whereas the E206K and K296E mutations showed strongly reduced enzymatic activity in the extracts. Upon incubation of the extracts at 37°C for ∼5 min before an assay at 25°C, E206A and K296A retained all and 10% of the activity, respectively, whereas the other mutants lost all of their activity.
These results show that the salt bridge between Glu206 and Lys296 is not required for the enzymatic activity of DHODA, but is important for the stability of the protein at increased temperature. The conversion of one of the amino acids of the salt bridge to opposite charge, as in the E206K and K296E single mutants, is very harmful to the function of DHODA, probably due to electrostatic repulsion over the subunit interface, and this effect is partially relieved by reestablishing a possibility of forming a salt bridge in opposite orientation to the E206K-K296E double mutant enzyme.
E206K-K296E mutant and wild-type DHODA lose activity upon dilution
The mutant E206K-K296E was purified as described in Materials and Methods. The activity was characterized by the use of DCIP (50 μM) as electron acceptor and by recording the disappearance of absorption at 600 nm as a measure of the initial reaction velocity. The kinetic parameters (KMapp = 7.2 ± 0.5 μM for DHO and a kcatapp = 22.4 ± 0.3 s−1) were very similar to those of wild-type DHODA (Björnberg et al. 1997). In addition, the affinity of the mutant enzyme for the product orotate (KD = 37 ± 2 μM) was similar to that of the wild-type enzyme (KD = 34 ± 2 μM) as revealed by recording the red-shift of the absorption spectrum during titration of enzyme solutions with orotate (Björnberg et al. 1997).
However, when the enzymes were diluted in 0.1 M Tris-HCl pH 8.0 prior to assay, the specific activity of the mutant enzyme gradually decreased as a function of time and approached a constant value (Fig. 2B ▶). The loss of activity could be fitted to first-order exponential decay function
Fig. 2.

Time course of the loss of enzyme activity upon dilution of wild-type and E206K-K296E double mutant enzymes. At time t = 0, the enzymes were diluted in 0.1 M Tris-HCl, pH 8.0 (25°C). At the indicated times following dilution, samples were withdrawn for determination of enzymatic activity. (A) Wild-type DHODA diluted to 9.8 μM (×, Ao = 31 ± 1 U/mg, but calculations of the inactivation rate according to eq. 1 could not be made due to the stability of the enzyme at this high protein concentration); 0.098 μM (▵, Ao = 28.5 ± 0.5 U/mg, Ar = 13.0 ± 0.5 U/mg, kobs = 0.033 ± 0.003 min−1), or 0.029 μM (○, Ao = 31.0 ± 0.7 U/mg, Ar = 4.9 ± 0.7 U/mg, kobs = 0.024 ± 0.002 min−1). (B) The E206K-K296E double mutant enzyme diluted to 9.8 μM (×, Ao = 29.3 ± 0.9 U/mg, Ar = 13.6 ± 0.3 U/mg, kobs = 0.146 ± 0.015 min−1), 0.98 μM (▵, Ao = 24.1 ± 0.6 U/mg, Ar = 2.9 ± 0.1 U/mg, kobs = 0.103 ± 0.005 min−1), or 0.098 μM (○, Ao = 27.1 ± 0.4 U/mg, Ar = 1.6 ± 0.2 U/mg, kobs = 0.091 ± 0.003 min−1). (C) The E206K-K296E double mutant enzyme diluted to 0.98 μM in 0.1 M Tris-HCl pH 8.0 (▵, same curve as in (B) or in 0.1 M Tris-HCl pH 8.0 containing 0.15 M NaCl (▴, Ao = 26.3 ± 0.4 U/mg, Ar = 11.1 ± 0.3 U/mg, kobs = 0.026 ± 0.002 min−1). The curves are the best fits to the data points according to eq. 1. The kinetic parameters calculated according to eq. 1 are given in Table 2.
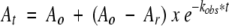 |
(1) |
in which At is the activity at time (t), Ar is the activity remaining at infinite time after dilution, Ao is the activity at time zero following dilution, and kobs is the rate constant for the decay reaction. As seen from the curves in Figure 2B ▶, the rate of inactivation did not depend much on the protein concentration. The inactivation rate constant kobs was close to 0.1 min−1, corresponding to a half-life of approximately 6 min. However, the level of remaining activity (Ar) was strongly dependent on the protein concentration, as shown by the individual inactivation curves in Figure 2B ▶. Such a behavior is most easily explained by assuming that the active dimeric mutant enzyme undergoes a slow and reversible dissociation into inactive monomers according to the following scheme, which also is supported by gel-filtration analyses (see below):
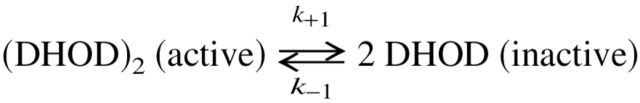 |
A number of kinetic parameters relating to the dissociation reaction could be determined from the decay curves in Figure 2 ▶, and these are given in Table 2.
