

Abstract
We conducted a Phase I clinical trial investigating the biologic activity of vaccination with irradiated autologous melanoma cells engineered to secrete human granulocyte–macrophage colony-stimulating factor in patients with metastatic melanoma. Immunization sites were intensely infiltrated with T lymphocytes, dendritic cells, macrophages, and eosinophils in all 21 evaluable patients. Although metastatic lesions resected before vaccination were minimally infiltrated with cells of the immune system in all patients, metastatic lesions resected after vaccination were densely infiltrated with T lymphocytes and plasma cells and showed extensive tumor destruction (at least 80%), fibrosis, and edema in 11 of 16 patients examined. Antimelanoma cytotoxic T cell and antibody responses were associated with tumor destruction. These results demonstrate that vaccination with irradiated autologous melanoma cells engineered to secrete granulocyte–macrophage colony-stimulating factor stimulates potent antitumor immunity in humans with metastatic melanoma.
There is compelling evidence that malignant melanoma can evoke specific humoral and cellular antitumor immune responses in some patients. The radial growth phase of primary melanoma is regularly associated with a significant dermal lymphocytic reaction, often resulting in partial tumor destruction (1). Clonal expansion of T cells occurs in primary regressing melanoma; lymphocytes explanted from such lesions demonstrate cytotoxicity toward autologous melanoma cells in vitro (2, 3). Although a brisk lymphocytic infiltrate in the vertical growth phase of primary melanoma occurs infrequently, this response is tightly correlated with prolonged survival and a reduced incidence of metastatic disease (4, 5). Melanoma that has spread to regional lymph nodes may occasionally elicit a striking lymphocytic reaction that is also highly associated with improved survival (6). Finally, in rare cases, widely disseminated melanoma may undergo spontaneous regression accompanied by a diffuse infiltrate of lymphocytes, plasma cells, and macrophages (7). Notwithstanding these provocative findings, however, it is clear that most patients fail to develop antimelanoma immune responses that are sufficiently potent to prevent lethal tumor progression.
The application of gene transfer technologies to investigative efforts in tumor immunology has led to the development of several strategies to enhance the frequency and intensity of antitumor immune responses (8). A large number of preclinical studies have convincingly demonstrated that engineering murine tumor cells to express a variety of immunostimulatory molecules can lead to enhanced tumor immunogenicity. Among the approaches using ex vivo modification of tumor cells, we have shown that vaccination with irradiated tumor cells engineered to secrete granulocyte–macrophage colony-stimulating factor (GM-CSF) stimulates potent, specific, and long-lasting antitumor immunity in multiple murine tumor model systems, including malignant melanoma (9). Immunization requires the participation of both CD4- and CD8-positive T lymphocytes and likely involves improved tumor antigen presentation by dendritic cells and macrophages recruited to the vaccination site. To evaluate whether this immunization strategy can also augment antitumor immunity in humans and to determine whether the elicited immune responses differ from those previously observed with traditional immunization schemes, we conducted a Phase I clinical trial of vaccination with autologous lethally irradiated melanoma cells engineered to secrete human GM-CSF in patients with metastatic melanoma.
METHODS
Clinical Protocol.
The details of the study design and methods of vaccine production have been presented previously (10–12). In brief, surgically resected tumors were processed to single-cell suspension by collagenase and mechanical digestion and were introduced into short-term culture. Replicating tumor cells were transduced with viral supernatants harvested from CRIP packaging cell lines transfected with MFG-S-human GM-CSF, irradiated with 15,000 cGy and cryopreserved in liquid nitrogen. Transduced cells were certified to be free of replication-competent retrovirus, endotoxin, mycoplasma, and other microbial contaminants. GM-CSF secretion was determined by ELISA (R & D Systems). A portion of the tumor culture for use in delayed-type hypersensitivity evaluation was irradiated but not transduced. Frozen cells were thawed and washed in Hanks’ balanced salt solution before injection; vaccines were administered intradermally (0.5 ml) and subcutaneously (0.5 ml) into normal skin on the limbs and abdomen on a rotating basis. Nontransduced cells were injected intradermally (0.5 ml) into normal skin at the time of beginning vaccination and then at monthly intervals to measure the generation of delayed-type hypersensitivity. Patient sera were tested regularly for replication-competent retrovirus; all samples were negative.
Immunologic Analyses.
