

Abstract
Although interleukin (IL) 17 has been extensively characterized, the function of IL-17F, which has an expression pattern regulated similarly to IL-17, is poorly understood. We show that like IL-17, IL-17F regulates proinflammatory gene expression in vitro, and this requires IL-17 receptor A, tumor necrosis factor receptor–associated factor 6, and Act1. In vivo, overexpression of IL-17F in lung epithelium led to infiltration of lymphocytes and macrophages and mucus hyperplasia, similar to observations made in IL-17 transgenic mice. To further understand the function of IL-17F, we generated and analyzed mice deficient in IL-17F or IL-17. IL-17, but not IL-17F, was required for the initiation of experimental autoimmune encephalomyelitis. Mice deficient in IL-17F, but not IL-17, had defective airway neutrophilia in response to allergen challenge. Moreover, in an asthma model, although IL-17 deficiency reduced T helper type 2 responses, IL-17F–deficient mice displayed enhanced type 2 cytokine production and eosinophil function. In addition, IL-17F deficiency resulted in reduced colitis caused by dextran sulfate sodium, whereas IL-17 knockout mice developed more severe disease. Our results thus demonstrate that IL-17F is an important regulator of inflammatory responses that seems to function differently than IL-17 in immune responses and diseases.
Since IL-17, also called IL-17A, was identified a decade ago from activated T cells, related cytokines, including IL-17B, C, D, E (also called IL-25), and F, have been reported and constitute the IL-17 cytokine family (1, 2). IL-17, the best-studied member in this family, is a proinflammatory cytokine that was historically associated with many inflammatory diseases, such as rheumatoid arthritis, asthma, lupus, and allograft rejection (1, 2). In vitro, mRNA expression of genes encoding several chemokines (CCL2, CCL7, CXCL1, and CCL20) and matrix metalloproteinase 3 (MMP3) and MMP13 was significantly up-regulated after IL-17 treatment (3). Moreover, IL-17 and TNF-α exhibit strong synergy in promoting inflammatory gene expression (4). In vivo, overexpressing IL-17 in the lungs resulted in chemokine up-regulation and tissue infiltration by leukocytes (3). Conversely, inhibition of IL-17 signaling led to impaired host defense against bacterial infection (5) and resistance to autoimmune diseases (3, 6–8).
IL-17 binds to and signals through IL-17RA, a member of the IL-17R family (9). Recently, it was reported that IL-17RA might form a heteromeric complex with IL-17RC (10). IL-17 activates NF-κB and the mitogen-activated protein kinase pathway (2). IL-6 induction by IL-17 in mouse embryonic fibroblasts (MEFs) is dependent on TNFR-associated factor 6 (TRAF6) (11). Recently, we identified Act1 adaptor protein as an immediate and essential signaling component of IL-17RA (12).
Recently, a new subset of Th cells named Th17 was identified (13, 14). The hallmark of Th17 subset is the production of IL-17. Interestingly, Th17 cells also express IL-17F, which shares the strongest homology to IL-17 (1, 2). IL-17 and IL-17F genes are localized in the same chromosome region, and we previously found that this locus undergoes chromatin remodeling associated with Th17 differentiation (15). Recently, an IL-17–IL-17F heterodimer was found to be expressed in Th17 cells together with IL-17 and IL-17F homodimers (16, 17). IL-17F did not bind to IL-17RA in vitro (18). However, an anti–IL-17RA antibody blocked IL-17F–mediated function, whereas IL-17RA–soluble protein did not (19). IL-17RC was recently discovered to serve as a receptor for IL-17F (20). However, the requirements of IL-17RA or IL-17RC in IL-17F signaling in vivo remain unclear.
IL-17F was reported to induce various cytokines, chemokines, and adhesion molecules in human airway epithelial cells, vein endothelial cells, and fibroblasts (21). IL-17F was found in the airways of allergic asthma patients upon allergen challenge (22). Interestingly, a mutation in the IL-17F gene was shown to be associated with human asthma and chronic obstructive pulmonary disease (21). This literature suggests a linkage of IL-17F with human asthma. To analyze the function of IL-17F in airway inflammation, adenoviral infection (23) or lipofectamine-mediated gene transfer (24) were adopted to acutely overexpress IL-17F in vivo, which resulted in the pulmonary recruitment of neutrophils.
In our current study, we have analyzed the expression, function, and signaling mechanisms of IL-17F. We demonstrate that IL-17–expressing T cells also produce IL-17F protein. IL-17F regulates the expression of inflammatory chemokines and cytokines, which is dependent on IL-17RA, Act1, and TRAF6 proteins. Lung-specific overexpresssion of IL-17F in mice leads to infiltration of lymphocytes and macrophages and mucus production. The examination of IL-17– and IL-17F–deficient animals in multiple models has revealed the distinct requirements of these two genes in different inflammatory responses. Thus, IL-17F plays a critical role in the regulation of inflammatory reactions.
RESULTS
Expression of IL-17F protein in IL-17–expressing T cells
Although IL-17F has been shown to be coexpressed with IL-17 in Th17 cells, whether other IL-17–expressing T cells in vivo also express IL-17F has not been clear. To allow measurement of IL-17F protein expression, we prepared a biologically active IL-17F–Ig fusion protein; we observed IL-6 or CXCL1 production in MEFs and peritoneal macrophages after IL-17F–Ig treatment (unpublished data). A polyclonal antibody against IL-17F was raised in rabbits. After affinity purification, we first tested the polyclonal antibody by intracellular staining of 293T cells transfected with a mouse IL-17F expression vector. Anti–IL-17F stained IL-17F but not the vector transfectant (Fig. 1 A). Moreover, this polyclonal antibody did not stain 293T cells expressing IL-17 (Fig. 1 A). Thus, the antibody we raised appears to be specific for IL-17F.
Figure 1.

IL-17F is produced by IL-17–expressing T cells. (A) 293T cells transfected with pcDNA–IL-17 or pcDNA–IL-17F, or vector only, were fixed and stained with the indicated antibodies. (B) CD4+ T cells from OT-II mice were differentiated into Th1, Th2, and Th17 lineages. On day 5 of culture, CD4+ T cells were restimulated with PMA and ionomycin and stained with appropriate antibodies. (C) Splenocytes were activated with PMA and Ionomycin for 5 h, and IL-17– and IL-17F–expressing cells were assessed by intracellular staining on CD4+ or TCRγδ+ gates. (D) IL-17F and IL-17F–expressing cells in CNS infiltrates of mice with EAE, and in the lamina propria and intestinal intraepithelial lymphocytes were analyzed by intracellular staining with CD4+ gating. CNS infiltrates were isolated from perfused mice on day 12 after the second immunization. Data are representative of at least two independent experiments with similar results (percentages are shown).
To analyze IL-17F protein expression in different Th subsets, we differentiated OT-II cells into Th1, Th2, and Th17 lineages. Using our anti–IL-17F antibody for intracellular staining, IL-17F protein was found expressed in Th17 cells that also expressed IL-17 but not in Th1 or Th2 cells (Fig. 1 B). The protein expression of IL-17F was consistent with its mRNA expression (unpublished data). Excessive amounts of IL-17 inhibited staining by anti–IL-17 but not anti–IL-17F (Fig. S1 A, available at http://www.jem.org/cgi/content/full/jem.20071978/DC1), further supporting the specificity of our antibody. After Th17 differentiation, almost all cells expressing IL-17 also expressed IL-17F (Fig. 1 B), indicating that they are regulated by a similar molecular mechanism. However, there appears to be an IL-17F single-positive population, suggesting the existence of heterogeneous subpopulations in the Th17 lineage.