Table 2.
Data and constants relating to the dimer to monomer dissociation equilibrium of DHODA
| Native DHODA | E206K-K296E mutant DHODA | E206K-K296E mutant DHODA with 0.15 M NaCl | |
| Ao (U/mg)a | 28.5 | 29.3 | 26.2 |
| Ar (U/mg)a | 13.0 | 13.6 | 11.1 |
| [Enzyme]totala (μM) | 0.098 | 9.76 | 0.976 |
| [Dimer]a (μM) | 0.0218 | 2.26 | 0.212 |
| [Monomer]a (μM) | 0.0519 (0.0745) | 5.24 (7.11) | 0.562 (0.867) |
| KDa (μM) | 0.123 μM (0.25 μM) | 12 μM (22 μM) | 1.49 μM (3.7 μM) |
| k+1b (min−1) | 0.024 ± 0.002 | 0.091 ± 0.003 | 0.026 ± 0.002 |
| KDc (μM) | 0.26 ± 0.05 | 37 ± 22 | 3.5 ± 0.6 |
| k−1d (μM−1 min−1) | 0.043 | 0.0010 | 0.0027 |
a The total enzyme concentrations is given as monomers. The data are taken from the decay experiments shown in Fig. 2 ▶. Monomer and dimer concentrations and the KD values are calculated as explained in Materials and Methods. The values of [Monomer] and KD in parentheses were calculated by assuming an identical initial specific activity. Ao = 35 U/mg in all cases.
b k+1 is assumed to be equal to kobs for the most dilute enzyme samples.
c Obtained from the data in Fig. 3 ▶ by eq. 3.
d Association rate constant derived by division of k+1 with KD above.
The remaining enzyme activity (Ar) at different enzyme concentrations could also be determined by letting the enzyme solution stand for a long time to reach equilibrium. The relationship between the remaining (equilibrium) activity and the enzyme concentration used is shown in Figure 3 ▶. The resulting saturation curve could be used to calculate the dissociation constant (KD) for the dimer to monomer dissociation reaction, which is given in the legend to the figure. From these data it appears that the dissociation constant KD = (k+1/k−1) for the E206K-K296E mutant enzyme in 0.1 M Tris-HCl pH 8.0 is about 37 μM at 25°C, and that the rate constant for the dissociation reaction k+1 was 0.093 min−1, assuming that kobs equals k+1 for the most dilute enzyme samples, which undergo almost complete dissociation. In turn, these parameters allow for calculation of a second-order rate constant k−1 for association of inactive monomers to active dimers of 0.0025 μM−1 min−1.
Fig. 3.

Remaining specific activity of DHODA as a function of enzyme concentration following dilution to different concentrations. •, wild-type DHODA diluted in 0.1 M Tris-HCl pH 8.0; ○, E206K-K296E double mutant diluted in 0.1 M Tris-HCl, pH 8.0; ⋄, E206K-K296E double mutant diluted in 0.1 M Tris-HCl, pH 8.0 containing 0.15 M NaCl. Following dilution at time t = 0, the samples were left to reach equilibrium for 200 min for the mutant DHODA and for 330 min for wild-type DHODA before assaying the remaining activity as described in Materials and Methods. The following parameters of the dissociation reaction were calculated according to eq. 3: Wild-type enzyme: KD = 0.26 ± 0.05 μM, SAMax = 35 ± 1 μmol min−1 mg−1; E206K-K296E double mutant: KD = 37 ± 22 μM, SAMax = 46 ± 17 μmol min−1 mg−1; E206K-K296E double mutant with 0.15M NaCl: KD = 3.5 ± 0.6 μM, SAMax = 46 ± 2 μmol min−1 mg−1.
The wild-type DHODA similarly lost activity upon dilution, however, at much lower protein concentrations than used to follow the inactivation of the E206K-K296E mutant enzyme (Fig. 2A ▶). The first-order rate constant for the inactivation k+1 was ∼0.03 min−1, corresponding to a half life t½ of about 22 min and independent of protein concentration to a first approximation. A measurement of the remaining enzyme activities at different enzyme concentrations (Fig. 3 ▶) revealed the constant for dissociation of active dimers to inactive monomers, KD = 0.26 μM. The dissociation rate constant k+1 was 0.024 min−1 (assumed again to equal kobs at the most dilute concentration of enzyme), and thus the second-order rate constant k−1 for association of inactive monomers to active dimers will be 0.043 μM−1 min−1. Thus, the rate of both the dissociation of active dimers to inactive monomers and the reverse association reaction was affected by the E206K-K296E mutations in the enzyme, but the association rate was more affected than the dissociation rate (Table 2).
