

Abstract
Phospholipase D (PLD), an important enzyme involved in signal transduction in mammals, is also secreted by many microorganisms. A highly conserved HKD motif has been identified in most PLD homologs in the PLD superfamily. However, the Ca2+-dependent PLD from Streptomyces chromofuscus exhibits little homology to other PLDs. We have cloned (using DNA isolated from the ATCC type strain), overexpressed in Escherichia coli (two expression systems, pET-23a(+) and pTYB11), and purified the S. chromofuscus PLD. Based on attempts at sequence alignment with other known Ca2+-independent PLD enzymes from Streptomyces species, we mutated five histidine residues (His72, His171, His187, His200, His226) that could be part of variants of an HKD motif. Only H187A and H200A showed dramatically reduced activity. However, mutation of these histidine residues to alanine also significantly altered the secondary structure of PLD. Asparagine replacements at these positions yielded enzymes with structure and activity similar to the recombinant wild-type PLD. The extent of phosphatidic acid (PA) activation of PC hydrolysis by the recombinant PLD enzymes differed in magnitude from PLD purified from S. chromofuscus culture medium (a 2-fold activation rather than 4–5-fold). One of the His mutants, H226A, showed a 12-fold enhancement by PA, suggesting this residue is involved in the kinetic activation. Another notable difference of this bacterial PLD from others is that it has a single cysteine (Cys123); other Streptomyces Ca2+-independent PLDs have eight Cys involved in intramolecular disulfide bonds. Both C123A and C123S, with secondary structure and stability similar to recombinant wild-type PLD, exhibited specific activity reduced by 10−5 and 10−4. The Cys mutants still bound Ca2+, so that it is likely that this residue is part of the active site of the Ca2+-dependent PLD. This would suggest that S. chromofuscus PLD is a member of a new class of PLD enzymes.
Keywords: Phospholipase D, Streptomyces chromofuscus, HKD motif, Ca2+-dependent enzyme, mutagenesis
Phospholipase D (PLD) activity has been identified in a variety of organisms including mammals (Exton 2000), plants (Wang 2000), yeast (Grossman et al. 1973), and bacteria (Okawa and Yamaguchi 1975; Imamura and Horiuti 1979; Shimbo et al. 1993). The enzyme catalyzes two reactions: (1) hydrolysis of the distal phosphodiester bond of phospholipids to generate phosphatidic acid (PA), and (2) transphosphatidylation with primary or secondary alcohols to generate a different phospholipid. The phosphotransferase activity has been used to generate a variety of phospholipids with defined chain lengths, unsaturation, isotopic labels, and so forth, from PC (Dawson 1967; Eible and Kovatchev 1981). In eukaryotes, further catabolism of PA to lysophosphatidic acid via a specific phospholipase A2 or to diacyglycerol via a PA phosphatase yields second messengers that are also involved in signal transduction. PLD in yeast has been implicated in sporulation (Ella et al. 1995), whereas PLD activities in bacteria appear to act as virulence determinants (McNamara et al. 1995).
Within the past five years, sequence alignments have identified a superfamily of PLD homologs that include most PLDs, other phosphodiesterases (e.g. Salmonella typhimurium endonuclease known as Nuc and Saccharomyces cerevisiae tyrosyl-DNA phosphodiesterase [Tdp1]), cardiolipin synthase, phosphatidylserine synthase, a bacterial toxin, and several poxvirus envelope proteins (Koonin 1996; Ponting and Kerr 1996; Zhao et al. 1997). Except for Nuc, all of the members of the PLD superfamily have four duplicated highly conserved regions (Ponting and Kerr 1996). Several highly conserved residues in these segments were proposed to be the catalytic residues. In particular, the duplicate HxK(x)4D (or HKD) motifs were identified as the common feature among the PLD superfamily homologs. Recent crystal structures of Nuc (Stuckey and Dixon 1999) and the PLD from Streptomyces sp. PMF strain (Leiros et al. 2000) have verified that the two HKD motifs in the Nuc dimer or the Streptomyces PLD monomer are clustered at the active site. One histidine was suggested to act as the nucleophile that attacks the phosphorus whereas the other histidine was suggested to act as a general acid to protonate the leaving group (Leiros et al. 2000).
Several bacterial PLDs from Streptomyces have been sequenced. PLD from S. antibioticus, S. cinnamoneus, S. halstedii, S. septatus, and Streptomyces sp. PMF strain show significant sequence similarity (Fig. 1 ▶) and similar enzymatic properties including high transphosphatidylation activity, relatively high optimum reaction temperatures, and Ca2+-independent activity (Juneja et al. 1988; Shimbo et al. 1993; Hatanaka et al. 2002). A different PLD, isolated from S. chromofuscus, is Ca2+-dependent and has very little sequence homology (Iwasaki et al. 1994) with the other Streptomyces PLD enzymes (Fig. 1 ▶). The primary amino acid sequence of S. chromofuscus PLD (Yoshioka et al. 1991) exhibits no HxK(x)4D motif. However, two sequences (e.g., 187HxK(x)3D193 and nearby 200HxK(x)7D210) in the same region of the protein as one of the HxK(x)4D motifs in the other Streptomyces PLDs might be variations of that catalytic motif.
Fig. 1.
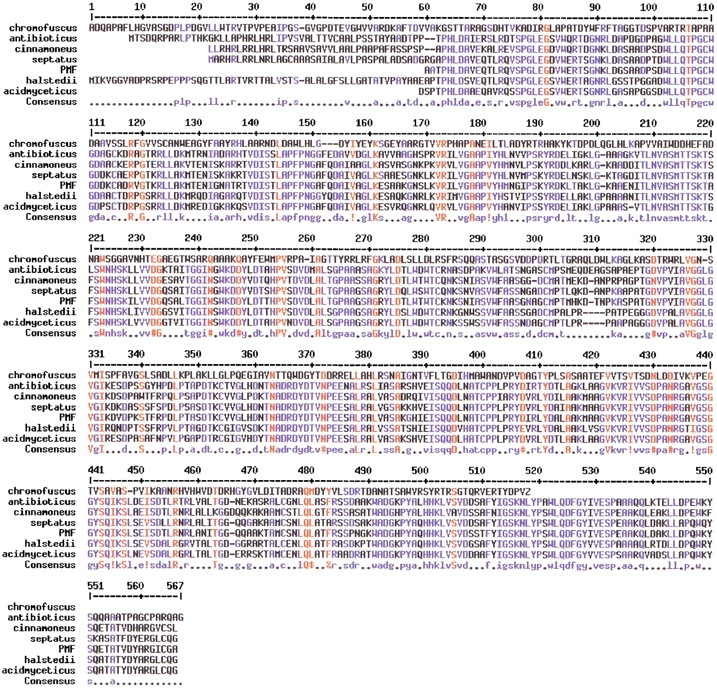
Alignment of PLD sequences from Streptomyces. All of the PLDs except that from S. chromofuscus are Ca2+-independent. Residues in red are conserved in all PLDs; residues in blue are conserved among the Ca2+-independent PLDs.