We also examined IL-17F expression in vivo. In addition to CD4+ T cells, it has been reported that IL-17 is also produced by γδ T cells (25). Also, during Mycobacterium tuberculosis infection, IL-17 production seems to be dominated by γδ T cells (26). In agreement with these studies, we found that splenic CD4+ and γδ T cells expressed IL-17 upon activation (Fig. 1 C). As seen in in vitro–generated Th17 cells, IL-17F was coexpressed with IL-17 in these cells, though there were some IL-17 single-positive cells.
In addition to Th17 cells generated in vitro, IL-17F was coexpressed with IL-17 in CD4+ T cells from the central nervous systems (CNS) of mice subject to the experimental autoimmune encephalomyelitis (EAE) model (Fig. 1 D). Interestingly, most of the T cells that expressed IL-17F also expressed IL-17, and there were few IL-17 or IL-17F single-positive cells. IL-17+ T cells have also been found constitutively in the lamina propria, dependent on IL-21, RORγt, and RORα (27–29). We found that IL-17hi CD4+ T cells in the lamina propria and intestinal intraepithelium expressed IL-17F (Fig. 1 D), but in these tissues, there existed a lot more IL-17 single-positive cells than Th17 cells generated in vitro and recovered from the CNS. Thus, analysis of IL-17F protein expression has revealed that IL-17F is generally coexpressed with IL-17 in T cells and has suggested the possibility of differential ratios of these two cytokines in different T cell states or in different tissues.
IL-17F regulates inflammatory gene expression via IL-17RA, Act1, and TRAF6
We previously used a microarray analysis to identify the molecular targets of IL-17 and observed the up-regulation of chemokines and MMPs by IL-17 (3). To address the biological function of IL-17F, we first treated MEFs with IL-17F. We found that IL-17F induced CXCL1, IL-6, CCL2, CCL7, and MMP13, similar to the action of IL-17 (Fig. 2 A). The levels of gene expression induced by IL-17F were typically more moderate than IL-17 at the concentration used (100 ng/ml). A detailed dose comparison of IL-17 versus IL-17F in IL-6 induction in MEFs also revealed that IL-17 is more potent than IL-17F (unpublished data). Moreover, thymic stromal lymphopoietin, a molecule involved in allergic asthma and atopic dermatitis (30), was induced by both IL-17 and IL-17F (Fig. 2 A). Because thymic stromal lymphopoietin is a potent inducer of Th2 responses, our result suggests a cross-regulatory mechanism of different Th subsets.
Figure 2.

IL-17F induces proinflammatory cytokines and chemokines in vitro via IL-17RA, TRAF6, and Act1. (A) MEFs were treated with 100 ng/ml IL-17 and IL-17F, and RNA samples were analyzed for expression of the indicated genes using real-time PCR. (B) MEFs from WT and IL-17RA KO mice were treated with 100 ng/ml IL-17 or IL-17F. After overnight incubation, cell supernatants were subjected for ELISA. After administration of recombinant IL-17 and IL-17F in WT and IL-17RA KO mice, neutrophil numbers were counted. The results shown are averaged from three animals in each group. (C) WT and TRAF6-deficient MEFs were treated with cytokines overnight. Culture supernatants were subjected for ELISA. (D) Real-time PCR was performed using cDNAs from WT and Act1-deficient MEFs treated with the indicated cytokines. p-values were calculated between cytokine treatment and medium alone (A) or between the groups indicated by a horizontal line (B–D) using an unpaired Student's t test. The experiments were repeated two to three times with consistent results. Results are mean ± SD. **, P < 0.005.
To address which molecules may be used by IL-17F to initiate the signaling cascade, we first prepared MEFs from IL-17RA–deficient mice. Because TNF-α– or IL-1β–mediated IL-6 or CXCL1 productions were normal in IL-17RA–deficient fibroblasts, IL-17 and IL-17F effects were both impaired in these cells (Fig. 2 B). Moreover, injection of IL-17 or IL-17F into mouse peritoneum resulted in the recruitment of neutrophils within 4 h (Fig. 2 B). In IL-17RA–deficient mice, however, IL-17 and IL-17F both lost their ability to recruit neutrophils (Fig. 2 B). These results indicate that IL-17RA is required for IL-17F as well as IL-17 signaling in vitro and in vivo.
IL-17 and IL-25 both use TRAF6 as their signaling intermediate (11, 31). Recently we reported Act1 as an immediate adaptor protein downstream of IL-17RA (12). We tested the requirement of these molecules in IL-17F–induced inflammatory gene expression. Induction of CXCL1 was impaired in TRAF6−/− MEFs upon IL-17F treatment (Fig. 2 C). Also, although WT MEFs up-regulated the expression of IL-6, CXCL-1, and the transcriptional regulators CCAAT/enhancer binding protein δ and IκBζ upon IL-17F treatment, Act1-deficient MEFs did not (Fig. 2 D). Our results thus indicate that similar to IL-17, IL-17F requires both Act1 and TRAF6 for its signal transduction leading to inflammatory gene expression.
IL-17F overexpression induces airway inflammation
IL-17F thus uses similar signaling mechanisms as IL-17 to regulate similar downstream genes. Because IL-17F has been implicated in airway inflammation, we treated the mouse lung epithelial cell line MLE12 with IL-17F. IL-17F induced several chemokines in MLE12, as we found in MEFs (Fig. 3 A). To understand the chronic effects of IL-17F function in vivo, we generated lung-specific transgenic mice that selectively overexpress IL-17F by epithelium cells using the Clara cell 10 (CC10) gene promoter (Fig. 3 B). Three founder lines were generated, and their offspring exhibited greatly increased IL-17F mRNA expression in the lung tissues (Fig. 3 C) and similar pathological symptoms. IL-17 mRNA expression in IL-17F transgenic mice remained unaffected (Fig. 3 C). Transgenic mice but not their control littermates developed abnormal lung pathology starting at 5 mo of age. Often, CC10–IL-17F transgenic mice developed substantial infiltration by macrophages (Fig. 3 E). Also, similar to IL-17 transgenic mice, IL-17F transgenic mice exhibit peribronchial and perivascular infiltration of CD4+ and B220+ lymphocytes (Fig. 3 D). However, we did not observe any neutrophil recruitment in transgenic mice. Occasionally, in severe cases, transgenic mice were found with a Charcot-Leyden–like crystal structure in the airway similar to that reported in the lung of IL-13 transgenic mice (32) (Fig. 3 F). Increased mucus production was observed in lung tissues from transgenic mice via periodic acid Schiff (PAS) staining (Fig. 3 G). The features of inflammatory pathology and mucus production in CC10–IL-17F mice are consistent with those observed in CC10–IL-17 mice (3), supporting the idea that these two cytokines may have similar biological functions in vivo. Also, the inflammatory genes up-regulated in MLE12 by IL-17F were also found significantly up-regulated in CC10–IL-17F mice (Fig. 3 H).