Peripheral blood mononuclear cells were obtained by centrifugation over Ficoll gradients. Tumor-infiltrating lymphocytes were prepared by mechanical digestion of metastatic deposits. For cytokine assays, lymphocytes were cocultured with irradiated autologous melanoma cells in 24-well dishes in 2 ml of DMEM plus 10% fetal calf serum, antibiotics, 2-mercaptoethanol, and glutamine. Media were harvested at day 8 and assayed for interleukin (IL)-3, IL-4, IL-5, IL-6, IL-10, GM-CSF, interferon (IFN)-γ, tumor necrosis factor (TNF)-α, and TNF-β production by ELISA, using the appropriate monoclonal antibodies (PharMingen). For cytotoxicity assays, lymphocytes were bulk stimulated with irradiated autologous tumor cells for 1 week in media plus 10 units per ml IL-2 and then tested by using standard techniques against 51Cr-labeled tumor targets (13).
For immunoblotting analysis, 0.5–2.0 mg of melanoma cell lysates (phosphate-buffered saline supplemented with 0.5% Nonidet P-40, soybean trypsin inhibitor, leupeptin, pepstatin, aminocaproic acid, and phenylmethylsulfonyl fluoride) was electrophoresed on SDS-polyacrylamide 4–12% gradient gels, transferred to Immobilon membranes (Millipore), and blocked overnight at room temperature in 5% nonfat dry milk in PBS. Membranes were probed overnight at 4°C in a 1:100 dilution of patient sera (in Tween 20/Tris buffered saline), washed, and incubated for 1 hour at room temperature with an antihuman IgG, Fcγ-specific antibody conjugated to alkaline phosphatase (Jackson ImmunoResearch). The membranes were developed then with nitroblue tetrazolium and 5-bromo-4-chloro-3-indoyl phosphate (BCIP) (Promega).
For flow cytometry analysis, at least 100,000 melanoma cells were incubated with a 1:100 dilution of patient serum (in 1% nonfat milk) for 3 hours on ice, washed, and then stained with a 1:100 dilution of antihuman IgG, Fcγ-specific antibody conjugated to fluorescein isothiocyanate.
RESULTS
Vaccine Production and Administration.
Thirty-three metastatic melanoma patients (stage IV) ranging from 32 to 82 years of age (18 women, 15 men) were enrolled in the clinical protocol (10). Twenty had received prior therapies. Patients underwent a surgical procedure to remove a metastatic lesion for vaccine preparation. Tumors were harvested from soft tissue (14 patients), lymph node (9 patients), lung (6 patients), liver (3 patients), and adrenal (1 patient). Two patients were excluded from the study after enrollment because of the absence of melanoma in the surgical specimen and two were excluded because vaccines could not be produced. In the remaining 29 patients, vaccines were successfully generated, achieving GM-CSF secretion rates ranging from 84 to 965 ng per 106 cells per 24 hours. The duration of vaccine preparation was generally 8 weeks (range 8–32). Three successive patient cohorts were immunized intradermally and subcutaneously with 107 irradiated tumor cells (each treatment) administered at 28-, 14-, or 7-day intervals (dose levels 1, 2, and 3, respectively) for a total of 84 days (total of 3, 6, or 12 vaccinations). Four patients at dose level 3 received additional vaccinations (up to a total of 24) after the first course of therapy. Three patients were withdrawn from study after tumor harvest and before vaccination because of rapid disease progression. Five patients were withdrawn from study after beginning vaccination because rapid disease progression prevented administration of the full course of immunizations. Twenty-one patients completed therapy (three at dose level 1, four at dose level 2, and fourteen at dose level 3), were fully evaluable for toxicity and biologic activity and comprised the study population reported here.
Toxicities.
Vaccination elicited erythema and induration at injection sites. Reactions were associated with local pruritus that was easily controlled with emollients. Grade 1 fatigue and nasal congestion were occasionally noted. No hepatic, renal, pulmonary, cardiac, hematologic, gastrointestinal, or neurologic toxicities were observed. No patient experienced vitiligo or autoimmune events.
Vaccination Reactions.
Injections of irradiated autologous GM-CSF-secreting melanoma cells evoked striking local reactions in all patients, with the intensity and duration of the responses generally increasing in proportion to the number of vaccines administered. Clinically, the reactions were characterized by substantial erythema (up to 35 cm in diameter) and induration (up to 14 cm in diameter). Occasionally, the reactions became hemorrhagic. Vaccination responses tended to peak at approximately 48 hours after cell injection, but the elicited induration could persist for several weeks, particularly after multiple immunizations. An intriguing observation was the frequent development of recall reactions at sites of previous vaccination. Several patients continued to experience these reactions intermittently in a mild form even after completion of therapy (for up to 2 years), although no clear precipitants were identified.
Vaccination sites in all patients were characterized histologically by an extensive infiltrate of dendritic cells, macrophages, eosinophils, and T lymphocytes that extended throughout the dermis and into the subcutaneous fat (Fig. 1A). The infiltrates at dose levels 2 and 3 were usually more cellular than those at dose level 1 and also resulted more frequently in the development of flame figures (collections of deposited eosinophil granules) and endothelial cell damage in the superficial venules of the upper dermis. Eosinophil degranulation in nerve sheaths, lymphocytic infiltration of hair follicles, and fat necrosis were observed in several patients as well.