In this work, the pld gene from the S. chromofuscus type strain (obtained from ATCC) was cloned into two overexpression vectors (pET-23a(+) and pTYB11) for expression in Escherichia coli. The protein sequence derived from this pld gene showed 87% identity with a published sequence for an S. chromofuscus PLD determined previously (Yoshioka et al. 1991). Recombinant S. chromofuscus PLD was overexpressed in E. coli and purified to homogeneity. Activities toward monomeric diC4PC and POPC vesicles in the absence and presence of the activator POPA were measured and compared to authentic PLD purified from S. chromofuscus growth media. Five histidine residues (H72, H171, H187, H200, and H226) that could be part of variants of an HKD motif (or that occur where one of the other HKD motifs occurs in other Streptomyces PLD enzymes) were mutated to assess any role in catalysis. None of these residues was shown to be essential for catalytic activity. However, mutation of the single cysteine (Cys123) in S. chromofuscus PLD to alanine or serine generated well-folded protein with greatly reduced activity. It is suggested that this unusual PLD from S. chromofuscus may carry out the phosphodiester cleavage by a different mechanism.
Results
Sequence of recombinant S. chromofuscus pld gene—variations with published sequence
The sequence of the pld gene cloned from the S. chromofuscus type strain had significant differences from the pld gene sequence published previously (the nucleotide sequence has been deposited with GenBank, accession number AF523823). The DNA alignment of the pld gene in pmPLD, which was prepared by using the forward primer based on the published DNA sequence, showed 87% identity to the published sequence. The translated amino acid sequences had 87% identity and 91% similarity (Fig. 2 ▶). The two strains, obtained from different sources, may not be identical, and this is likely to be the source of the differences in the PLD sequence. In any case, the changes in the S. chromofuscus PLD sequence do not yield protein with higher homology to other PLD superfamily members. Native PLD purified from S. chromofuscus medium has two active forms, intact PLD and a proteolytically clipped but associated form of the protein (Geng et al. 1999; Stieglitz et al. 1999). The larger N-terminal part (amino acids 1–350) contains the catalytic machinery, and the smaller C-terminal segment (amino acids 351–510) has regulatory elements. In comparing the two S. chromofuscus sequences, 49 of the 64 amino acid changes (77%) were in the larger N-terminal segment, consistent with a random distribution of the changes.
Fig. 2.

Sequence of S. chromofuscus rPLD with differences from the previously published sequence for S. chromofuscus PLD indicated on the line below. N-PLD would have the first four residues as TTGT instead of ADQA.
All 14 of the histidines in the original S. chromofuscus clone are present as well as two additional histidines (His87 and His175). Both His187 and His200 are conserved; however, Lys189 was replaced by Arg and Lys202 by an Ala. Thus, the sequence containing His200 is altered so that its resemblance to an HKD motif is even less than in the previously published S. chromofuscus PLD sequence. Comparing the two S. chromofuscus sequences to other Streptomyces PLD sequences fails to align any His or to identify an HKD motif, although one of the HKD motifs is found in the sequence between residues 165 and 224 in all the other PLDs. This might suggest that His clustered in this region of the S. chromofuscus PLD would be good targets for mutagenesis. All the other Streptomyces PLDs have eight conserved Cys likely to be involved in disulfide bond formation, whereas S. chromofuscus PLD has only a single Cys (Cys123). Of the very few aligned and conserved residues, there are seven Gly, two Pro, and very few charged amino acids (two Arg, one Lys, and three Glu).
Overexpression and purification of PLD
The overexpression of the S. chromofuscus PLD gene in BL21(DE)3plysS is about 5%, as judged by intensities on the SDS-PAGE of the crude lysed cell supernatant (Fig. 3A ▶), much lower than many other recombinant genes expressed in E. coli. This lower level of overexpression made the purification difficult. However, the M1-PLD could be purified to more than 90% by an adaptation of the protocol used by Imamura and Horiuti (1979). Precipitation with 40–80% acetone isolated most of the recombinant PLD protein. The native S. chromofuscus PLD has been suggested to possess a hydrophobic site distinct from its catalytic site, and could be adsorbed onto palmitoyl-cellulose resin through this hydrophobic site (Imamura and Horiuti 1979). Chromatography of M1-PLD using this resin removed most of the E. coli proteins from PLD preparations (Fig. 3A ▶). QFF chromatography further purified the PLD protein to more than 90% purity. Loss of material during the QFF chromatography was substantial, perhaps because of different ions bound to Ca2+ sites or some residual Triton X-100 that was used to elute the protein from the palmitoyl-cellulose column. However, the protein obtained was very pure and quite stable. A summary of recoveries at each step in the purification is presented in Table 1.
Fig. 3.
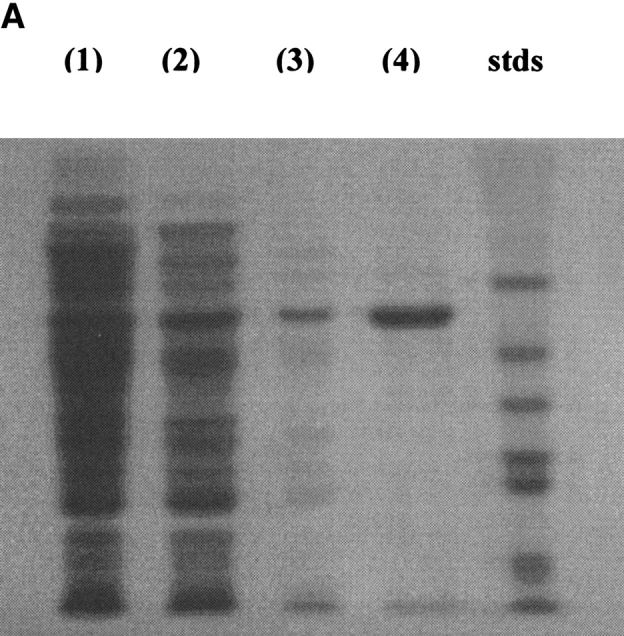
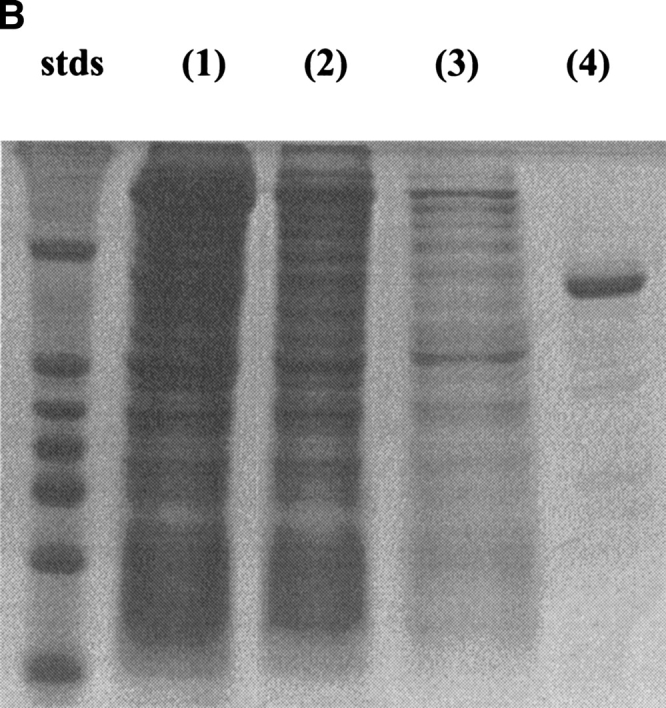
SDS-PAGE showing overexpression and purification of rPLD from pET expression (A) and H226A from the IMPACT fusion protein expression system (B). (A): (lane 1) protein content of the crude cell extract, (lane 2) protein content after acetone precipitation, (lane 3) protein content after the palmitoyl-cellulose column, and (lane 4) protein content after QFF chromatography. Molecular masses for the standard proteins are 66, 45, 36, 29, 24, and 20.1 kD. (B): (lane 1) protein content of lysed cell extract, (lane 2) protein content of flow-through from the chitin column, (lane 3) protein content of wash from the column, and (lane 4) protein eluted after incubation of column with DTT.