Figure 3.

CC10–IL-17F mice develop lung inflammation and mucus hyperplasia. (A) MLE12 were treated with 100 ng/ml IL-17F for 4 h and subjected to real-time PCR analysis. (B) Schematic illustration of the construct in the IL-17F transgenic mice. hGH, human growth hormone. (C) RT-PCR detection of IL-17 and IL-17F mRNA in the lung. (D) Immunohistochemistry of lung sections from IL-17F transgenic mice. Frozen lung tissues were stained with CD4, CD8, B220, and CD11C. (E and F) H&E staining of lung sections from 5–7-mo-old IL-17F transgenic mice. High power magnification shows the accumulation of mixed inflammatory cells, macrophages, and Charcot-Leyden–like crystals. (G) PAS staining of transgenic mouse lung shows mucus overproduction. (H) RT-PCR of homogenized lung from IL-17F transgenic mice and age-matched WT. Data shown were repeated at least twice with similar results. Results are mean ± SD. Bars: (D and E) 100 μm; (F and G) 300 μm.
Generation of IL-17– and IL-17F–deficient mice
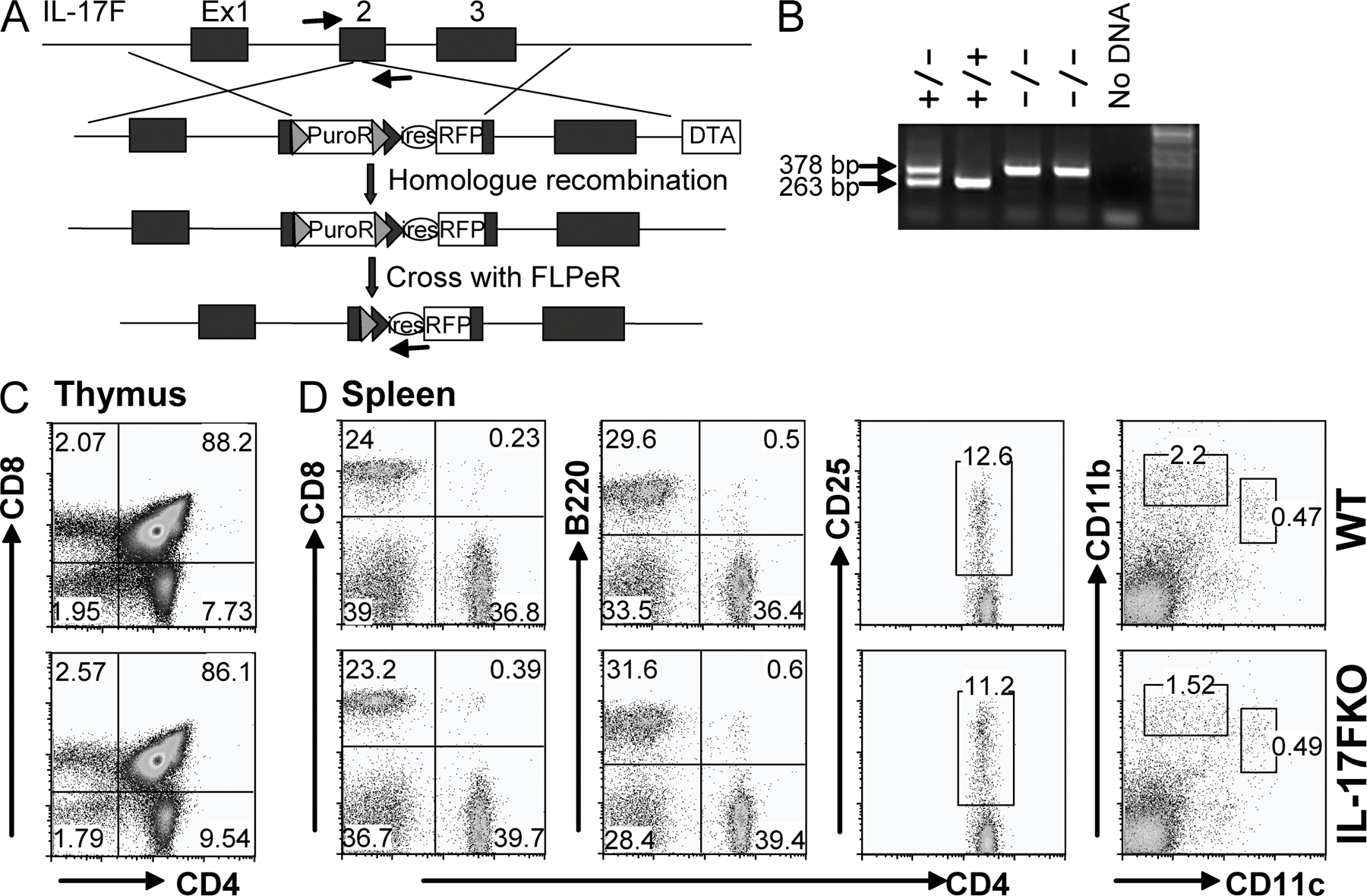
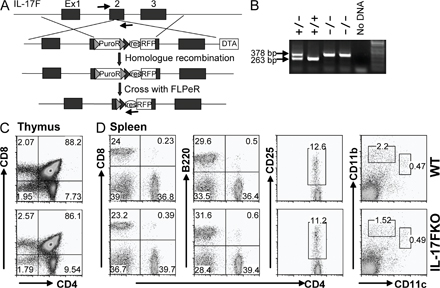
To determine whether IL-17 and IL-17F have distinct functions in vivo, we generated and analyzed mice deficient in IL-17 or IL-17F. IL-17–deficient mice were generated by gene targeting in embryonic stem cells by replacement of part of exon 2 with a luciferase-IRES-eGFP cassette without disrupting the splicing boundary (Fig. S2, A and B, available at http://www.jem.org/cgi/content/full/jem.20071978/DC1). The loxP-flanked neomycin cassette was removed by crossing the heterozygous mice with CMV-Cre mice, and the resulting mice were intercrossed to generate homozygous KO mice. Compared with WT mice, IL-17 KO mice exhibited normal lymphoid compartments in both the thymus and spleen but moderately decreased percentages of CD4+ T cells in the spleen (P = 0.03; Fig. S2, C and D). IL-17F KO was generated by an insertional inactivation of IL-17F exon 2 with an IRES-mRFP-polyA cassette (Fig. S3 A). After removal of the puromycin-resistant gene in the mouse germline, heterozygous IL-17F KO mice were intercrossed to make homozygous animals, and PCR analysis of tail genomic DNA was used to analyze the genotypes (Fig. S3 B). IL-17F−/− mice developed normally. Compared with WT mice, IL-17F KO mice exhibited normal CD4+, CD8+, and CD4+CD8+ ratios in the thymus, and similar CD4+, CD8+, and B220+ populations in the spleen (Fig. S3, C and D). CD11b+ macrophage/granulocyte, CD11c+CD11blo dendritic cell, and CD4+CD25+ regulatory T cell numbers were also compatible in IL-17F KO mice (Fig. S3 D).