Figure 1.
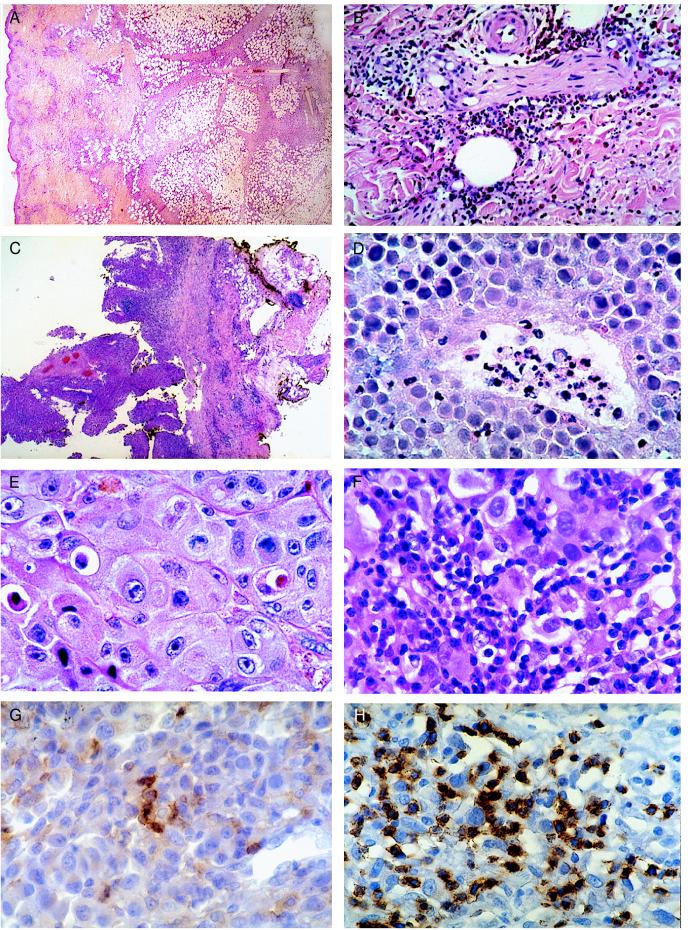
(A) Injection site of irradiated GM-CSF secreting melanoma cells. Note the extensive inflammatory reaction throughout all layers of the skin and the marked fibrosis in the subcutaneous fat. (B) Injection site of irradiated nontransfected melanoma cells after vaccination. Note the prominent infiltrate composed primarily of lymphocytes and eosinophils. (C) Melanoma metastasis after vaccination showing extensive necrosis and fibrosis. (D) Vasculopathy in metastasis after vaccination. (E) Absence of infiltrate in metastasis pretreatment. (F) Diffuse infiltrate of T lymphocytes and plasma cells in metastasis after vaccination. (G) CD4-positive T cell reaction in metastasis after vaccination. (H) CD8 positive T cell reaction in metastasis after vaccination.
Delayed-Type Hypersensitivity Reactions.
Although injections of irradiated autologous nontransfected melanoma cells failed to elicit significant responses in all patients at the time of beginning treatment, these injections evoked strong responses in all patients after several vaccinations were administered. Clinically, these delayed-type hypersensitivity reactions were characterized by extensive erythema (up to 10 cm) and induration (up to 6 cm), which peaked at 48 hours and then gradually resolved. Histopathologically, the reactions were characterized by dense infiltrates of T lymphocytes and degranulating eosinophils extending throughout the dermis (Fig. 1B). The infiltrates at dose levels 2 and 3 were usually greater than those at dose level 1.
Eosinophilia.
In addition to the striking involvement of eosinophils in the reactions to injections of both irradiated GM-CSF-secreting and irradiated nontransfected melanoma cells, significant increases in the numbers of peripheral blood eosinophils (but not other leukocytes) were also observed after immunization, with mean peak eosinophil counts of 705 ± 715, 515 ± 102, and 928 ± 571 per mm3 for dose levels 1, 2, and 3, respectively. The duration of eosinophilia tended to vary as a function of dose, with elevated counts persisting for several weeks more frequently at dose level 3 than at dose level 1 or 2.