Table 1.
Summary of the purification of PLD fromS. chromofuscus (ATCC strain) cloned from pET-23a(+) and IMPACT systems
| Purification step | Protein (mg) | Specific activity (μmole min−1 mg−1) | Total units | Yield (%) |
| pET-23a(+) | ||||
| crude | 199 | 1.9 | 377 | 100 |
| acetone ppt. | 77.7 | 3.5 | 273 | 72.5 |
| Pal-C | 21.2 | 11.2 | 237 | 63.0 |
| QFFa | 1.65 | 13.0 | 21.5 | 5.7 |
| IMPACT | ||||
| crude | 331 | 0.36 | 119 | 100 |
| chitin column | 6.0 | 12.0 | 72.0 | 60.5b |
a Only those fractions from the QFF column that had a single band on SDS-PAGE were combined. This reduces the yield but generates very pure protein.
b The yield is referenced to the specific activity of the fusion protein in the crude supernatant and the total protein. Although the fusion protein has some activity, it is reduced compared to that of M1-PLD overexpressed in the pET system. Therefore, the yield is likely to be lower than estimated.
The low yield of M1-PLD protein from expression in BL21(DE)3plysS prompted us to subclone the pld gene into the fusion protein expression vector, pTYB11. The overexpression of the intein-PLD fusion protein in ER2566 was ∼10%, as judged by the intensity of the band for the fusion protein in the crude supernatant (Fig. 3B ▶). The specific activity of this fusion protein in the crude cell extract was 0.04 to 0.4 μmole min−1 mg−1, a value much lower than in the pET system, indicating that the addition of the intein to PLD greatly reduced catalytic activity. The intein protein, with an inserted chitin-binding domain, allows facile purification of the fusion protein from E. coli proteins by use of an affinity column of chitin beads. After DTT induced cleavage of PLD from the intein, the PLD protein was more than 85% pure (Fig. 3B ▶). To obtain the highest purity (>90%) PLD, protein from the chitin column could be chromatographed on a QFF column. The yield was ∼6 mg per liter after cleavage from the intein (Table 1), higher than that obtained for PLD cloned in pET-23a(+). This method of generating rPLD also yields a protein that does not have an N-terminal Met, but rather has the N-terminal residue of the secreted protein (A in the case of rPLD). A comparison of the two recombinant enzymes can assess whether the N-terminus can affect activity or other physical properties of the protein.
Kinetic characterization of recombinant PLD
The activities of M1-PLD and rPLD toward diC4PC, a good water-soluble substrate (Geng et al. 1998, 1999), were determined. Both recombinant enzymes absolutely require Ca2+ for activity. They also catalyzed a phosphodiestrase reaction—the phosphomonoester product PA was not a substrate for either recombinant enzyme. In the presence of 5 mM Ca2+, M1-PLD had a Vmax of 12.4 μmole min−1 mg−1 (measured by pH-stat) and a Km of 0.16 mM. The Vmax of rPLD was slightly higher (15.0 μmole min−1 mg−1) whereas Km was the same. The Vmax for N-PLD with its corrected N-terminal residues (TTGT rather than ADQA) was comparable to that of rPLD. These kinetic parameters for recombinant PLD were very similar to the values obtained from authentic PLD purified from S. chromofuscus media (Geng et al. 1999). Therefore, replacing the N-terminal amino acid of mature PLD (alanine in rPLD) with methionine or changing other residues at the N terminus has no significant effect on activity of the enzyme activity toward soluble substrate (diC4PC).
The product PA has been shown to activate authentic PLD toward PC vesicles (Geng et al. 1998). Therefore, the effect of 10 mole% PA in PC SUVs was examined with the three recombinant PLDs. The extent of PA activation (Table 2) under these conditions (10 mM total phospholipid, pH 7.5) was lower (approximately twofold) than that of authentic PLD isolated from S. chromofuscus growth media, which showed a four to sixfold enhancement of activity under the same conditions (Geng et al. 1998).
Table 2.
Comparison of PLD mutant thermal stability, relative activity toward diC4PC, extent of PA activation toward PC SUVs, and IC50 for Ca2+ (as measured by quenching of intrinsic fluorescence) to recombinant PLD enzymes
| PLD | Tm (°C)a | diC4PC relative activityb | Specific activity POPC SUVsc | PA activationd | IC50 (Ca2+) (mM)e |
| M1-PLD | 57.5 | 1.00 | 8.6 | 1.6 | |
| rPLD | 19.8 | 2.0 | 0.10 | ||
| N-PLD | 59 | 1.07 | 23.8 | 1.85 | |
| H72N | 59 | 0.22 | 2.9 | 1.9 | |
| H171A | 58 | 0.17 | 0.31 | 2.3 | |
| H187A | 59 | 2.6 × 10−5 | <0.0001 | ||
| H187N | 56 | 0.67 | 15.7 | 1.4 | 0.35 |
| H200A | 57.5 | 5.3 × 10−5 | 0.0054 | ||
| H200N | 56 | 0.49 | 11.4 | 2.0 | |
| H226A | 55 | 0.28 | 0.12 | 12.0 | 0.04 |
| C123A | 58 | 7.3 × 10−6 | 0.066 | ||
| C123S | 58 | 1.4 × 10−4 | 0.00087 | 1.4 | 0.088 |
a Tm is the midpoint in the loss of secondary structure (as measured by ellipticity at 222 nm) upon heating; errors in Tm values are typically 1–2°.
b Specific activities toward 5 mM diC4PC in 5 mM Ca2+, 20 mM Tris HCl at pH 8.0, were measured using 31P NMR spectroscopy at either 25 or 37°C as indicated. The specific activity of recombinant M1-PLD is 9.9 μmole min−1 mg−1 at 25°C and 43.9 μmol min−1 mg−1 at 37°C.
c PLD activity toward 10 mM POPC SUVs as measured by 1H NMR spectroscopy for the vesicles prepared in 5 mM Ca2+ and 50 mM imidazole, pH 7.7.
d The extent of activation is the enzyme specific activity toward POPC/POPA (9 mM:1 mM) SUVs compared to activity toward pure POPC (10 mM) SUVs.
e IC50 is the concentration of Ca2+ responsible for 50% of the initial reduction of the intrinsic fluorescence of PLD.