As a first step to analyze these animals, IL-17F KO, IL-17 KO, and WT control mice were immunized with KLH in CFA. 7 d after immunization, the animals were killed and spleens were collected and restimulated with KLH. In ELISA analysis, IL-17F and IL-17 could not be detected in IL-17F KO and IL-17 KO splenocyte cultures, respectively (Fig. 4 A). Intracellular cytokine staining also revealed no IL-17 and IL-17F expression in the corresponding animals (Fig. S1 B). Levels of IL-17F were also reduced in IL-17 KO cells, whereas IL-17F deficiency did not lead to a substantial reduction of IL-17 expression (Fig. 4 A). IL-22 expression levels were moderately reduced in both IL-17F KO and IL-17 KO samples (Fig. 4 A). In addition to T cell responses, KLH-specific Ig levels in sera from immunized IL-17F KO, IL-17 KO, and WT mice were determined by ELISA (Fig. 4 B). Compared with WT mice, IL-17–deficient mice exhibited lower levels of IgM, total IgG, and all IgG subclasses tested, consistent with a previous paper on 2,4,6-trinitrochlorobenzene–challenged IL-17–deficient mice (33), whereas IL-17F deficiency resulted in a significant increase of IgG2a levels, though IFN-γ levels were not increased in these mice. These data suggest that IL-17F and IL-17 play important but perhaps differential roles in humoral immunity.
Figure 4.

Analysis of T and B cell responses in IL-17 and IL-17F KO animals. IL-17F KO, IL-17 KO, and WT control mice were immunized with KLH in CFA (three per group). 7 d later, the mice were killed and spleens and blood were collect. (A) Splenocytes from the immunized mice were restimulated with KLH for 3 d, and cytokine expression was measured by ELISA. (B) KLH-specific antibodies were measured in the sera by ELISA. The sera were subject to a threefold serial dilution, and the antibody concentrations are shown as the mean for each group. Results are mean ± SD.
Differential requirements of IL-17 and IL-17F in autoimmune responses
To examine the functions of IL-17 and IL-17F in vivo, we first used EAE, a Th17-mediated CNS inflammation disease model. EAE was induced in WT, IL-17 KO, and IL-17F KO mice. IL-17–deficient mice were significantly delayed in disease onset and progression, whereas IL-17F KO mice only showed moderately improved recovery compared with both WT and IL-17 KO mice (Fig. 5 A). This indicates that IL-17 but not IL-17F is required for the initiation of neuronal inflammation. When we examined infiltrating mononuclear cells in the CNS, CD4+ cells were greatly reduced in both IL-17F KO and IL-17 KO compared with WT mice, whereas CD11b+ cells were slightly increased in IL-17F KO animals (Fig. 5 B). We then assessed the molecular basis for the reduced recruitment of T cells into the CNS in the mice deficient in IL-17F or IL-17 by real-time RT-PCR to assess chemokine expression in CNS tissue after induction of EAE. The expression of the chemokines CCL2 and CCL7 was greatly reduced in IL-17F KO compared with WT CNS, but more profoundly in IL-17 KO (Fig. 5 C). CXCL1 expression was severely impaired in IL-17 KO but not in IL-17F KO in comparison with WT animals (Fig. 5 C). To further understand whether IL-17F or IL-17 deficiency affects the generation of autoreactive T cells, we assessed cytokine expression in splenocytes from the EAE mice upon myelin oligodendrocyte glycoprotein (MOG) peptide restimulation by ELISA. IFN-γ expression levels were slightly increased in IL-17F KO but greatly elevated in IL-17 KO compared with WT cells (Fig. 5 D). In comparison with WT splenocytes, IL-22 levels were slightly but not significantly decreased in IL-17 but not in IL-17F KO mice, whereas IL-17 and IL-17F expression were not significantly altered in IL-17F KO and IL-17 KO, respectively.
Figure 5.

IL-17 but not IL-17F is required for the initiation of EAE. (A) EAE was induced in IL-17F KO, IL-17 KO, and WT control mice. Data shown are a combination of two independent experiments (n = 10 for each group). (B) Infiltrates in the CNS from the EAE mice were isolated after perfusion on day 12 after the second immunization, and CD4+ and CD11b+ cells were assessed by FACS. Horizontal bars indicate mean values. Data shown represent two independent experiments with similar results. (C) Chemokine expression in the CNS from the EAE mice was measured by real-time RT-PCR. (D) Splenocytes from the EAE mice were stimulated with MOG peptide, and cytokine expression levels were measured by ELISA. Data shown represent two independent experiments with similar results. (E) WT, IL-17 KO, and IL-17F KO mice were immunized with MOG/CFA and administered with pertussis toxin on the next day. 7 d after immunization, the mice were killed and IL-17–, IL-17F–, and IFN-γ–producing cells in the spleen and draining lymph nodes were analyzed by intracellular staining. Data shown are gated on CD4+ cells and are averaged from three to four mice in each group. Results are mean ± SD. *, P < 0.05; and **, P < 0.005 using the Student's t test. n.s., not significant.
To better understand the differential effects of IL-17 and IL-17F on EAE pathogenesis, we immunized mice with MOG in CFA and analyzed cytokine expression by intracellular staining on day 7. At this early phase, the frequencies of IFN-γ–expressing CD4+ T cells were elevated in the spleen but decreased in the draining lymph nodes in IL-17 KO compared with WT mice, whereas no difference was found in cells from IL-17F KO mice (Fig. 5 E). The percentages of IL-17+ CD4+ cells were slightly decreased in both the spleens and draining lymph nodes from IL-17F KO in comparison to WT mice (Fig. 5 E). In IL-17 KO mice, the frequencies of IL-17F+ CD4+ cells were not altered (Fig. 5 E). Our data thus indicate an effect of IL-17 but not IL-17F deficiency in the priming phase, possibly resulting in a different distribution of IFN-γ+ cells.
Differential functions of IL-17 and IL-17F in acute and chronic allergic airway responses
Because IL-17F has been implicated in allergic inflammation in humans and mice, we subjected WT, IL-17 KO, and IL-17F KO mice to intranasal challenge by an allergenic fungal proteinase (FAP) derived from Aspergillus oryzae and analyzed these mice 18 h later. In this acute allergic response, consistent with a previous report (34), significant numbers of neutrophils were elicited by FAP in bronchoalveolar lavage fluid (BALF) from WT mice as determined by CD11b and Gr-1 staining (Fig. 6 A), or by May-Grünwald-Giemsa staining of lavage cells (not depicted). IL-17F KO mice exhibited substantially reduced numbers of neutrophils. However, IL-17 KO mice had comparable numbers of neutrophils as the WT mice (Fig. 6 A). Interestingly, IL-17RA KO mice had a similar reduction in neutrophils as IL-17F KO mice (Fig. 6 A), indicating a requirement of IL-17RA in IL-17F–mediated neutrophilia in response to allergen challenge. To gain insight into the molecular mechanism underlying IL-17F–mediated neutrophil recruitment, we assessed the expression of neutrophil-attracting chemokines in the whole lungs by real-time PCR. Although many chemokines were elevated by allergen treatment in WT and two different KO mice, expression levels of CXCL5, a neutrophil-recruiting chemokine and one of the IL-17F target genes, were significantly reduced in IL-17F KO but not IL-17 KO mice (Fig. 6 B). This indicates that IL-17F may regulate neutrophil recruitment in vivo by promoting CXCL5 induction in the lung in response to allergen.