Because eosinophilia in many model systems is T cell-dependent (14), we investigated whether vaccination induced alterations in peripheral blood T cell cytokine production. For these studies, peripheral blood mononuclear cells, obtained at varying times during treatment, were cultured with autologous irradiated nontransfected melanoma cells in the absence of supplemental growth factors; culture supernatants were harvested at day 8 and assayed for cytokine content by ELISA (Table 1). In nine of ten patients studied, vaccination elicited substantial levels of T cell-derived IL-5, IL-3, and GM-CSF, in contrast to the variable production of IL-4, IL-6, IL-10, and TNF-β and the negligible induction of IFN-γ. The enhanced T cell secretion of IL-3, IL-5, and GM-CSF as a consequence of vaccination likely contributed, at least in part, to the augmented production of eosinophils, as these molecules have been shown to enhance the proliferation of eosinophilic precursors in vitro and in vivo (15). Moreover, the persistence of distinctive cytokine profiles for several months after completing treatment suggests that immunization stimulated the development of memory T cells.
Table 1.
Cytokines produced by peripheral blood lymphocytes
| Treatment day | IL-3, pg/ml | IL-4, pg/ml | IL-5, ng/ml | IL-6, ng/ml | IL-10, pg/ml | GM-CSF, pg/ml | IFN-γ, pg/ml | TNF-β, pg/ml |
|---|---|---|---|---|---|---|---|---|
| Tumor | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Day −6 | 0 | 0 | 0 | 2.78 | 0 | 0 | 50 | 0 |
| Day 28 | 163 | 0 | 6.45 | 3.71 | 20 | 724 | 0 | 50 |
| Day 56 | 411 | 45 | 15.81 | 1.91 | 204 | 804 | 0 | 0 |
| Day 150 | 831 | 45 | 21.17 | 2.23 | 127 | 1,027 | 0 | 0 |
Irradiated autologous nontransfected melanoma cells (1 × 104) were cultured for 8 days with 1 × 106 peripheral blood mononuclear cells obtained at various times during treatment. Culture supernatants were harvested and analyzed by ELISA. “Tumor” represents the cytokines produced by the autologous irradiated nontransfected melanoma cells in the absence of lymphocytes. Vaccinations were administered on days 0, 28, and 56.
Immune Responses in Metastases.
To determine whether vaccination generated antimelanoma immune responses capable of inducing antitumor effects, we examined the host reactions to metastatic lesions resected before and after completing therapy. Metastatic lesions procured before the beginning of immunization revealed in all patients either the absence of host reactivity or only a modest inflammatory reaction present focally within the tumor (Fig. 1E). Metastatic lesions resected after the completion of immunization, however, demonstrated a profound immune response in 11 of 16 patients in which tissue could be obtained (Fig. 1C). These responses were found in metastatic lesions (up to 10 cm in diameter) derived from a variety of sites including skin, subcutaneous tissue, lymph node, lung, spleen, and intestine.
One important characteristic of the antimelanoma immune reaction in each of the 11 responding patients was the diffuse infiltration of tumor masses by large numbers of T lymphocytes and plasma cells (Fig. 1F). Many CD4- and CD8-positive T lymphocytes were organized into rosettes around dying melanoma cells (satellitosis), a morphologic pattern indicative of lymphocyte-induced tumor apoptosis (Fig. 1 G and H). Plasma cells accounted for nearly 50% of the inflammatory cells and were intimately associated with the T lymphocytes and melanoma cells. A second intriguing feature of the antimelanoma response, observed in four patients, was the targeted destruction of the tumor vasculature, whereby lymphocytes, eosinophils, and neutrophils were closely associated with dying tumor blood vessels (Fig. 1D). Overall, the chronic inflammatory reactions evident in these 11 patients resulted in substantial tumor destruction (at least 80%) and the development of significant edema and fibrosis throughout the resected metastases. Of the five patients failing to develop inflammatory infiltrates in metastatic lesions as a consequence of vaccination, two were treated at dose level 1 and three had rapidly progressive disease resulting in death shortly after completion of therapy. No significant differences in the metastatic immune responses were observed between dose levels 2 and 3.
Characterization of Antimelanoma Cellular and Humoral Immunity.