Effect of salt and the product orotate on the inactivation of E206K-K296E mutant DHODA
The inactivation of the E206K-K296E mutant DHODA was influenced by the salt concentration of the dilution buffer (Fig. 2C ▶). The presence of 0.15 M NaCl reduced the dissociation rate constant k+1 (kobs) to 0.026 min−1 and the equilibrium constant for the dissociation process KD to 3.5 μM (Fig. 3 ▶). The association rate constant k−1 was increased to 0.0074 μM−1 min−1, and thus the salt influenced both the rates of association of monomers and dissociation of dimers (Table 2). As shown in Table 3, the product of the enzymatic reaction, orotate, also protected the enzyme against inactivation upon dilution, although the kinetic behavior was not analyzed in detail.
Table 3.
Protection of DHODA against dilution with the reaction product orotate
| Specific activity (U/mg) with orotate (0.5 mM) | Specific activity (U/mg) Control without orotate | |
| Wild-type DHODA (0.029 μM) | 21.6 ± 0.6 | 1.8 ± 0.1 |
| (3.4 ± 0.2) | ||
| E206K-K296E mutant | 14.6 ± 0.1 | 0.7 ± 0.1 |
| DHODA (0.098 μM) | (1.5 ± 0.1) |
The enzymes at the indicated concentrations were incubated at 25°C in Tris-HCl pH 8.0 with 0.5 mM orotate or without orotate. The remaining activity was assayed after 80 min, for the mutant enzyme, or 330 min, for the wild-type enzyme. Care was taken to have the same final concentration of orotate (50 μM) in the assays. The control activities without orotate in the assays are given in parentheses.
In one experiment, 1M NaCl (final concentration) was added to a solution of E206K-K296E mutant DHODA that had previously been diluted and allowed to dissociate and inactivate for 3 h in the absence of salt. Following addition of NaCl, the enzyme slowly regained activity as a function of time, but the kinetics of the reactivation process have not been analyzed in detail. In addition, an experiment was made in which orotate (final concentration 430 μM) was added to a solution of the E206K-K296E mutant enzyme (9.8 μM) which previously had been allowed to dissociate for 3 h to gain a specific activity of 13.8 ± 0.2 U/mL. Following the addition of orotate, the specific activity of this enzyme solution gradually increased to 27.6 ± 1.6 U/mg over a period of 30 min and remained constant thereafter.
Gel-filtration behavior of mutant and wild-type DHODA
The wild-type and E206K-K296E mutant enzymes behaved very similarly during gel-filtration chromatography on a Superose 12 column and eluted as single symmetric peaks of a size similar to that of bovine serum albumin (Mr 67.000, i.e., corresponding to the size of a dimeric DHODA molecule) when they were loaded on the column at high concentration, 3.9 mg/mL (114 μM) in the 200 μL sample (data not shown). When the concentration of enzyme in the samples was reduced to 0.14 mg/mL (4 μM), the wild-type enzyme retained its elution behavior, but the mutant enzyme developed a pronounced shoulder on its elution profile corresponding to the occurrence of lower-molecular-weight species in the sample (Fig. 4A ▶). This shoulder was not seen if the gel filtration was made in Buffer A containing 1 M NaCl (data not shown), and it became less pronounced when 600 μM orotate was included in the enzyme samples that were loaded on the column (Fig. 4B ▶). This behavior during gel filtration is in agreement with the notion that the dimeric E206K-K296E double mutant enzyme has an increased tendency to dissociate to (inactive) monomers upon dilution, and that factors (NaCl and orotate) which protect the enzyme from loss of activity during dilution also stabilize the dimeric form of the enzyme.
Fig. 4.