Figure 6.

IL-17 KO and IL-17F KO mice exhibited differential acute and chronic allergic responses. (A) Recruitment of neutrophils by FAP. WT, IL-17 KO, IL-17F KO, and IL-17RA KO mice were challenged once intranasally with either PBS or OVA with FAP. 18 h later, BALF cells were collected and stained with Gr.1 by FACS. Data shown are a combination of two independent experiments with similar results (n = 6–8 mice per group). (B) Expression of chemokines. Whole-lung mRNA was collected and subjected to quantitative real-time RT-PCR. Data are expressed as the fold induction relative to PBS-challenged animals. (C–E). WT, IL-17, and IL-17F KO mice were subjected to an asthma model. (C) Cellular profiles in BAL fluid upon OVA challenge. Cells were harvested from BALF, stained by May-Grünwald-Giemsa, and counted under a microscope. Eos, eosinophil; Lymp, lymphocyte; Mac, macrophage; Neu, neutrophil. Data shown represent two independent experiments with consistent results (n = 5). (D) Major basic protein 1 (MBP-1) and eosinophil peroxidase (EPO) expression in BAL cells. Real-time RT-PCR was performed using cDNA derived from BAL cells. CD11b expression in BAL cells was evaluated by FACS. Statistical analysis was performed between the indicated group and WT. (E) Expression of type 2 cytokines in lung lymph nodes and spleen. 3 d after culture in the presence of OVA, the levels of IL-4, IL-5, and IL-13 were measured by ELISA. Data shown in B, D, and E were repeated twice with similar results. Results are mean ± SD. *, P < 0.05; and **, P < 0.01 using the Student's t test. n.s., not significant.
Because IL-17 and IL-17F exhibited differential activities during innate responses to allergen challenge in the lung, we further investigated their roles in a chronic asthma model. WT, IL-17 KO, and IL-17F KO mice were immunized twice i.p. with OVA in aluminum hydroxide and subsequently challenged by intranasal administration of OVA protein in PBS. IL-17 KO mice receiving OVA had fewer BALF cells and eosinophils (Fig. 6 C), which was consistent with previous studies using IL-17 or IL-17RA KO animals (33, 35). On the other hand, there was no significant difference in total BALF cell numbers between WT and IL-17F KO mice, but BALF cells from IL-17F KO mice had significantly increased eosinophils when compared with IL-17 KO mice (Fig. 6 C). FACS analysis revealed that eosinophils in IL-17F KO mice exhibited increased phenotypes of CD11bintSSClo, suggesting enhanced degranulation (Fig. 6 D) (36). In support of this idea, RT-PCR analysis indicated that BALF cells from IL-17F KO mice expressed higher levels of major basic protein 1 and eosinophil peroxidase (Fig. 6 D) but not neutrophil-specific elastase (not depicted). These patterns were also observed by RT-PCR using total lung tissue RNAs (unpublished data).
Eosinophil degranulation is regulated by Th2 cytokines (36). We asked whether IL-17F deficiency might have an impact on type 2 cytokine expression. Lung lymph node cells from OVA-challenged IL-17F KO mice produced significantly higher levels of IL-4, IL-5, and IL-13 than cells from WT and IL-17 KO mice upon ex vivo OVA restimulation (Fig. 6 E). Splenocytes from IL-17 KO mice showed a greatly reduced production of IL-4, IL-5, and IL-13, whereas those from IL-17F KO mice exhibited enhanced IL-5 and IL-13 production (Fig. 6 E). These analyses indicate that even though IL-17 positively regulates asthmatic allergic responses, especially Th2 cytokine production, IL-17F has a negative role and IL-17F deficiency leads to greater Th2 cytokine expression and enhanced eosinophil function.
Differential function of IL-17 and IL-17F in dextran sulfate sodium (DSS)–induced colitis
The results on CNS and lung inflammation indicate differential functions of IL-17 and IL-17F that target different tissues. To further ascertain this finding, we subjected WT, IL-17 KO, and IL-17F KO mice to a colitis model induced by oral feeding with DSS (a compound from Gram-positive bacteria Leuconostoc species). Upon DSS treatment, WT mice developed acute colitis symptoms indicated by bloody diarrhea on days 2–6, with a peak on days 3–4 and loss of body weight from day 5. On day 8, the experimental mice were killed, and colon tissues were collected for histological and gene expression analysis. Compared with WT mice, IL-17 KO mice exhibited more severe diarrhea and more blood in the feces with increased weight loss, whereas IL-17F KO mice had much milder symptoms and less weight loss (Fig. 7 A and not depicted). As revealed by histological analysis, colon tissues from IL-17 KO mice exhibited the most severe epithelial lesion and crypt basal separation, consistent with a previous study using an anti–IL-17 monoclonal antibody (37). In colon tissues from IL-17F KO mice, only mild damage was observed (Fig. 7, B and C). Accordingly, there are more leukocytes in IL-17–deficient than WT lamina propria (Fig. 7 B), consisting of a majority of macrophages together with neutrophils and lymphocytes, which was consistent with a previous study (38). In a sharp contrast, IL-17F deficiency led to greatly reduced infiltrates in the lamina propria (Fig. 7 B).
Figure 7.

IL-17 and IL-17F differentially regulate DSS-induced colitis. (A) Acute colitis was induced in WT, IL-17 KO, and IL-17F KO mice by oral feeding of DSS (n = 5–8 per group). Weight loss during colitis progression is shown. (B) Mice with colitis were killed on day 8, and the middle segment of the colon was fixed, sectioned, and stained with H&E. Bar, 1 mm. (C) Clinical scores are indicated based on microscopic examination of an epithelial lesion in the colon. (D) Chemokine mRNA expression was assessed in colon tissues by real-time RT-PCR. Data were expressed as the relative abundance of Actb. The experiments were repeated at least two to three times with consistent results. Results are mean values. *, P < 0.05; and **, P < 0.005 using the Student's t test.
We investigated the molecular basis for differential recruitment of inflammatory cells in the experimental animals by analysis of chemokine gene expression in colons using real-time RT-PCR. Unlike CXCL1, which was similarly reduced in IL-17 KO and IL-17F KO mice, CCL2, CCL5, and CCL7 mRNA expression were elevated in IL-17–deficient mice upon DSS treatment when compared with WT mice, whereas their expression was dramatically decreased in IL-17F KO mice (Fig. 7 D). Collectively, these data suggest that in intestinal inflammation caused by DSS, IL-17 plays a protective role, whereas IL-17F may exacerbate the inflammation.
DISCUSSION
Th17 and other IL-17–expressing T cells have recently emerged as crucial regulators of inflammatory responses. However, although IL-17 has been relatively well studied, the function of IL-17F is poorly understood. In this study, we describe the expression, signaling pathway, and in vivo function of IL-17F. Our study for the first time indicates that although IL-17F has very similar regulation and function as IL-17 in vitro, analysis of animals deficient in either of the two genes has revealed their distinct functions in inflammatory responses.