Characterization of the histidine mutants
There are six histidines in the S. chromofuscus PLD sequence between residues 170 and 230 that are in the same region of the sequence as one of the HKD motifs in the Ca2+-independent PLDs: 171HAPA, 175HEILTLAD, 187HGRYKTD, 200HAAAPVVA, 212HEIANDT, and 226HTEGVE. His175 does not occur in the published S. chromofuscus PLD sequence, and the sequence following His212 is not conserved between the two S. chromofuscus enzymes (nor does it contain a Lys or Arg). None of the other sequences match the expected HKD motif (in fact, none of the histidines were conserved in sequence alignments of the S. chromofuscus PLD with the other known PLDs). However, because this Streptomyces PLD requires Ca2+ (which could replace the role of cationic lysine in the HKD motif) for catalytic activity, and because the Asp residue was not found at the active site in the crystal structure of Streptomyces sp. PMF PLD (Leiros et al. 2000), several of these His-containing sequences could easily be variations that lead to a similar active site. If the catalytic mechanism of S. chromofuscus PLD is similar to that of the other Streptomyces enzymes, then one histidine would act as a nucleophile (forming a phosphatidylhistidine intermediate) whereas the other histidine would act as a general acid (Stuckey and Dixon 1999; Leiros et al. 2000).
Four histidine mutants were initially prepared: H171A, H187A, H200A, and H226A. Except for H226A, overexpression of the histidine mutants in the pET-23a(+) system was dramatically less than for wild-type PLD. Expression levels were sufficiently low that the mutant proteins could not be adequately purified. However, expressing the His mutant proteins cloned in the pTYB11 fusion protein system generated sufficient protein that was easy to purify. These four His mutants exhibited activities lower than wild-type PLD, so that the 31P NMR assay was used to compare specific activities (Table 2). H187A and H200A showed dramatic decreases in specific activity, with H187A essentially inactive and H200A <0.01% as active as wild-type PLD. H171A and H226A exhibited activities that were only 17 and 28% that of wild-type PLD. Interestingly, the PA activation, 2-fold with wild-type recombinant PLD, was dramatically increased to 12-fold for H226A. The extent of PA activation for this mutant was two times higher than what was observed for the native protein isolated from S. chromofuscus (Geng et al. 1998).
The loss of activity for H187A and H200A could indicate these histidines are critical catalytic residues. However, the mutant proteins could also be misfolded. To assess this possibility, CD spectra of each mutant were obtained and used to compare secondary structure elements. As shown in Figure 4 ▶, the secondary structure of H187A was quite different from wild type. This inactive mutant showed a large increase in α-helix and decreased percentage of β-sheet. The H200A mutation showed a similar trend with a smaller increase in helix content. For comparison, both H171A and H226A mutants had the same secondary structure as the three "wild-type" proteins (19.9 ± 0.8% α-helix, 22.8 ± 0.4% β-sheet, 17.4 ± 0.03% β-turn, and 39.9 ± 0.4% random coil). The histidine side chain could be critical for structural elements, and replacement of the imidazole ring with a methyl group would not allow any hydrogen bonding interactions to occur. Therefore, Asn, whose side chain can adopt a conformation that places functional groups in the positions of the imidazole nitrogens, was introduced in place of His to form H187N and H200N. As shown in Table 2, both of these mutants have specific activities within a factor of two of the wild-type PLD. Therefore, neither of these His could act as a nucleophile in the PLD mechanism. However, these two do have an important role in maintaining the structure of the protein.
Fig. 4.

Comparison of secondary structure for rPLD (solid bars), H187A (//// bars), H187N (shaded bars), H200A (\\\\ bars), and H200N (open bars) as estimated from the CD spectrum using ellipticities in the region 190–260 nm. The error bars on the elements for wild-type PLD represent standard deviations for multiple determinations of secondary structure using rPLD, M1-PLD, and N-PLD.
A fifth histidine, His72, was also mutated to Asn. Although it did not appear to be part of an HKD motif, it had all three amino acids but in a slightly different sequence: HTVKAD. As with the other four His residues, the H72N mutant was active; hence, it is unlikely to have a catalytic role similar to active site His in the Ca2+-independent PLDs.
The binding of the active histidine mutants to PC sonicated vesicles in the absence and presence of metal ion and to PA and PC/PA SUVs was examined (Table 3). None of the proteins bound tightly to PC SUVs in the absence of Ba2+, but with this ion (10 mM), essentially all the protein could be partitioned to the PC vesicles. Because all mutants exhibited about the same affinity for PA and PA/PC vesicles, the difference in PA activation between wild-type PLD and H226A cannot reflect enhanced PA binding.
Table 3.
Binding of recombinant PLD and mutants to phospholipid small unilamellar vesicles
| %PLD bound to vesiclesa | ||||
| PC SUVs | PA SUVs | PC/PA (9:1) | ||
| PLD | +EDTA | +Ba2+ | +EDTA | +EDTA |
| rPLD | 14 | 100 | 100 | 70 |
| H226A | 8 | 100 | 92 | 77 |
| H171A | 18 | 64 | 100 | 86 |
| H187N | 24 | 90 | 92 | 61 |
| C123A | 5 | 75 | 100 | 54 |
a SUVs composed of 2 mM POPC or POPC/POPA (1.8 mM:0.2 mM) in 10 mM Tris HCl at pH 8.0, with either EDTA (5 mM) or Ba2+ (10 mM) in the vesicle binding buffer. PLD concentration was 48 μg/ml.
The higher PA activation of H226A could be caused by differences in Ca2+ binding. To assess this, we examined the intrinsic fluorescence of rPLD and the H226A and H187N mutant proteins as a function of added Ca2+ (Fig. 5 ▶). These three active PLDs have a range of PA activations: 2.0, 1.4, and 12.2 for rPLD, H187N, and H226A, respectively (Table 2). All three show a biphasic fluorescence response with ∼15% decrease at lower Ca2+ concentrations followed by a slower loss of intensity (additional 10–15%) that is the smallest for H187N. This is different from the response of authentic PLD purified from the S. chromofuscus growth medium, which loses >80% of its fluorescence intensity in the same concentration range with a midpoint around 0.06 mM, then exhibits an increase in fluorescence with Ca2+ in the millimolar range (Stieglitz et al. 1999). Nonetheless, the fluorescence indicates at least two Ca2+ ions bind to both authentic and recombinant PLDs. The IC50 values for Ca2+ for the tight site (Table 2) are 0.10, 0.35, and 0.04 mM for rPLD, H187N, and H226A. The response of the PLD fluorescence to binding of Ca2+ to the weaker site is smallest for H187N, the enzyme with the smallest PA activation. This may suggest that differences in PA activation are related to binding of Ca2+ to the weak site, and that His187 may have a role in Ca2+ binding.
Fig. 5.

Intrinsic fluorescence intensity at 337 nm of PLD (24 μg/mL) as a function of added Ca2+: (open circles) rPLD; (filled circles) H226A; (filled triangles) H187N; and (filled squares) C123S.