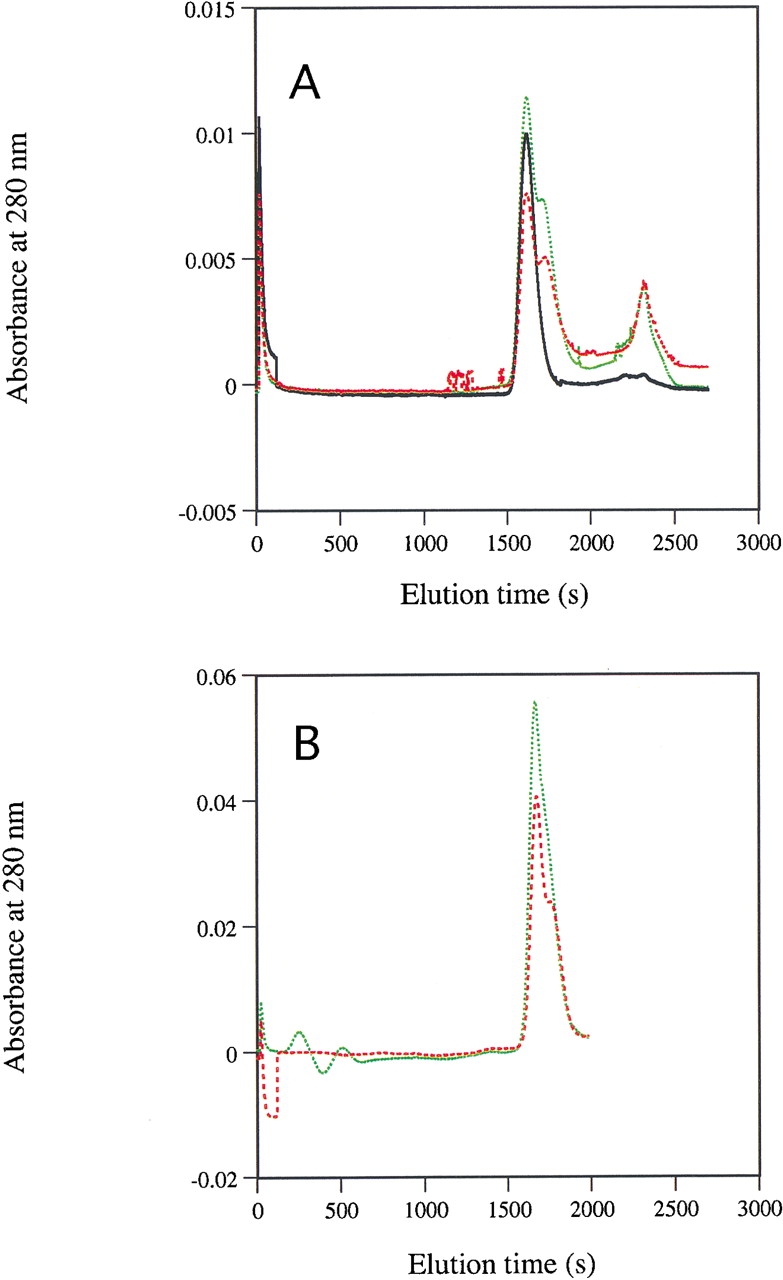
Gel-filtration behavior of wild-type and E206K-K296E double mutant DHODA enzymes under dilute conditions. (A) Wild-type DHODA at 138 μg/mL in the applied sample (black), E206K-K296E double mutant DHODA at 138 μg/mL in the applied sample (red broken line), and E206K-K296E double mutant DHODA at 277 μg/mL in the applied sample (green broken line). The absorbance peak eluting at ∼2350 sec is likely due to small differences between the applied sample and the elution buffer; it did not contain any flavin. (B) E206K-K296E double mutant DHODA at 185 μg/mL in the applied sample (red broken line) and E206K-K296E double mutant DHODA at 185 μg/mL in the applied sample which also contained 600 μM of orotate (green broken line). The part of the elution curve following 2000 sec in B was not shown because of the very strong absorbance from orotate in this region of the profile. The samples were run on a Superose 12 column as described in Materials and Methods.
We considered the possibility that the flavin cofactor might be lost upon dissociation of the dimeric enzyme, which would explain the loss of activity upon dilution. However, in all gel filtration experiments, the flavin absorption followed the protein peaks and could never be detected in the fractions that contained the low-molecular-weight material. Furthermore, the relative fluorescence intensity of the flavin did not change as a function of enzyme concentration in the interval used, which was expected if FMN dissociated from the enzyme upon dilution, because fluorescence of the enzyme-bound cofactor is strongly quenched (F. Nielsen and K. Jensen, unpubl.).
Discussion
The results of this study strongly indicate that the dimeric dihydroorotate dehydrogenase A (DHODA) dissociates reversibly into inactive monomers upon dilution. This assumption is supported by the kinetics of the loss of enzymatic activity following dilution (Fig. 2 ▶), by the increasing (and saturating) specific activity as a function of increasing protein concentration (Fig. 3 ▶), and by the behavior of mutant and wild-type enzyme during gel filtration (Fig. 4 ▶), as well as the recovery of enzymatic activity upon addition of salt or orotate. The dissociation reaction was more pronounced in the double mutant E206K-K296E in which the polarity of two salt bridges between the two subunits of the dimer (Rowland et al. 1997b) have been inverted. The equilibrium constants for the dimer to monomer dissociation reaction could be estimated both from the end points of the kinetic analyses shown in Figure 2 ▶, that is, from the initial activity Ao and the equilibrium activity Ar, and from analysis of equilibrium specific activity as a function of protein concentration (Fig. 3 ▶). The values are given in Table 2. The crude determinations of KD from Ao and Ar by the first method tended to generate smaller values than were found by the second method. We ascribe this (minor) discrepancy to the fact that the first method depends on the assumption that all enzyme exists as dimers prior to dilution. This may not be the case, and hence Ao is likely to be underestimated—an error that leads to underestimation of the concentration of monomers at equilibrium and consequently also to (falsely) low values of KD. Another qualification to our calculations is our assumption that the monomers are totally devoid of activity. As illustrated in Figure 2B ▶, this may not be the case, since the decay curve for the most dilute solution (0.1 μM) of the E206K-K296E double mutant enzyme approached an equilibrium specific activity of 1.6 U/mg, when close to zero specific activity was expected from the KD value (37 μM). Therefore, we cannot exclude the possibility that the monomer has an enzymatic activity, which corresponds to 5%, or less, of that of the dimeric enzyme.