IL-17F is expressed in Th17 cells and other types of IL-17–expressing T cells in vivo. Previous papers describe similar regulation of IL-17 and IL-17F expression by cytokines IL-23, TGF-β, IL-6, and IL-21, as well as transcription factors RORγt, RORα, and STAT3 (8, 27–29, 39, 40). Moreover, we found that IL-17 and IL-17F gene promoters share the same pattern of chromatin remodeling in differentiated Th17 cells (15). Several conserved noncoding sequences in this locus, including CNS2 (29), likely mediate the coordinated expression of IL-17 and IL-17F. However, our analysis also revealed different ratios of IL-17 and IL-17F expression in different T cell populations in vitro and in vivo. This suggests differential cytokine expression in differentiated Th17 cells. What accounts for this differential regulation is unknown at this point. The biological or pathological significance of this regulation is also unclear. In Th2 cells, we previously found that the inducible co-stimulator–c-Maf pathway only controls IL-4 but not IL-5 or IL-10 expression in the effector stage, and inducible co-stimulator deficiency selectively abrogates IL-4–dependent IgE production but not IL-5–mediated airway eosinophilia (41, 42). Moreover, expression of IL-22 by Th17 cells appears to be more dependent on IL-23 than IL-17 and IL-17F (43). More studies are necessary in the future to comprehend the differential regulation of Th17 cytokines. Interestingly, our recent work revealed that RORα mutant T cells had a selective defect in IL-17 but not IL-17F or IL-22 production (29).
Compared with IL-17, IL-17F has weaker activity in inducing proinflammatory molecules in vitro. However, this activity is meaningful as in vivo, transgenic overexpression of IL-17F led to pathological airway phenotypes. Why, then, do we need two similar cytokines? The situation is even more complex considering the heterodimeric IL-17A/F molecule with intermediate activity in vitro as compared with the IL-17 and IL-17F homodimers (16). One possible explanation is that these cytokines may differentially use different cytokine receptors that are differentially expressed or alternatively spliced. IL-17, IL-17F, and IL-17A/F depend on IL-17RA for signal transduction (16). How IL-17RA mediates IL-17F signaling is unclear at this point. Previous literature showed that IL-17F did not bind to IL-17RA in vitro (18). We also found normal binding of IL-17F–Ig to IL-17RA−/− macrophages (unpublished data). Thus, IL-17RA may not directly mediate binding of IL-17F to the cell surface but rather may regulate its signaling with other receptor components. IL-17 signaling also requires IL-17RC, which forms a complex with IL-17RA (10). In our preliminary analysis, we found that siRNA reduction of IL-17RC expression in MEFs also decreased IL-17F–induced gene expression (unpublished data), suggesting the involvement of IL-17RC in IL-17F signaling. These results are consistent with a recent paper demonstrating the binding of IL-17F to IL-17RC (20). Further studies are necessary to elucidate the receptors used by these cytokines. Although IL-17 and IL-17F may potentially bind to different receptors on the target cells, they appear to use the same signaling components. In the current study, we for the first time show the requirements of Act1 and TRAF6 in IL-17F induction of downstream inflammatory genes.
In vitro, IL-17F appears to have a weaker proinflammatory function. One may predict that IL-17F may be less important than IL-17 in vivo. This may be true in the EAE model in which IL-17F is not required for the initiation of EAE with only a minor role in maintaining inflammation in the CNS. Our results using IL-17F KO mice are consistent with our unpublished data that anti–IL-17F did not ameliorate EAE (unpublished data). In contrast, IL-17 KO mice, similar to mice treated with anti–IL-17 (3, 8), exhibited greatly delayed onset and progression of EAE. Thus, IL-17 is thus a dominating pathogenic factor in EAE. However, our current study using mice deficient in either the IL-17 or IL-17F gene has for the first time provided a surprising insight into the differential function of these two cytokines in immune responses. During T cell priming in vivo in response to KLH or MOG peptide immunization, IL-17– but not IL-17F–deficient mice exhibited increased IFN-γ expression in the spleen. Because IL-17 has not been found to regulate Th cell differentiation (39, 44), this regulation may be indirect, for example, by acting on myeloid cells. On the other hand, IFN-γ expression was found reduced in draining lymph nodes in these animals, suggesting a possibility that IL-17 regulates the localization of Th1 cells. In IL-17F–deficient animals, IL-17 expression was reduced in the spleen although not significantly in the lymph nodes. However, in the late phases of EAE, such a defect was not found, suggesting that IL-17F regulation of IL-17 expression could be overcome by chronic immune responses. Furthermore, IgG2a production was consistently up-regulated only in IL-17F–deficient animals. Because IFN-γ production was not affected significantly in the same setting, it is possible that IL-17F may regulate B cell responses to IFN-γ, which is known to be important for IgG2a switching. B cells express the receptors for IL-17F (unpublished data). Whether they respond to IL-17F and what effect will be caused need to be investigated further. Nonetheless, our data suggest differential but clear involvement of IL-17 and IL-17F in the early phases of immune responses.
Evidence such as its presence in asthmatic CD4 T cell clones (22) or its mutations in asthma patients (45) suggests that IL-17F might be relevant to asthma. Our current study using IL-17 and IL-17F KO animals indicates that IL-17 is not required for neutrophil recruitment in innate responses to an allergen; instead, IL-17F appears more prominent in recruiting neutrophils. However, when we examined the effect of IL-17 or IL-17F deficiency on an asthma model, IL-17 KO mice, as predicted from the literature, exhibited reduced Th2 cytokine expression, whereas IL-17F KO mice had enhanced clinic symptoms such as elevated type 2 cytokines and eosinophil functions compared with WT mice. Thus, IL-17 and IL-17F may have opposite functions in chronic allergic airway diseases. The contrasting effects of IL-17 and IL-17F have also been observed in acute experimental colitis induced by DSS. IL-17F KO mice were protected, whereas IL-17 deficiency increased the colon damage. At this stage, it is difficult to comprehend the functional differences of IL-17 and IL-17F in these models. Perhaps the receptors for them are differentially expressed on target cells or these cytokines transduce differential signals that may, in turn, activate feedback antiinflammatory mechanisms. Further studies will be needed, and our data represent a first step toward a better understanding of IL-17 and IL-17F functions in vivo.
In summary, in addition to IL-17, IL-17F is expressed in Th17 cells and IL-17–expressing γδ T cells. These two cytokines may use the same signaling components, such as IL-17RA, Act1, and TRAF6, to induce similar downstream inflammatory genes. In vivo, transgenic overexpression of IL-17 or IL-I7F in the lung led to similar pathological phenotypes. However, our analysis using mice deficient in either gene has revealed their distinct functions in different types of inflammatory responses. Our results have revealed complex mechanisms underlying tissue inflammation. A further understanding of these mechanisms may shed light on cytokine targeting in the treatment of inflammatory diseases such as allergic asthma and multiple sclerosis.
MATERIALS AND METHODS
Reagents.
Mouse IFN-γ, IL-4, and IL-17 staining antibodies; IFN-γ, IL-4, IL-17, and IL-6 ELISA antibodies; and CD4, CD8, B220, and isotype control antibodies used in immunohistochemistry analysis were purchased from BD Biosciences. CD11c antibody was purchased from Endogen. IL-6, TGF-β, IL-1β, and TNF-α were purchased from PeproTech, and LPS was obtained from Sigma-Aldrich. IL-23, IL-17, and IL-17F cytokines and ELISA antibodies for CXCL1 and IL-17F were obtained from R&D Systems. For the generation of IL-17F–Ig protein, cDNA sequences encoding IL-17F were amplified by PCR and cloned into the DES-Ig vector (46). The secreted Ig fusion proteins from stably transfected S2 cells were purified by a protein A column after the CuSO4 induction of fusion proteins expression. Polyclonal antibody against IL-17F was made by PRF&L by immunizing rabbits with purified IL-17F–Ig fusion proteins.