Mutants of Cys123
S. chromofuscus PLD has a single cysteine residue that is not involved in disulfide bond formation, as the protein is a monomer (Geng et al. 1998). To assess the role of this unique residue in the protein, two mutants, C123A and C123S, were prepared. Both mutants had secondary structure and thermostability similar to wild-type PLD but were very inactive. C123A had 0.0007% and C123S 0.014% the activity of wild-type PLD toward diC4PC. Clearly, the sulfhydryl group of cysteine has a key role in the catalytic mechanism. Because the serine mutant still has greatly reduced activity, the role of the sulfhydryl is not likely to be just hydrogen bonding. Although unlikely, Cys123 could be a ligand for the catalytically necessary Ca2+. The intrinsic fluorescence of C123S showed a decrease with added Ca2+, consistent with the behavior of wild-type PLD (Fig. 5 ▶). In the wild-type PLD and all the mutants, the loss of intensity in this initial phase was 13–17%, with an average IC50 value of 0.13 ± 0.06 mM (0.075 ± 0.013 if H187N is excluded). The relatively inactive C123S exhibited the same behavior as active H187N, indicating that replacement of cysteine by serine did not affect Ca2+ binding. Therefore, the cysteine must have another role in PLD cleavage of phospholipids.
C-terminal truncation of PLD
Previous studies showed that intact S. chromofuscus PLD could be cleaved into two fragments by proteinases secreted into the S. chromofuscus medium (Geng et al. 1999). The tightly associated fragments had a higher specific activity but lacked PA activation. Separation of the two pieces generated an N-terminal fragment that lost activity over a day or two. Therefore, an attempt was made to construct a C-terminally truncated PLD. The pld gene was truncated by 530 bp and overexpressed with the intein protein. The activity of the crude extract containing the intein-truncated PLD was tested, and no PLD activity was detected. For comparison, the fusion protein of wild type, H171A, or H226A had detectable PLD activity in the crude extract (typically 0.04 μmole min−1 mg−1). Unfortunately, after DTT induced cleavage of the fusion protein, no truncated PLD could be detected eluting from the column. This observation suggested that the truncated PLD was misfolded. Perhaps the intact PLD sequence is necessary for the protein to fold into a stable structure.
Discussion
The PLD from S. chromofuscus is unique in that it is a phospholipase, but shows virtually no sequence homology with other protein members of the PLD superfamily. The only region of similarity to the other members of PLD superfamily is a local PLD "X" region that has nothing to do with the active site (Iwasaki et al. 1994). Even compared to other PLD proteins isolated from Streptomyces it is quite different. It has no identifiable HKD motifs and is a Ca2+-dependent phosphodiesterase. Furthermore, it exhibits an interesting kinetic activation by product PA (Geng et al. 1998), and, when secreted by S. chromofuscus and cleaved by protease that is also secreted, it has higher activity. The clipped PLD is also fusogenic (Stieglitz et al. 2001). To understand its unusual kinetics and interactions with membranes, a more detailed characterization of the enzyme is necessary.
The present work reports the sequencing and overexpression of the S. chromofuscus PLD gene cloned from the ATCC type strain. The gene was amplified with synthetic primers designed from the sequence of an S. chromofuscus PLD published previously (Yoshioka et al. 1991). Therefore, in our original recombinant PLD the N and C terminus were constructed to be the same as the previous strain. However, when the recombinant PLD gene, which contained the signal sequence, was sequenced by primer walking, it was noted that the real N-terminal amino acids were TTGT instead of ADQA. Furthermore, there were 64 amino acid changes aside from this change when the two S. chromofuscus sequences were compared. Because the specific activity of our purified recombinant PLD is similar to the PLD activity reported for the one sequenced previously, none of these changes must be critical for activity. In particular, the N terminus must have little influence on catalysis. Both Vmax and Km were comparable to those for PLD purified from S. chromofuscus medium.
The structure of Streptomyces sp. PMF PLD shows 494 amino acids visible in the electron density map. Two histidines in S. chromofuscus PLD, His187 and His200, are located approximately in the same region as His170 (one of the HKD motif His) in the Streptomyces sp. PMF PLD. In particular, His187 is located in a HxR(x)3D region that could be a variation of the HxK(x)4D motif. Mutations of these and two other nearby histidines (His171 and His226) can assess the likelihood that they are required for catalysis. Of the five histidines mutated (including His72), none is required for catalysis, although two are critical for the protein to adopt the right secondary structure. Although the enzyme contains other histidines, none of these is likely to be part of an HKD motif.
If the S. chromofuscus PLD does not have an HKD motif, is there any other feature of the protein that is critical for activity? The unique cysteine of this PLD appears to be very necessary for catalytic activity. The catalytic mechanism of two members of the PLD superfamily, cabbage PLD and E. coli phosphatatdylserine synthase have been studied (Stanacev and Stuhne-Sekalec 1970; Raetz et al. 1987). A two-step catalytic mechanism involving a covalent phosphorylenzyme intermediate was proposed and confirmed for a tyrosyl-DNA phosphodiesterase in the PLD superfamily (Interthal et al. 2001). If S. chromofuscus works by a similar mechanism, it is possible that a different group acts as a nucleophile to attack the phosphodiester bond. Our results raise the possibility that cysteine functions as such in the S. chromofuscus PLD. This enzyme would then be the first of a new class of PLD enzymes which carry out the phospholipid cleavage reaction in a Ca2+-dependent fashion, have no HKD motifs, and for which a cysteine is required for catalysis.
The availability of rPLD and the ability to make mutants has also provided insight into PA activation of the enzyme. The PA activation of recombinant wild-type PLD and most of the mutants was lower than that of the authentic PLD purified from the growth medium. The one interesting exception was H226A, which displayed a 12-fold activation that is twice as high as that for authentic PLD. The extent of PA activation did not correlate with the IC50 for Ca2+ or the affinity of the protein for PA. Although a more detailed analysis of this effect will require a structure for the S. chromofuscus PLD, it is possible that the higher PA activation of H226A might arise from the conformation change when the small alanine side chain replaces the imidazole ring, and this makes the activator site more accessible to PA.
Material and methods
Chemicals
Phospholipids including diC4PC, POPC, and POPA were purchased from Avanti as chloroform solutions and used without further purification. Palmitoyl chloride, chloroform, acetone, and pyridine were purchased from Aldrich. SDS-PAGE molecular weight markers, NaCl, dithiothreitol, and cellulose were purchased from Sigma. Q-Sepharose fast flow resin was obtained from Pharmacia. Restriction enzymes and plasmids pTYB11 and ER2566 were purchased from New England BioLabs. The pET-23a(+) vector, ligation kit, BL21(DE)3plysS, and Novablue E. coli strains were purchased from Novagen. The Advantage GC genomic PCR kit was purchased from Clonetech. The DNA extraction and Quik-Change site-directed mutagenesis kits were obtained from Stratagene. Oligonucleotide primers were purchased from Operon Technology.
Media and plates
LB broth containing 10 g of tryptone, 5 g of yeast extract, and 10 g of NaCl per liter with the pH adjusted to 7.2 was used for growth of E. coli suspensions. LB plates had 100 μg/mL carbenicillin and 34 μg/mL chloroamphenicol when used for recombinant plasmid transformed BL21(DE)3plysS and had 100 μg/mL of carbenicillin when used for growth of transformed Novablue and ER2566 cells.