Despite the qualifications described above, it is clear that the double E206K-K296E mutation has destabilized the dimeric form of DHODA by a factor of approximately 100, and that the rate of association of monomers to dimers was more strongly affected (∼40-fold reduced) than the dissociation of dimers to monomers, which occurred 3–4 times faster for the mutant enzyme than the wild type. The fact that the dimeric form was stabilized, rather than destabilized, by sodium chloride indicates that the hydrophobic interactions between dimers are more important than the electrostatic forces, which are expected to be weakened by the presence of salt.
The dissociation constant KD for the dimer to monomer conversion reaction for the wild-type enzyme was estimated to be 0.26 μM, corresponding to a ΔG°d = 8.98 kcal mol−1 for the wild-type DHODA and 37 μM for the E206K-K296E mutant enzyme, corresponding to a ΔG°d = 6.04 kcal mol−1. This means that inversion of the two salt bridges in the E206K-K296E mutant enzyme has destabilized the dimer formation to ΔΔG°d = 2.94 kcal mol−1. The architecture of the intersubunit salt bridge in the wild-type enzyme is shown in Figure 1B ▶. (Unfortunately, we have not been able to elucidate the structure of the mutant enzyme, because we were unable to obtain crystals despite several attempts.) It is clear that Lys296 and Glu206 are part of a complex network of hydrogen bonds that includes the side chain -OH of Tyr298, the backbone carbonyl oxygen of Asp204 and several water molecules. Most of these hydrogen bonds are likely to be perturbed in the double mutant enzyme, and it is therefore not possible to assign the decrease in stability of the dimeric form of the mutant enzyme to a single intermolecular interaction.
It can be concluded, however, that the salt bridge is not needed for the catalytic function of DHODA, because the mutant enzymes, in which one of the residues involved in salt bridge formation was replaced by alanine, were also catalytically active, although they tended to dissociate and precipitate under certain conditions. In addition, the loss of enzymatic activity upon dissociation was not due to loss of the prosthetic FMN from the monomers, and thus our results show that the catalytic function of the two active sites is dependent on the presence of the neighboring subunit of the dimer, which perhaps protects the active sites against access of solvent. Although the two active sites are located at some distance from the subunit interface, the loops that cover the active sites are in contact with the neighboring subunit in both the dimeric and the heterotetrameric Class 1 dihydroorotate dehydrogenases DHODA (Rowland et al. 1997b; 1998) and DHODB (Rowland et al. 2000). Removal of the neighboring subunit is likely to influence the conformation of these loops so that they no longer adopt a structure suited for optimal enzyme function. In the monomeric Class 2 DHODs from human (Liu et al. 2000) and Escherichia coli (Nørager et al. 2002), an N-terminal domain responsible for communication with the respiratory quinones and attachment to membranes interacts with the DHOD core domain at a region similar to the one which is involved in dimerization in the Class 1 enzymes.
Materials and methods
Materials
Orotic acid, (S)-dihydroorotate, and markers for gel filtration were purchased from Sigma.
General instrumentation
A Zeiss Specord S10 diode-array photometer was used for the enzyme assays and absorption spectra.
Construction of mutant DHODAs
The mutations were introduced in the pyrDa gene using plasmid pFN1 (Nielsen et al. 1996b) as template for polymerase chain reactions (PCRs) with mutant primers as described by Björnberg et al. (1997). The mutated pyrDa genes were cloned in pUHE23-2 (Deuschle et al. 1986) to produce plasmids similar to expression vector for the wild-type enzyme, pFN1 (Nielsen et al. 1996b), and DNA sequencing revealed that they contained no other mutation than the desired one(s). The plasmids were transformed into the Escherichia coli strain SØ6645, which lacks its native dihydroorotate dehydrogenase due to a deletion of the chromosomal pyrD gene and requires exogenous pyrimidine for growth. The strain SØ6645 also contains an F‘lacIq episome, which causes overproduction of the lacI repressor and keeps transcription of the cloned pyrDa gene at a low level until induction by addition of isopropyl-β-D-thiogalactoside (IPTG).