Transient transfection.
IL-17 and IL-17F cDNA were PCR amplified and cloned into the pcDNA 3.1+ vector (Invitrogen), followed by sequencing confirmation. 293T cells were transfected by calcium phosphate transfection.
Cell isolation and culture.
Naive CD4+ T cells from OT-II mice purified by AutoMACS (Miltenyi Biotec) were cultured at a ratio of 1:2 with irradiated B6 splenic APCs. Th1, Th2, and Th17 differentiation were performed as described previously (39). IL-17R−/− mice obtained from Amgen were used to generate MEFs using standard approaches. TRAF6−/− MEFs were provided by T. Mak (University of Toronto, Toronto, Canada) and grown in high glucose DMEM supplemented with 10% FBS. For the measurement of IL-6 or CXCL1, cells were treated with cytokines overnight, and the culture supernatants were analyzed by ELISA. For RNA analysis, MEFs were treated for 4 h in serum-free DMEM before stimulation with various cytokines and homogenization in TRIzol (Invitrogen). RNA samples from IL-17F–treated Act1-deficient MEFs were provided by X. Li and C. Liu (Cleveland Clinic, Cleveland, OH). Lamina propria and intestinal intraepithelial lymphocytes were isolated from the small intestine as previously described (47).
RT-PCR.
cDNA was synthesized using RNase H RT (Invitrogen) analyzed in triplicates by using iQ SYBR Green Supermix in an iCycler (both from Bio-Rad Laboratories). The expression levels of each gene were normalized to the reference gene Actb expression level using a standard curve method. The primer sets for real-time PCR are as follows: IL-17F, (F) 5′-CTGGAGGATAACACTGTGAGAGT-3′ and (R) 5′-TGCTGAATGGCGACGGAGTTC-3′. Other primers were described previously (3, 12, 36).
Recombinant cytokine administration.
IL-17R−/− and WT mice were administered i.p. with 0.5 μg of recombinant mouse IL-17 or IL-17F (R&D Systems) per mouse in 500 μl of endotoxin-free PBS. The peritoneal lavage fluid was carefully collected after 4 h. Cell pellets were resuspended in PBS/EDTA and total cell numbers were counted. Differential cell counting was performed on cytospin preparations stained with May-Grünwald-Giemsa.
Generation of IL-17F transgenic mice.
To generate IL-17F transgenic mice, the CC10 promoter sequence (a gift from J. Elias, Yale University, New Haven, CT) was ligated to full-length mouse IL-17F cDNA followed by an internal ribosomal entry site (IRES)–GFP cassette (provided by K. Murphy, Washington University, St. Louis, MO). Human growth hormone intronic and polyadenylation sequences were inserted into the CC10–IL-17F–IRES–GFP–pBluescript II construct using the BamHI and NotI restriction enzyme sites. The IL-17F transgene construct was isolated by digestion with NotI and SapI and was microinjected into B6 mice at the Transgenic Facility at the University of Washington. Three IL-17F transgenic founders were obtained and were maintained by breeding with B6 mice. Lungs were infused with 4% paraformaldehyde, and sections from these samples were made and stained with hematoxylin and eosin (H&E) and PAS. For immunohistochemistry, the lungs were inflated with a 1:1 mixture of PBS/optimum cutting temperature (OCT) compound (Tissue-Tek; Sakura Finetek), embedded in OCT, and quickly frozen in a dry ice/isopropyl alcohol bath.
Generation of IL-17 and IL-17F KO mice.
IL-17–deficient mice were generated by replacement of partial IL-17 exon 2 with a luciferase-IRES-eGFP cassette without disrupting splicing boundaries, using the 129/TC1 embryonic stem cell line. The targeting vector applied NeoR as positive and thymidine kinase as negative selection markers. IL-17F KO was generated by insertion of an IRES-mRFP-polyA cassette into IL-17F exon 2. An Frt-flanked puromycin resistance gene and a diphtheria toxin gene served as positive and negative selection markers, respectively. Targeted embryonic stem clones for IL-17 or IL-17F were selected and injected into C57BL/6 blastocysts to generate chimeras. High percentage chimeras were bred with C57BL/6 for germline transmission. IL-17+/− mice were obtained after deletion of the NeoR cassette by crossing with the CMV-Cre strain, and IL-17F+/− mice were obtained after excision of the PuroR cassette by crossing with FLPeR strain. Homozygous KO and WT animals on the same 129 × C57BL/6 F1 mixed background were bred and used in experiments. The genotyping primers for IL-17 KO were as follows: (F) 5′-TCAACCGTTCCACGTCACCCTGGAC-3′ and (R) 5′-TCAGCATTCAACTTGAGCTCTCATGC-3′, amplifying a 299-bp WT band and/or a 414-bp KO band. The genotyping primers for IL-17F KO were as follows: (F) 5′-ACATTGCCCACCACCAGGGCTC-3′, (R1) 5′-CCCATGGGGAACTGGAGCGGTTC-3′, and (R2) 5′-TTCGGCCAGTAACGTTAGG-3′. The primers F and R1 surrounding the insertion breakpoint in exon 2 amplify a 263-bp WT band, whereas the primers F and R2 flanking the FRT site amplify a 378-bp KO band.
KLH immunization.
6–8-wk-old mice (three per group) were immunized with 0.5 mg/ml KLH emulsified in 0.5 mg/ml CFA at the base of the tail (100 μl/mouse). 7 d after immunization, the mice were killed and analyzed individually. Splenocytes from the immunized mice were restimulated with 50 μg/ml KLH and analyzed by intracellular staining or ELISA for cytokine expression. Sera from immunized mice were collected, and KLH-specific antibodies were measured by using ELISA. In brief, serum samples were added in a threefold serial dilution onto plates precoated with 10 μg/ml KLH. KLH-specific antibodies were detected with biotinylated goat anti–mouse IgM and rat anti–mouse IgG, IgG1, IgG2a, and IgG2b antibodies (SouthernBiotech).
Intranasal challenge of allergen.
7 μl (7 μg) FAP derived from Aspergillus oryzae (Sigma-Aldrich) was added to 43 μl (21.5 μg) of OVA immediately before intranasal administration. Anesthetized mice were instilled intranasally with 50 μl FAP/OVA (34). Mice were killed 18 h later for BALF cellular analysis. Neutrophils were counted after cytospin and May-Grünwald-Giemsa staining, or were stained with Gr.1 and analyzed by FACS. Part of the lungs was collected for quantitative mRNA analysis.
Asthma induction.
Mice were immunized twice at 2-wk intervals with 0.2 ml saline containing 100 μg OVA in aluminum hydroxide. Mice were sensitized at day 14 and rechallenged intranasally three more times at days 25, 26, and 27 with 100 μg OVA. 1 d after the last challenge, mice were killed and BALF was collected. BALF was analyzed for cellular composition and cytokine concentrations. Parts of BAL cells and lungs were homogenized in TRIzol for RNA extraction. Spleens and mediastinal lymph node cells from the asthma mice were further cultured with OVA for 3 d, and supernatants were analyzed for cytokine expression by ELISA.