DNA isolation
Frozen pellets (∼1 g) of the S. chromofuscus cells grown from the ATCC #23616 strain were used to prepare genomic DNA by pronase lysis; proteins were salted out using the standard NaCl solution. The genomic DNA was recovered by ethanol precipitation and resuspended in 10 mM Tris buffer (pH 8.0), 0.1 mM EDTA.
Cloning of S. chromofuscus (ATCC #23616) pld gene into pET-23a(+)
Two oligonucleotide primers were designed based on the published S. chromofuscus PLD DNA sequence (Yoshioka et al. 1991). Initially, the forward primer was designed to amplify the whole precursor protein including the signal sequence. 5′-attatt atatatCATATGgtgctggccggcccgctcgccgccgccctt-3′ (containing an NdeI site as indicated by the capitals) and 5′-ataaaatttGAATTCct actcggggtcgtaggtgcgctcgaccctct-3′ (containing an EcoRI site) were used to amplify the 1575 bp pld gene with high-fidelity pfu DNA polymerase. The 30-cycle PCR products were cut with NdeI and EcoRI and ligated to the NdeI- and EcoRI-digested pET-23a(+). The constructed plasmid (pssPLD), was transformed into BL21(DE)3plysS. Upon induction with IPTG, little recombinant PLD was expressed, although PLD activity could be detected in the lysed cells (no PLD activity was detected prior to induction). Considering the potential toxicity of PLD (the enzyme is relatively nonspecific [Geng et al. 1999] and can hydrolyze a variety of phospholipids in the E. coli membrane to PA that not only changes the local charge of the membrane but can be fusogenic as well) and the possibility that the E. coli cells may not recognize the S. chromofuscus signal sequence, we removed the signal sequence codons using 5′-attatatatCATATGgccgaccaggcgcccgccttcctgcacggcgtc-3′ as the forward primer based on the published DNA sequence of mature PLD. Overexpression of the plasmid, pmPLD, containing that signal-less gene yielded mature protein (designated M1-PLD) with an initial methionine residue. To sequence the whole recombinant pld gene in pmPLD, several oligonucleotide primers were designed by the primer walking method. The Molecular Medicine Unit, Beth Israel Deaconess Medical Center, carried out the DNA sequencing. Later sequencing of the pld insert in pssPLD showed the initial sequence following the start of the mature protein to be TTGT rather than ADQA as in the published S. chromofuscus PLD (Yoshioka et al. 1991). However, because the specific activity of M1-PLD was comparable to that of protein purified from S. chromofuscus, the N-terminally modified material was used for further studies.
Subcloning the pld gene into pTYB11
The IMPACT-CN fusion protein expression system from New England Biolabs was also used for expression of PLD. Two oligonucleotides, 5′-ggtggttGCTCTTCCAACgccgaccaggcgcccgcctt cctgcacggcg-3′ (containing a SapI site), and 5′-ataaaatttGAAT TCctactcggggtcgtaggtgcgctcgaccctct-3′ (containing an EcoRI site) were used to amplify the S. chromofuscus pld gene (1533 bp) from pmPLD with pfu DNA polymerase. The 30-cycle PCR products were double digested with SapI-EcoRI and ligated to similarly digested pTYB11 to form pfPLD. Upon ligation, the SapI site was lost in the recombinant plasmid. Translation yielded the PLD protein fused to an intein containing a chitin-binding domain. Positive clones were identified by restriction digestion with NcoI (for which there is a single site in the pTYB11 vector and not in the cloned pld gene) and EcoRI. The recombinant PLD sequence was further confirmed by automatic double-strand DNA sequencing. The plasmid containing this fusion protein, pfPLD, was used for constructing single-site mutations. A modified gene that would code for PLD containing the true first four amino acids as determined from sequencing the insert in pssPLD was also inserted into the pTYB11 vector to produce pfNPLD. The SapI primer used to construct that pld gene from pfPLD was 5′-ggtggttGCTCTTCC AACaccaccgggacccccgccttcctgcacggcg-3′
Overexpression and purification of recombinant PLD cloned in pET-23a(+)
The recombinant plasmid pmPLD was transformed into BL21(DE)3pLysS competent cells for expression. A single colony of BL21(DE)3pLysS containing pmPLD plasmid was grown in 5 mL of LB medium containing carbenicillin and chloroamphenicol until OD600 reached 0.6. Cell pellets from the 5-mL culture were resuspended in 1 mL fresh LB medium and used to inoculate 2 L fresh medium. Cultures were grown with rapid shaking (200 rpm) at 37°C to OD600 ∼0.7. Overexpression of M1-PLD was induced by the addition of IPTG to a final concentration of 0.4 mM. After induction, cultures were incubated at 28°C for 8 h (continued growth at 37°C after induction yielded much less M1-PLD in the soluble fraction). Cells were harvested by centrifugation and stored at −80°C until needed. The crude cell extract had high PLD activity and a band ∼55 kD on SDS-PAGE, whereas the BL21(DE)3pLysS/pET-23a(+) cell extract (cells containing the plasmid without the pld gene inserted) did not have the corresponding band on SDS-PAGE or any detectable PLD activity.
The PLD purification procedure was a variation of that described by Imamura and Horiuti (1979). Frozen pellets (6 g) were thawed at room temperature and resuspended in 20 mM Tris HCl (pH 8.0). The suspensions were sonicated for 10 × 30 sec on ice, and the supernatant was separated from cell debris by centrifugation (12,400g for 30 min). The solubilized protein solution (∼45 mL) was treated first with acetone (30 mL). After centrifugation (12,400g for 30 min), the supernatant was mixed with 150 mL acetone (final concentration 80% v/v) to precipitate the PLD. The mixture was centrifuged again (same conditions) and the supernatant (containing little PLD activity) was decanted. The precipitate was dried at room temperature and then resuspended in 10 mL of 10 mM Tris HCl (pH 8.0) containing 1% KCl. Insoluble material was removed by centrifugation; the supernatant was applied to a 1.5 cm × 4 cm column of palmitoyl-cellulose synthesized as described previously (Imamura and Horiuti 1979). The column was washed with 1 mM Tris-HCl buffer (pH 8.0) until the effluent showed low absorbance at 280 nm. M1-PLD was eluted with the same buffer containing 0.2% (w/v) Triton X-100. Fractions with PLD activity were pooled and directly applied to a Q-sepharose fast-flow column equilibrated with 20 mM Tris HCl (pH 8.0). PLD was eluted with a gradient of 0–0.5 M KCl in 20 mM Tris HCl (pH 8.0). The purity of each fraction was monitored by SDS-PAGE. Fractions with more than 90% purity were pooled and concentrated to 1.0 mg/mL. The PLD stock solution could be stored for 4 wk at 4°C without loss of activity.