Complementation of a ΔpyrD mutation
The ability of the mutant pyrD gene harbored in plasmid pUHE23-2 to complement the chromosomal pyrD deletion was tested by plating the strain SØ6645 transformed with the plasmids on agar plates containing glucose (0.2%), casamino acid (0.2%), and thiamine (2 μg/mL) and incubating them at either 25°C or 37°C. The size of colonies was inspected after approximately 24 and 48 h and compared with similar control plates also containing uracil (20 μg/mL). The complementation test was carried out in the absence and in the presence of the inducer IPTG (50 μM), but only the results in the absence of induction were used, because the induction of DHODA mutant—as well as wild-type enzymes—strongly inhibited colony growth even in the presence of uracil.
Crude extract analyses
Ten-mL cultures of the transformed strains were grown in LB-broth medium (Miller 1972) at 25°C or 37°C, with vigorous shaking. The cultures were induced at an OD436 of about 0.5 by addition of IPTG (0.75 mM) and grown to stationary phase overnight. The cultures were harvested by centrifugation for 10 min at 5000 rpm at 5°C (Sorvall SS-34). Cells were suspended in 1 mL of Buffer A (50 mM potassium phosphate, pH 6.0, 10% glycerol, and 1 mM EDTA) and disrupted by ultrasonic treatment during cooling to produce a crude extract. The extract was further fractionated by centrifugation as above. The pellets and supernatant as well as the crude extracts were analyzed for activity using the DCIP assay described below and for protein content by 12.5% SDS-PAGE. Protein concentrations were determined by the Lowry method.
Purification of wild-type and E206K-K296E double mutant DHODAs
Purification of wild-type DHODA was carried out as described (Nielsen et al. 1996b). The E206K-K296E double mutant enzyme was purified similarly, but the transformed strain was grown at 25°C. During purification, chromatography on the hydroxylapatite column was omitted and, instead, the enzyme solution was subjected to chromatography twice on a DE52-column. It appeared to be ≥ 95% pure by SDS gel electrophoresis. The concentration of purified enzymes was estimated from the absorption spectra using the extinction coefficient of FMN of 11.5 mM−1 cm−1 at 456 nm as described (Björnberg et al. 1997).
Enzymatic activity
The oxidation of DHO was measured with DCIP as electron acceptor and monitored by recording the decrease of absorption at 600 nm due to the reduction of dichloroindophenol (DCIP), ɛ600 = 20 mM−1 cm−1 (Karibian 1978; Björnberg et al. 2001). The UV lamp was turned off during these measurements to avoid photobleaching of DCIP. The assay mixture (1 mL) contained 0.1 M Tris-HCl, pH 8.0, 50 μM DCIP, 0.5 mM (S)-DHO and was started by adding the enzyme. One unit corresponds to 1 μmol of orotate produced by the enzyme per min at 25°C.
Gel filtration
Gel-filtration FPLC using a BioRad BioLogic Workstation was performed on a 25-mL Superose-12 column (Pharmacia) at room temperature. A volume of 200 μL enzyme solution was loaded on the column at a flow-rate of 0.5 mL/min, and fractions of 0.5 mL were collected. Buffer A (50 mM potassium phosphate, pH 6.0, 10% glycerol, and 1 mM EDTA) was used as running buffer in most cases, but in some cases 1 M NaCl was included in the buffer, or 0.6 mM orotate was included in the applied enzyme sample.
Loss of enzymatic activity caused by dilution
A concentrated enzyme stock (5–10 mg/mL, 150–300 μM) was diluted to the concentration given in figures and tables at time t = 0, usually in 0.1 M Tris-HCl, pH 8.0 at 25°C. Subsequently, at short intervals, aliquots of the diluted enzyme solution were added to a cuvette and the activity was assayed as described above. During these experiments the specific activity of the enzyme gradually approached an equilibrium level of residual activity (Ar) that was retained for many hours. To determine this level of residual activity at several concentrations of enzyme, the enzyme stock was diluted to various concentrations for 200 min for the E206K-K296E double mutant enzyme or 330 min for the wild-type enzyme to acquire the final equilibrium level of activity (Ar).