The functional properties of the lymphocytes infiltrating the metastatic lesions were examined in two patients and found to be comparable. When single-cell suspensions of the excised metastases were introduced into culture in the presence of low doses of IL-2 (10 units per ml), the inflammatory cells lysed all of the residual viable melanoma cells. To quantify cytotoxicity in a more formal way, we established additional primary cultures of the explanted metastases in which the nonadherent inflammatory cells were removed; viable melanoma cells could be propagated in this way. When the tumor-infiltrating lymphocytes (bulk cultured for 1 week with the autologous melanoma cells plus 10 units per ml of IL-2) were tested in a standard 4-hour cytotoxicity assay against the explanted melanoma cells, highly significant lysis of the autologous melanoma cells, but not of melanoma cells derived from another patient (differing major histocompatability complex profile), was observed (Fig. 2). In contrast, peripheral blood lymphocytes failed to lyse either of these targets (not shown). The tumor-infiltrating lymphocytes also demonstrated the ability to produce a broad range of cytokines in response to the autologous melanoma cells, a property that likely contributed to the enhanced T cell cytotoxicity and the prominent antitumor plasma cell response. Analysis of the conditioned medium obtained by coculturing the tumor-infiltrating lymphocytes and autologous melanoma cells revealed substantial levels of IL-4, IL-5, IL-6, IL-10, GM-CSF, and IFN-γ, but not TNF-β (Table 2). This cytokine profile indicates the coordinate expression of gene products that are associated with both Th1 and Th2 cells and suggests that multiple lymphocyte effector mechanisms can result in potent antitumor immune responses. Moreover, the substantial secretion of IL-10 is provocative, given the widely held view that this molecule is primarily immunosuppressive (16).
Figure 2.

Tumor-infiltrating lymphocytes obtained after vaccination specifically lyse melanoma cells derived from the resected metastasis. A 4-hour 51Cr release assay against the autologous tumor (K008) and a second melanoma line (E:T ratio of 50:1) derived from another patient (K012, differing major histocompatability complex class I profile) was performed.
Table 2.
Cytokines produced by tumor-infiltrating lymphocytes
| Source | IL-4, pg/ml | IL-5, ng/ml | IL-6, ng/ml | IL-10, pg/ml | GM-CSF, pg/ml | IFN-γ, pg/ml |
|---|---|---|---|---|---|---|
| TILs | 166 | 7.7 | 2.9 | 2095 | 241 | 171 |
| Metastasis | 0 | 0 | 1.12 | 8.4 | 0 | 0 |
Tumor-infiltrating lymphocytes (TILs) were cultured for 1 week with autologous melanoma cells in the presence of 10 units/ml IL-2; the supernatant was harvested and analyzed by ELISA. “Metastasis” refers to tumor cells cultured alone.
To determine whether the plasma cell infiltration of the metastatic lesions resulted in the generation of antibodies recognizing melanoma cells, we performed immunoblotting analysis using autologous melanoma cell lysates and sera obtained at various times during vaccination. Immunization stimulated the enhanced production of IgG antimelanoma antibodies in seven patients examined thus far (Fig. 3). The reactivity of postvaccination sera was characterized both by increased recognition of proteins detected by preimmunization sera and the recognition of proteins not detected by preimmunization sera. The induction of IgG antibodies recognizing surface melanoma cell determinants was also demonstrated in these patients by flow cytometry analysis; significant augmentation of reactivity to cultured melanoma cells as a function of vaccination was observed (Table 3). The specificity profiles of the antibodies elicited by immunization suggest the existence of several independent antigens.
Figure 3.

Melanoma cell lysates were immunoblotted with autologous serum. Lanes: A, pretreatment; B, 1 month after starting vaccination; C, 2 months after starting vaccination; D, 3 months after starting vaccination. Increased reactivity was specific for autologous cells, as testing of allogeneic cell lysates revealed different patterns of reactivity.
Table 3.
Vaccination stimulated antimelanoma antibodies
| Tumor | Serum
|
||||||
|---|---|---|---|---|---|---|---|
| K008 | K016 | K017 | K023 | K027 | K029 | K032 | |
| K008 M | ++ | + | + | ++ | +++ | ++ | + |
| K016 V | ND | 0 | ND | ND | ND | ND | ND |
| K017 V | 0 | ND | + | ND | ND | + | ND |
| K023 V | ND | ND | ND | 0 | ++ | 1/2+ | ND |
| K023 M | 1/2+ | ND | ND | + | ND | 0 | + |
| K027 M | +++ | + | 0 | 0 | 0 | 0 | + |
| K029 V | ++ | 0 | 1/2+ | 0 | + | 0 | 0 |
| K029 M | + | 0 | 1/2+ | 0 | + | 0 | 0 |
Melanoma cell lines were stained with sera obtained before and after vaccination, developed with an antihuman IgG Fcγ-specific antibody and analyzed by flow cytometry. Changes in reactivity as a function of vaccination are reported. M, cell line established from a metastasis removed following treatment; V, cell line established from a metastasis used to prepare the vaccine; +++, strong shift between pre- and post-immunization sera; ++, intermediate shift; +, small shift; 1/2+, borderline shift; 0, no shift; ND, not determined.
Clinical Outcome.
According to standard clinical criteria, one partial response (shrinkage of subcutaneous lesions), one mixed response, and three minor responses were observed. Three patients remain free of disease with followup of 36, 36, and 20 months; two were rendered disease-free by surgery (pathologic examination showed brisk lymphocyte and plasma cell infiltration with extensive tumor necrosis); one underwent radiation therapy for a scapular metastasis during vaccine preparation. Before beginning immunization, these patients had developed multiple new metastatic lesions.