Overexpression and purification of recombinant PLD cloned in pTYB11
The recombinant plasmid pfPLD (or pfNPLD) was transformed into ER2566 (supplied with the IMPACT-CN kit) for expression. A single colony of ER2566 containing the recombinant plasmid was grown in 5 mL of LB medium with 100 μg carbenicillin until OD600 reached 0.6. The cell pellet from the 5 mL culture was resuspended in 1 mL fresh LB medium and used to inoculate 1 L fresh LB medium (containing the carbenicillin). The culture was grown at 37°C with rapid shaking (200 rpm) to OD600 ∼0.7, then IPTG was added to a final concentration of 0.4 mM. Incubation of the culture at 15°C overnight yielded the optimum of fusion protein. Cells were harvested by centrifugation and stored at −80°C until needed.
Frozen cell pellets were thawed at room temperature, resuspended in 48 mL 20 mM Tris HCl (pH 8.0), then sonicated for 10 × 30 sec on ice, and centrifuged (12,400g for 30 min) to remove cell debris. The supernatant was slowly (0.5 mL/min) loaded onto an affinity column of chitin beads (∼20 mL) equilibrated with 0.5 M NaCl in 20 mM Tris HCl (pH 8.0). After loading, the column was washed with the same buffer containing 1 M NaCl until the OD280 reached zero. The protein splicing reaction was induced by the addition of 3 bed volumes of DTT (50 mM) to the column, which was then stoppered and incubated for 16 h. Cleaved PLD (designated rPLD from pfPLD and N-PLD from the plasmid with the N-terminal four residues corrected) was eluted from the resin with 20 mM Tris HCl (pH 8.0). Fractions containing PLD activity were pooled and concentrated to 1–6 mg/mL. The purity was more than 85% as monitored by SDS-PAGE. If further purification was needed, a QFF column was used.
C-terminal truncation of PLD
A new reverse oligonucleotide primer (5′-tgacggccagGAATTC gtggggcaggccgagca-3′ containing an EcoRI site) was designed based on the sequenced S. chromofuscus pld gene and the N-terminal sequence of "PLD20" (Geng et al. 1999). PCR with the original forward primer and the new reverse primer and pfPLD produced a 1003 bp fragment after 30 amplification cycles using high-fidelity pfu DNA polymerase. The PCR products were doubly digested with SapI and EcoRI and ligated to the digested pTYB11. Positive clones were identified by restriction mapping as described for the intact pld gene.
Site-specific mutagenesis of PLD
Five of 16 histidine residues, His72, His171, His187, His200, and His226, were mutated singly to alanine and in some cases asparagine using QuikChange methodology (Braman et al. 1996), pfPLD, and the appropriate primers (and their complements). Primers used included the following (the altered code is shown in capitals): 5′-gtcggccttgaccgtGTTgtcggaggcgg-3′ (H72N); 5′-cctccgtccgctcgGCGgcccccgcgcacgag-3′ (H171A); 5′-ctaccgcgtc cgggctggccggtacaagac-3′ (H187A); 5′-ctaccgcgtccggaacggccggt acaagac-3′ (H187N); 5′-gacctccaggcgctgGCTgccgccgccccggtcgt ggcgat-3′ (H200A); 5′-gacctccaggcgctgAACgccgccgccccggtc-3′ (H200N); 5′-ggcgccgagaacGCGaccgagggcgtcgagggc-3′ (H226A). Cys123 was also mutated to alanine (5′-ccgcttcggcgtcgtctccGCC gccaactgggaggccg-3′) and to serine (5′-ccgcttcggcgtcgtctccTCC gccaactgggaggccg-3′). All recombinant genes were sequenced (by the Molecular Medicine Unit, Beth Israel Deaconess Medical Center) to confirm the desired mutation. Mutant PLDs were expressed as intein-fusion proteins and cleaved and purified as for rPLD and N-PLD.
Preparation of small unilamellar vesicles
An appropriate aliquot of phospholipid dissolved in chloroform was dried under nitrogen gas for 1 h to remove organic solvent. The resulting film was dissolved in 5 mL water, frozen on dry ice, and lyophilized overnight. The phospholipid film was solubilized in 50 mM imidazole in D2O (pH 7.7), containing 5 mM Ca2+ in the case of kinetic assays and 5 mM EDTA or 10 mM Ba2+ for vesicles to be used in binding assays. The lipid suspension was sonicated on ice using a Branson W-150 sonifier with a 1-cm-diameter probe until maximum clarity was achieved. Vesicles were used the same day they were prepared.
PLD activity assays
Two methods were used to measure PLD specific activity. A pH-stat assay was used to measure the specific activity of wild-type PLD toward 5 mM diC4PC in the presence of 5 mM Ca2+. Hydrolysis of diC4PC to diC4PA was monitored with a Radiometer pH-stat model VIT90 as described previously (Geng et al. 1999) using 2 mM NaOH as the titrant. For each phospholipid concentration, assays were run in duplicate or triplicate. For assays comparing mutant activities with diC4PC or with POPC unilamellar vesicles as the substrate, 31P or 1H NMR spectra were acquired to monitor the release of diC4PA (31P) or choline (1H) products (Geng et al. 1998, 1999).
Circular dichroism spectroscopy
CD spectra of M1-PLD, PLD, N-PLD, and mutants were obtained at 25°C using an AVIV circular dichroism model 202 spectrometer with a 1-cm pathlength cylindrical quartz cell and averaging times of 1 sec. A baseline scan from 300 to 190 nm of the cell filled with buffer was subtracted from the spectra obtained in the presence of protein. The percentage of secondary structure elements was deconvoluted using the program CDNN (Bohm et al. 1992; Andrade et al. 1993). Temperature-induced changes in the ellipticity (measured in millidegrees) at 222 nm were monitored at 1° intervals from 25 to 100°C. The temperature was maintained to within ±0.2° with 1-min equilibration time between each increase in temperature. The thermal melting transition, Tm, of the protein was determined by plotting the derivative of the [θ]222 versus temperature scan; the Tm is the maximum in the derivative spectrum.
PLD intrinsic fluorescence
Fluorescence studies of PLD (24 μg/mL in 150 mM HEPES at pH 8.0) to monitor Ca2+ binding (Stieglitz et al. 1999) were carried out at 25°C using a Shimadzu RF5000U fluorimeter with excitation at 290 nm and emission monitored at 337 nm.
Vesicle-binding assays
Solutions of PLD (48 μg/mL in 10 mM Tris HCl at pH 8.0) incubated with SUVs of POPC or POPC/POPA (9:1) and a total phospholipid concentration of 2 mM were centrifuged through an Amicon centricon-100 filter to separate free from vesicle-bound enzyme. Filtrates were lyophilized and analyzed as described previously (Stieglitz et al. 1999) for the amount of free PLD (PLDf) by SDS-PAGE. The percentage of PLD bound to vesicles was estimated as (PLDo − PLDf)/PLDo, where PLDo is the total amount of PLD.
Acknowledgments
This work has been supported by NIH grant GM26762.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0225302.