Data treatment
According to Neet and Ainslie’s theory for hysteretic enzymes (Neet and Ainslie Jr. 1980), the decrease of enzymatic activity following dilution could be described by equation 1:
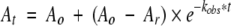 |
in which At is the activity at time (t), Ar is the activity remaining at infinite time after dilution, and Ao is the activity at time zero following dilution. It was possible to describe the relationship between the remaining activity (Ar) and enzyme concentration as a dimer (DHOD2) to monomer (DHOD) equilibrium, assuming that all activity derived from the dimeric form of the enzyme and that the monomeric form was inactive. To make the analysis we assumed that the enzyme sample contained only fully active dimers at t = 0, and that it gradually distributed itself into inactive monomers and active dimers during the dissociation reaction. Under these assumptions, we could calculate the concentration of inactive monomer by the expression [DHOD] = [E]total × (Ao−Ar)/Ao and the concentration of active dimer [DHOD2] = 0.5 [E]total × Ar/Ao, since it takes two monomers to form a dimer. ([E]total is the total enzyme concentration expressed as monomers). The dissociation constant (KD) for the equilibrium could then be calculated from the activity decay curves after equilibrium was reached by the equation:
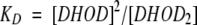 |
(2) |
The KD value for each enzyme could also be determined from the saturation of remaining specific activity (Ar/[E]total) as a function of increasing protein concentration using the equation:
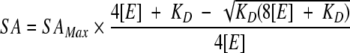 |
(3) |
where "E" represents the total enzyme concentration expressed as monomers.
The data were fitted to the equations using the BIOSOFT program UltraFit for Macintosh.
The free energy of dissociation of dimers to monomer subunits was obtained from the equation:
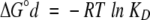 |
where R is the gas constant (1.987 cal K−1 mol−1).
Acknowledgments
We thank Lise Schack for skilled technical assistance and Dr. Anne Mølgaard for assistance in the preparation of Figure 1 ▶. The financial support from the Danish National Science Council, the Danish National Research Foundation, and the Carlsberg Foundation is greatly appreciated. B.A.P. was supported by NIH Grant GM61087.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0220302.
References
- Argyrou, A. and Washabaugh, M.W. 1999. Proton transfer from the C5-proR/proS positions of L-dihydroorotate: General-base catalysis, isotope effects, and internal return. J. Am. Chem. Soc. 121 12054–12062. [Google Scholar]
- Argyrou, A., Washabaugh, M.W., and Pickart, C.M. 2000. Dihydroorotate dehydrogenase from Clostridium oroticum is a Class 1B enzyme and utilizes a concerted mechanism of catalysis. Biochemistry 39 10373–10384. [DOI] [PubMed] [Google Scholar]
- Björnberg, O., Rowland, P., Larsen, S., and Jensen, K.F. 1997. The active site of dihydroorotate dehydrogenase A from Lactococcus lactis investigated by chemical modification and mutagenesis. Biochemistry 36 16197–16205. [DOI] [PubMed] [Google Scholar]
- Björnberg, O., Grüner, A.C., Roepstorff, P., and Jensen, K.F. 1999. The activity of Escherichia coli dihydroorotate dehydrogenase is dependent on a conserved loop identified by sequence homology, mutagenesis and limited proteolysis. Biochemistry 28 2899–2908. [DOI] [PubMed] [Google Scholar]
- Björnberg, O., Jordan, D.B., Palfey, B.A., and Jensen, K.F. 2001. Dihydrooxonate is a substrate of dihydroorotate dehydrogenase (DHOD) providing evidence for involvement of cysteine and serine residues in base catalysis. Arch. Biochem. Biophys. 391 286–294. [DOI] [PubMed] [Google Scholar]
- Deuschle, U., Kammerer, W., Gentz, R., and Bujard, H. 1986. Promoters of E. coli. A hierarchy of in vivo strength indicates alternate structures. EMBO J. 5 2987–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraaije, M.W. and Mattevi, A. 2000. Flavoenzymes: Diverse catalysts with recurrent features. TIBS 25 126–132. [DOI] [PubMed] [Google Scholar]
- Gao, G., Nara, T., Nakajima-Shimada, J., and Aoki, T. 1999. Novel organization and sequences of five genes encoding all six enzymes for de novo pyrimidine biosynthesis in Trypanosoma cruzi. J. Mol. Biol. 285 149–161. [DOI] [PubMed] [Google Scholar]
- Jensen, K.F. and Björnberg, O. 1998. Evolutionary and functional families of dihydroorotate dehydrogenases. Paths to Pyrimidines 6 20–28. [Google Scholar]
- Jiang, W., Locke, G., Harpel, M.R., Copeland, R.A., and Marcinkeviciene, J. 2000. Role of Lys100 in human dihydroorotate dehydrogenase: Mutagenesis studies and chemical rescue by external amines. Biochemistry 39 7990–7997. [DOI] [PubMed] [Google Scholar]