DISCUSSION
The Phase I study presented here demonstrates that vaccination with irradiated autologous melanoma cells engineered to secrete GM-CSF consistently augments antitumor cellular and humoral immunity in patients with metastatic melanoma and should be considered for further clinical evaluation to define potential therapeutic efficacy. The most convincing evidence that this immunization scheme enhances antimelanoma immunity is the finding that distant metastases were frequently infiltrated by large numbers of T lymphocytes and plasma cells after, but not before, vaccination. Antimelanoma immune reactions were found in metastases, including bulky lesions, derived from a variety of sites and were documented pathologically in one patient to be persistent 5 months after the completion of therapy in all eight sites of metastatic disease (not shown). Immunohistochemical analysis demonstrated that both CD4- and CD8-positive T cells were in direct contact with dying melanoma cells. Analysis of the infiltrating T lymphocytes and plasma cells suggested several potential antitumor effector mechanisms including lymphocyte-mediated cytotoxicity, cytokine production, and antibody formation. Antimelanoma immune responses were more intense at dose levels 2 and 3 than at dose level 1; no clear relationship to the level of GM-CSF secretion could be delineated, though.
Pathologic assessment revealed that the coordinated activation of T lymphocytes and plasma cells resulted in destruction of at least 80% of the tumor cells in the infiltrated metastases. In most cases, however, these antitumor immune responses failed to induce clinical regressions; rather, the necrotic tumor masses were largely replaced by inflammatory cells, edema, and extensive fibrosis. These findings underscore the limitations of relying exclusively on traditional measurements of tumor shrinkage in assessing the antitumor activity of this vaccination scheme. Additional studies are required to clarify the mechanisms underlying the resistance of the residual tumor cells.
Many other strategies to enhance the frequency and intensity of antimelanoma immune responses are under active clinical investigation. These include vaccination with autologous, hapten-modified tumor cells in conjunction with bacillus Calmette–Guérin and cyclophosphamide (17); immunization with allogeneic melanoma cells in a variety of forms, including intact cells or shed antigens with bacillus Calmette–Guérin, viral-modified cell lysates, and cell lysates admixed with complex adjuvants (18–22); and vaccination with defined melanoma antigens such as the ganglioside GM2 or peptides derived from the MAGE and melanocyte differentiation protein families (23–26). Whereas differences in patient population and immunologic evaluation render it difficult to compare the results of these vaccination schemes with the findings reported here, several similarities with the studies using hapten-modified tumor cell vaccinations nonetheless are evident. These similarities include the frequent infiltration of distant metastases with T lymphocytes, the stimulation of antimelanoma cytotoxic T cells, and the expression of IL-4, IFN-γ, and IL-10 in the inflamed metastases (17, 27–31). However, the prominent plasma cell infiltration, IgG antibody response, extensive fibrosis, and vasculopathy observed in the current work have not been described with hapten-modified tumor cell vaccines. Further investigations are required to characterize these differences more thoroughly and to determine whether the mechanisms underlying the two vaccination strategies involve distinct or overlapping pathways.
Several additional features of the antitumor immune responses elicited here underscore distinctive properties of this immunization scheme. First, all patients developed impressive admixtures of dendritic cells, macrophages, eosinophils, and T lymphocytes at vaccination sites. The dramatic influx of dendritic cells and macrophages supports the hypothesis, derived from studies in experimental murine model systems, that GM-CSF functions to improve tumor antigen presentation by increasing the numbers and activities of host-derived professional antigen presenting cells (9). All patients also developed, as a function of vaccination, intense infiltrates of T lymphocytes and eosinophils in response to injections of irradiated nontransfected melanoma cells. Whereas the antigens stimulating these delayed-type hypersensitivity reactions remain to be determined (and in particular it remains to be delineated whether they represent previously reported or novel melanoma targets and/or components of the culture media), the prominent eosinophil component distinguishes these infiltrates from the classical tuberculin-type reactions generated by other vaccination schemes (32). This histopathology, moreover, bears a remarkable resemblance to that observed in allergic disease and parasitic infection, suggesting intriguing connections among these forms of immunity (33).
Pathologic examination of metastases resected after vaccination delineated the context in which eosinophils could mediate antitumor activity. Degranulating eosinophils (along with neutrophils and lymphocytes) were found in association with damaged endothelium within the tumor vasculature; this vasculopathy resulted in significant zonal necrosis within the tumor mass. Although the mechanisms underlying the targeting and destruction of the tumor vasculature remain to be fully clarified, it is tempting to speculate that tumor-reactive T cells initially recognize tumor antigens presented by endothelial cells and then secrete cytokines that lead to the local recruitment and activation of eosinophils. The frequent finding of eosinophilia in response to parasites residing in small blood vessels (34), together with the antitumor effects observed here, raises the possibility that eosinophils function in a general way to guard the vasculature.