References
- Andrade, M.A., Chacon, P., Merelo, J.J., and Moran, F. 1993. Evaluation of secondary structure of proteins from UV circular dichroism spectra using an unsupervised learning neural network. Protein Eng. 6 383–390. [DOI] [PubMed] [Google Scholar]
- Bohm, G., Muhr, R., and Jaenicke, R. 1992. Quantitative analysis of protein far UV circular dichroism spectra by neural networks. Protein Eng. 5 191–195. [DOI] [PubMed] [Google Scholar]
- Braman, J., Papworth, C., and Greener, A. 1996. Site-directed mutagenesis using double-stranded plasmid DNA templates. Methods Mol. Biol. 57 31–44. [DOI] [PubMed] [Google Scholar]
- Dawson, R.M.C. 1967. The formation of phosphatidylglycerol and other phospholipids by the transferase activity of phospholipase D. Biochem. J. 102 205–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eible, H. and Kovatchev, S. 1981. Preparation of phospholipids and their analogues by phospholipase D. Methods Enzymol. 72 632–639. [DOI] [PubMed] [Google Scholar]
- Ella, K.M., Dolan, J.W., and Meier, K.E. 1995. Characterization of a regulated form of phospholipase D in the yeast Saccharomyces cerevisiae Biochem. J. 307 799–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Exton, J.H. 2000. Phospholipase D. Ann. N.Y. Acad. Sci. 905 61–68. [DOI] [PubMed] [Google Scholar]
- Geng, D., Chura, J., and Roberts, M.F. 1998. Activation of phospholipase D by phosphatidic acid. Enhanced vesicle binding, phosphatidic acid-Ca2+ interaction, or an allosteric effect? J. Biol. Chem. 273 12195–12202. [DOI] [PubMed] [Google Scholar]
- Geng, D., Baker, D.P., Foley, S.F., Stieglitz, K., and Roberts, M.F. 1999. A 20-kDa domain is required for phosphatidic acid-induced allosteric activation of phospholipase D from Streptomyces chromofuscus. Biochim. Biophys. Acta 1430 234–244. [DOI] [PubMed] [Google Scholar]
- Grossman, S., Cobley, J., Hogue, P.K., Kearney, E.B., and Singer, T.P. 1973. Relation of phospholipase D activity to the decay of succinate dehydrogenase and of covalently bound flavin in yeast cells undergoing glucose repression. Arch. Biochem. Biophys. 158 744–753. [DOI] [PubMed] [Google Scholar]
- Hatanaka, T., Negishi, T., Kubota-Akizawa, M, and Hagishita, T. 2002. Study on thermostability of phospholipase D from Streptomyces sp. Biochim. Biophys. Acta 1598 156–164. [DOI] [PubMed] [Google Scholar]
- Imamura, S. and Horiuti, Y. 1979. Purification of Streptomyces chromofuscus phospholipase D by hydrophobic affinity chromatography on palmitoyl cellulose. J. Biochem. 85 79–95. [DOI] [PubMed] [Google Scholar]
- Interthal, H., Pouliot, J.J., and Champoux J. 2001. The tyrosyl-DNA phosphodiesterase Tdp1 is a member of the phospholipase D superfamily. Proc. Natl. Acad. Sci. U.S.A. 98 12009–12014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki, Y., Nakano, H., and Yamane, T. 1994. Phospholipase D from Streptomyces antibioticus: Cloning, sequencing, expression, and relationship to other phospholipases. Appl. Microbiol. Biotechnol. 42 290–299. [DOI] [PubMed] [Google Scholar]
- Juneja, L.R., Kazuoka, T., Yamane, T., and Shimizu, S. 1988. Kinetic evaluation of phosphatidylcholine to phosphatidylethanolamine by phospholipase D from different sources. Biochim. Biophys. Acta 960 334–341. [DOI] [PubMed] [Google Scholar]
- Koonin, E.V. 1996. A duplicated catalytic motif in a new superfamily of phosphohydrolases and phospholipid synthases that includes poxvirus envelope proteins. Trends Biochem. Sci. 21 242–243. [PubMed] [Google Scholar]
- Leiros I., Secundo, F., Zambonelli, C., Servi, S., and Hough, E. 2000. The first crystal structure of a phospholipase D. Struct. Fold Des. 8 655–667. [DOI] [PubMed] [Google Scholar]
- McNamara, P.J., Cuevas, W.A., and Songer, J.G. 1995. Toxic phospholipases D of Corynebacterium pseudotuberculosis, C. ulcerans and Arcanobacterium haemolyticum: Cloning and sequence homology. Gene 156 113–118. [DOI] [PubMed] [Google Scholar]
- Okawa, Y. and Yamaguchi, T. 1975. Studies on phospholipases from Streptomyces. II. Purification and properties of Streptomyces hachijoensis phospholipase D. J. Biochem. 78 363–372. [DOI] [PubMed] [Google Scholar]
- Ponting, C.P. and Kerr, I.D. 1996. A novel family of phospholipase D homologues that includes phospholipid synthases and putative endonucleases: Identification of duplicated repeats and potential active site residues. Protein Sci. 5 914–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raetz, C.R., Carman, G.M., Dowhan, W., Jiang, R.T., Waszkuc, W., Loffredo, W., and Tsai, M.D. 1987. Phospholipids chiral at phosphorus. Steric course of the reactions catalyzed by phosphatidylserine synthase from Escherichia coli and yeast. Biochemistry 26 4022–4027. [DOI] [PubMed] [Google Scholar]
- Shimbo, K., Iwasaki, Y., Yamane, T., and Ina, K. 1993. Purification and properties of phospholipase D from Streptomyces antibioticus. Agric. Biol. Chem. 57 1946–1948. [Google Scholar]
- Stanacev, N.Z. and Stuhne-Sekalec, L. 1970. On the mechanism of enzymatic phosphatidylation. Biosynthesis of cardiolipin catalyzed by phospholipase D. Biochim. Biophys. Acta 210 350–352. [DOI] [PubMed] [Google Scholar]
- Stieglitz, K., Seaton, B., and Roberts, M.F. 1999. The role of interfacial binding in the activation of Streptomyces chromofuscus phospholipase D by phosphatidic acid. J. Biol. Chem. 274 35367–35374. [DOI] [PubMed] [Google Scholar]
- Stieglitz, K., Seaton, B., and Roberts, M.F. 2001. Binding of proteolytically processed phospholipase D from Streptomyces chromofuscus to phosphatidylcholine membranes facilitates vesicle aggregation and fusion. Biochemistry 40 13954–13963. [DOI] [PubMed] [Google Scholar]
- Stuckey, J.A. and Dixon, J.E. 1999. Crystal structure of a phospholipase D family member. Nat. Struct. Biol. 6 278–284. [DOI] [PubMed] [Google Scholar]
- Wang, X. 2000. Multiple forms of phospholipase D in plants: The gene family, catalytic and regulatory properties, and cellular functions. Prog. Lipid Res. 39 109–149. [DOI] [PubMed] [Google Scholar]
- Yoshioka, K., Mizoguchi, M., Takahara, M., Imamura, S., Beppu, T., and Horinuchi, S. 1991. DNA having the genetic information of phospholipase D and its uses. European patent 0435725B1.
- Zhao, Y., Stuckey, J.A., Lohse, D.L., and Dixon J.E. 1997. Expression, characterization, and crystallization of a member of the novel phospholipase D family of phosphodiesterases. Protein Sci. 6 2655–2658. [DOI] [PMC free article] [PubMed] [Google Scholar]