- Jordan, D.B., Bisaha, J.J., and Picollelli, M.A. 2000. Catalytic properties of dihydroorotate dehydrogenase from Saccharomyces cerevisiae: Studies on pH, alternate substrates, and inhibitors. Arch. Biochem. Biophys. 378 84–92. [DOI] [PubMed] [Google Scholar]
- Kahler, A.E., Nielsen, F.S., and Switzer, R.L. 1999. Biochemical characterization of the heteromeric Bacillus subtilis dihydroorotate dehydrogenase and its isolated subunits. Arch. Biochem. Biophys. 37 191–201. [DOI] [PubMed] [Google Scholar]
- Karibian, D. 1978. Dihydroorotate dehydrogenase (Escherichia coli). Methods Enzymol. 51 58–63. [DOI] [PubMed] [Google Scholar]
- Kraulis, P.J. 1991. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallography 24 946–950. [Google Scholar]
- Liu, S., Neidhardt, E.A., Grossman, T.H., Ocain, T., and Clardy, J. 2000. Structures of human dihydroorotate dehydrogenase in complex with antiproliferative agents. Structure 8 25–33. [DOI] [PubMed] [Google Scholar]
- Marcinkeviciene, J., Tinney, L.M., Wang, K.H., Rogers, M.J., and Copeland, R.A. 1999. Dihydroorotate dehydrogenase B of Enterococcus faecalis. Characterization and insights into chemical mechanism. Biochemistry 38 13129–13137. [DOI] [PubMed] [Google Scholar]
- Marcinkeviciene, J., Jiang, W., Locke, G., Kopcho, L.M., Rogers, M.J., and Copeland, R.A. 2000. A second dihydroorotate dehydrogenase (Type A) of the human pathogen Enterococcus faecalis: Expression, purification and steady-state kinetic mechanism. Arch. Biochem. Biophys. 277 178–186. [DOI] [PubMed] [Google Scholar]
- Merritt, E.A. and Bacon, D.J. 1997. Raster3D photorealistic molecular graphics. Methods Enzymol. 277 505–524. [DOI] [PubMed] [Google Scholar]
- Miller, J.H. 1972. Experiments in molecular genetics, p. 433. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY.
- Nagy, M., Lacroute, F., and Thomas, D. 1992. Divergent evolution of pyrimidine biosynthesis between anaerobic and aerobic yeasts. Proc. Natl. Acad. Sci. 89 8966–8970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neet, K.E., and Ainslie, Jr., G.R. 1980. Hysteretic enzymes. Methods Enzymol. 64 192–226. [DOI] [PubMed] [Google Scholar]
- Nielsen, F.S., Andersen, P.S., and Jensen, K.F. 1996a. The B-form of dihydroorotate dehydrogenase from Lactococcus lactis consists of two different subunits, encoded by the pyrDb and pyrK genes, and contains FMN, FAD, and [FeS] redox centres. J. Biol. Chem. 271 29359–29365. [DOI] [PubMed] [Google Scholar]
- Nielsen, F.S., Rowland, P., Larsen, S., and Jensen, K.F. 1996b. Purification and characterization of dihydroorotate dehydrogenase A from Lactococcus lactis, crystallization and preliminary X-ray diffraction studies of the enzyme. Protein Sci. 5 857–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nørager, S., Jensen, K.F., Björnberg, O., and Larsen, S. 2002. E. coli dihydroorotate dehydrogenase reveals structural and functional differences between different classes of dihydroorotate dehydrogenases. Structure (in press). [DOI] [PubMed]
- Palfey, B.A., Björnberg, O., and Jensen, K.F. 2001. Insight into the chemistry of flavin reduction and oxidation in Escherichia coli dihydroorotate dehydrogenase obtained by rapid reaction studies. Biochemistry 40 4381–4390. [DOI] [PubMed] [Google Scholar]
- Pascal, R.A., Trang, N.L., Cerami, A., and Walsh, C. 1983. Purification and properties of dihydroorotate oxidase from Crithidia fasciculata and Trypanosoma brucei. Biochemistry 22 171–178. [DOI] [PubMed] [Google Scholar]
- Rowland, P., Björnberg, O., Nielsen, F.S., Jensen, K.F., and Larsen, S. 1998. The crystal structure of Lactococcus lactis dihydroorotate dehydrogenase A complexed with the reaction product ortate throws light on its enzymatic function. Protein Sci. 7 1269–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland, P., Larsen, S., Nielsen, F.S., Björnberg, O., and Jensen, K.F. 1997a. Properties of dihydroorotate dehydrogenase A from Lactococcus lactis. Crystallization and three dimensional structure of the enzyme. In Flavins and flavoproteins 1996 (eds. K.J. Stevenson, V.l. Massey, and C.H. Williams, Jr.), pp. 927–930. University of Calgary Press. UBC Press, Vancouver, Canada..
- Rowland, P., Nielsen, F.S., Jensen, K.F., and Larsen, S. 1997b. The crystal structure of the flavin containing enzyme dihydroorotate dehydrogenase A from Lactococcus lactis. Structure 5 239–250. [DOI] [PubMed] [Google Scholar]
- Rowland, P., Nørager, S., Jensen, K.F., and Larsen, S. 2000. Structure of dihydroorotate dehydrogenase B: Electron transfer between two flavin groups bridged by an iron-sulphur cluster. Structure 8 1227–1238. [DOI] [PubMed] [Google Scholar]