Finally, the provocative association between the presence of brisk intratumoral lymphocytic infiltrates and improved survival of patients with malignant melanoma (4–7, 28) raises the possibility that antitumor immune responses modulate the natural history of this disease. The discovery of immunization strategies that stimulate potent antimelanoma immunity, particularly in individuals who spontaneously fail to generate intratumoral lymphocytic infiltrates, should allow the testing of this idea with randomized clinical trials conducted in the setting of minimal residual disease after surgical resection. The results presented here strongly suggest that vaccination with irradiated autologous melanoma cells engineered to secrete GM-CSF is a compelling candidate for such evaluation.
Acknowledgments
We thank Esther Brisson, David Wemple, Dr. Jeffrey S. Ross (Albany Medical College, Albany, NY), and Stephen Conley for their excellent help with the histological specimens. This work was supported by the Claudia Adams Barr Foundation, the Cancer Research Institute/Partridge Foundation, a Young Markey Scientist Award (G.D.), the Connell–O’Reilly Laboratory, and Cell Genesys.
ABBREVIATIONS
- GM-CSF
granulocyte–macrophage colony-stimulating factor
- IL
interleukin
- TNF
tumor necrosis factor
- IFN
interferon
References
- 1. Clark W, From L, Bernardino E, Mihm M. Cancer Res. 1969;29:705–726. [PubMed] [Google Scholar]
- 2.Ferradini L, Mackensen A, Genevee C, Bosq J, Duvillard P, Avril M-F, Hercend T. J Clin Invest. 1993;91:1183–1190. doi: 10.1172/JCI116278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mackensen A, Carcelain G, Viel S, Raynal M-C, Michalaki H, Triebel F, Bosq J, Hercend T. J Clin Invest. 1994;93:1397–1402. doi: 10.1172/JCI117116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clark W, Elder D, Guerry D, Braitman L, Trock B, Schultz D, Synnestvedt M, Halpern A. J Natl Cancer Inst. 1989;81:1893–1904. doi: 10.1093/jnci/81.24.1893. [DOI] [PubMed] [Google Scholar]
- 5.Clemente C, Mihm M, Bufalino R, Zurrida S, Collini P, Cascinelli N. Cancer (Phila) 1996;77:1303–1310. doi: 10.1002/(SICI)1097-0142(19960401)77:7<1303::AID-CNCR12>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 6.Mihm M, Clemente C, Cascinelli N. Lab Invest. 1996;74:43–47. [PubMed] [Google Scholar]
- 7.Bulkley G, Cohen M, Banks P, Char D, Ketcham A. Cancer (Phila) 1975;36:485–494. doi: 10.1002/1097-0142(197508)36:2<485::aid-cncr2820360227>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 8.Dranoff G, Mulligan R C. Adv Immunol. 1995;58:417–454. doi: 10.1016/s0065-2776(08)60624-0. [DOI] [PubMed] [Google Scholar]
- 9.Dranoff G, Jaffee E, Lazenby A, Golumbek P, Levitsky H, Brose K, Jackson V, Hamada H, Pardoll D, Mulligan R C. Proc Natl Acad Sci USA. 1993;90:3539–3543. doi: 10.1073/pnas.90.8.3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Soiffer R, Lynch T, Mihm M, Jung K, Kolesar K, Liebster L, Lam P, Duda R, Mentzer S, Singer S, et al. Hum Gene Ther. 1997;8:111–123. doi: 10.1089/hum.1997.8.1-111. [DOI] [PubMed] [Google Scholar]
- 11.Ellem K A O, O’Rourke M G E, Johnson G R, Parry G, Misko I S, Schmidt C W, Parsons P G, Burrows S R, Cross S, Fell A, et al. Cancer Immunol Immunother. 1997;44:10–20. doi: 10.1007/s002620050349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Simons J W, Jaffee E M, Weber C E, Levitsky H I, Nelson W G, Carducci M A, Lazenby A J, Cohen L K, Finn C C, Clift S M, et al. Cancer Res. 1997;57:1537–1546. [PMC free article] [PubMed] [Google Scholar]
- 13.Coligan J E, Kruisbeek A M, Margulies D H, Shevach E M, Strober W. Current Protocols in Immunology. New York: Wiley; 1991. [Google Scholar]
- 14.Coffman R L, Seymour B W, Hudak S, Jackson J, Rennick D. Science. 1989;245:308–310. doi: 10.1126/science.2787531. [DOI] [PubMed] [Google Scholar]
- 15.Arai K-I, Lee F, Miyajima A, Miyatake S, Arai N, Yokota T. Annu Rev Biochem. 1990;59:783–836. doi: 10.1146/annurev.bi.59.070190.004031. [DOI] [PubMed] [Google Scholar]
- 16.Moore K W, O’Garra A, de Waal Malefyt R, Vieira P, Mosmann T R. Annu Rev Immunol. 1993;11:165–190. doi: 10.1146/annurev.iy.11.040193.001121. [DOI] [PubMed] [Google Scholar]
- 17.Berd D, Murphy G, Maguire H C, Mastrangelo M J. Cancer Res. 1991;51:2731–2734. [PubMed] [Google Scholar]
- 18.Oratz R, Cockerill C, Speyer J, Harris M, Roses D, Bystryn J-C. J Biol Response Modif. 1989;8:355–358. [PubMed] [Google Scholar]
- 19.Mitchell M S, Harel W, Kempf R A, Hu E, Kan-Mitchell J, Boswell W D, Dean G, Stevenson L. J Clin Oncol. 1990;8:856–869. doi: 10.1200/JCO.1990.8.5.856. [DOI] [PubMed] [Google Scholar]
- 20.Hersey P, Edwards A, Coates A, Shaw H, McCarthy W, Milton G. Cancer Immunol Immunother. 1987;25:257–265. doi: 10.1007/BF00199156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morton D L, Foshag L J, Hoon D S B, Nizze J A, Wanek L A, Chang C, Davtyan D G, Gupta R K, Elashoff R, Irie R F. Ann Surg. 1992;216:463–482. doi: 10.1097/00000658-199210000-00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Slinghuff C, Vollmer R, Siegler H. J Surg Oncol. 1988;39:139–147. doi: 10.1002/jso.2930390302. [DOI] [PubMed] [Google Scholar]
- 23.Livingston P, Wong G, Adluri S, Tao Y, Padavan M, Parente R, Hanlon C, Calves M, Helling F, Ritter G, et al. J Clin Oncol. 1994;12:1036–1044. doi: 10.1200/JCO.1994.12.5.1036. [DOI] [PubMed] [Google Scholar]
- 24.Mukherji B, Chakraborty N, Yamasaki S, Okino T, Yamase H, Sporn J, Kurtzman S, Ergin M, Ozols J, Meehan J, et al. Proc Natl Acad Sci USA. 1995;92:8078–8082. doi: 10.1073/pnas.92.17.8078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marchand M, Weynants P, Rankin E, Arjenti F, Belli F, Parmiani G, Cascinelli N, Bourlond A, Vanwijck R, Humblet Y, et al. Int J Cancer. 1995;63:883–885. doi: 10.1002/ijc.2910630622. [DOI] [PubMed] [Google Scholar]
- 26.Jager E, Ringhoffer M, Dienes H P, Arand M, Karbach J, Jager D, Ilsemann C, Hagedorn M, Oesch F, Knuth A. Int J Cancer. 1996;67:54–62. doi: 10.1002/(SICI)1097-0215(19960703)67:1<54::AID-IJC11>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 27.Berd D, Maguire H, Mastrangelo M J, Murphy G. Cancer Immunol Immunother. 1994;39:141–147. doi: 10.1007/BF01533378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berd D, Maguire H C, Schuchter L M, Hamilton R, Hauck W W, Sato T, Mastrangelo M J. J Clin Oncol. 1997;15:2359–2370. doi: 10.1200/JCO.1997.15.6.2359. [DOI] [PubMed] [Google Scholar]
- 29.Lattime E, Mastrangelo M J, Bagastra O, Li W, Berd D. Cancer Immunol Immunother. 1995;41:151–156. doi: 10.1007/BF01521340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sensi M, Farina C, Maccalli C, Lupetti R, Nicolinin G, Anichini A, Parmiani G, Berd D. J Clin Invest. 1997;99:710–717. doi: 10.1172/JCI119215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murphy, G., Radu, A., Kaminer, M. & Berd, D. (1993) J. Invest. Dermatol. 100, Suppl., 335S–341S. [DOI] [PubMed]
- 32.Barth A, Hoon D S B, Foshag L J, Nizze J A, Famatiga E, Okun E, Morton D L. Cancer Res. 1994;54:3342–3345. [PubMed] [Google Scholar]
- 33.Hsu S, Hsu H, Penick G, Lust G, Osborne J. J Allergy Clin Immunol. 1974;54:339–349. [Google Scholar]
- 34.Gleich G J, Adolphson C R. Adv Immunol. 1986;39:177–253. doi: 10.1016/s0065-2776(08)60351-x. [DOI] [PubMed] [Google Scholar]