EAE induction.
6–8-wk-old female mice were immunized subcutaneously at the dorsal flanks with 150 μg of MOG peptide in CFA at days 0 and 7. 500 ng/mouse of pertussis toxin was given i.p. at days 1 and 8. Signs of EAE were assigned scores on a scale of 1–5 as follows: 0, none; 1, limp tail or waddling gait with tail tonicity; 2, wobbly gait; 3, hind limb paralysis; 4, hind limb and forelimb paralysis; 5, death. p-values were calculated using the Student's t test by comparing the disease scores. To analyze CNS infiltrates, both the brain and spinal cord were collected from perfused mice, and mononuclear cells were prepared by Percoll gradient.
DSS-induced colitis.
6–10-wk-old age-matched WT, IL-17 KO, and IL-17F KO male mice were fed with 3.5% DSS in their drinking water for 5 d. The mice were weighed daily. On day 8, the mice were killed and colon samples were collected. After multiple flushes with PBS, a 3-mm-long piece was sampled from each colon in the middle and fixed in 4% paraformaldehyde. Microscopic sections were stained with H&E, and disease severity was evaluated based on epithelial injury. Colonic crypt epithelial injury was scored from 0 (no injury) to 10 (100% crypt loss). Three 2-mm pieces from the proximal, middle, and distal portions of the colon were collected and homogenized in TRIzol reagent for RNA expression analysis.
Statistical analysis.
Results were expressed as mean ± SD. Differences between groups were calculated for statistical significance using the unpaired Student's t test. P ≤ 0.05 was considered as significant. The animal experiments were performed using protocols approved by the Institutional Animal Care and Use Committee at the University of Texas M.D. Anderson Cancer Center.
Online supplemental material.
Fig. S1 shows the specificity of anti–IL-17F polyclonal antibody. Fig. S2 and S3 describe the generation of IL-17– and IL-17F–deficient mice, respectively. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20071978/DC1.
Supplementary Material
Acknowledgments
We thank Sandy Rivera, Jan Parker-Thornburg, and Steve Kuschert for their help in the generation of KO animals; Dr. Jack Elias for the CC10 promoter construct; Dr. Tak Mak for TRAF6−/− MEFs; Drs. Xiaoxia Li and Caini Liu for providing RNA samples from IL-17F–treated Act1-deficient MEFs; Dr. Andy Farr (University of Washington, Seattle, WA) and David Corry (Baylor College of Medicine, Houston, TX) for their helpful suggestions; and the entire Dong laboratory for their help and discussion.
This work was supported in part by grants from the National Institutes of Health (to C. Dong and Z. Zhu). S.H. Chang received an Arthritis Foundation postdoctoral fellowship, R. Nurieva is an American Heart Association Scientist Development award recipient, and C. Dong received a Cancer Research Institute Investigator award and an American Lung Association Career Investigator award. K.S. Schluns and C. Dong are both Trust Fellows of the M.D. Anderson Cancer Center.
The authors have no conflicting financial interests.
Abbreviations used: BALF, bronchoalveolar lavage fluid; CC10, Clara cell 10; CNS, central nervous system; DSS, dextran sulfate sodium; EAE, experimental autoimmune encephalomyelitis; FAP, allergenic fungal proteinase; H&E, hematoxylin and eosin; MEF, mouse embryonic fibroblast; MMP, matrix metalloproteinase; MOG, myelin oligodendrocyte glycoprotein; PAS, periodic acid Schiff; TRAF6, TNFR-associated factor 6.
X.O. Yang and S.H. Chang contributed equally to this study.
References
- 1.Kolls, J.K., and A. Linden. 2004. Interleukin-17 family members and inflammation. Immunity. 21:467–476. [DOI] [PubMed] [Google Scholar]
- 2.Moseley, T.A., D.R. Haudenschild, L. Rose, and A.H. Reddi. 2003. Interleukin-17 family and IL-17 receptors. Cytokine Growth Factor Rev. 14:155–174. [DOI] [PubMed] [Google Scholar]
- 3.Park, H., Z. Li, X.O. Yang, S.H. Chang, R. Nurieva, Y.H. Wang, Y. Wang, L. Hood, Z. Zhu, Q. Tian, and C. Dong. 2005. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol. 6:1133–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ruddy, M.J., G.C. Wong, X.K. Liu, H. Yamamoto, S. Kasayama, K.L. Kirkwood, and S.L. Gaffen. 2004. Functional cooperation between interleukin-17 and tumor necrosis factor-alpha is mediated by CCAAT/enhancer-binding protein family members. J. Biol. Chem. 279:2559–2567. [DOI] [PubMed] [Google Scholar]
- 5.Ye, P., F.H. Rodriguez, S. Kanaly, K.L. Stocking, J. Schurr, P. Schwarzenberger, P. Oliver, W. Huang, P. Zhang, J. Zhang, et al. 2001. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J. Exp. Med. 194:519–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakae, S., A. Nambu, K. Sudo, and Y. Iwakura. 2003. Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. J. Immunol. 171:6173–6177. [DOI] [PubMed] [Google Scholar]
- 7.Bush, K.A., K.M. Farmer, J.S. Walker, and B.W. Kirkham. 2002. Reduction of joint inflammation and bone erosion in rat adjuvant arthritis by treatment with interleukin-17 receptor IgG1 Fc fusion protein. Arthritis Rheum. 46:802–805. [DOI] [PubMed] [Google Scholar]
- 8.Langrish, C.L., Y. Chen, W.M. Blumenschein, J. Mattson, B. Basham, J.D. Sedgwick, T. McClanahan, R.A. Kastelein, and D.J. Cua. 2005. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med. 201:233–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yao, Z., W.C. Fanslow, M.F. Seldin, A.M. Rousseau, S.L. Painter, M.R. Comeau, J.I. Cohen, and M.K. Spriggs. 1995. Herpesvirus Saimiri encodes a new cytokine, IL-17, which binds to a novel cytokine receptor. Immunity. 3:811–821. [DOI] [PubMed] [Google Scholar]
- 10.Toy, D., D. Kugler, M. Wolfson, T. Vanden Bos, J. Gurgel, J. Derry, J. Tocker, and J. Peschon. 2006. Cutting edge: interleukin 17 signals through a heteromeric receptor complex. J. Immunol. 177:36–39. [DOI] [PubMed] [Google Scholar]
- 11.Schwandner, R., K. Yamaguchi, and Z. Cao. 2000. Requirement of tumor necrosis factor receptor–associated factor (TRAF) 6 in interleukin 17 signal transduction. J. Exp. Med. 191:1233–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang, S.H., H. Park, and C. Dong. 2006. Act1 adaptor protein is an immediate and essential signaling component of IL-17 receptor. J. Biol. Chem. 281:35603–35607. [DOI] [PubMed] [Google Scholar]
- 13.Dong, C. 2006. Diversification of T-helper-cell lineages: finding the family root of IL-17-producing cells. Nat. Rev. Immunol. 6:329–334. [DOI] [PubMed] [Google Scholar]
- 14.Weaver, C.T., L.E. Harrington, P.R. Mangan, M. Gavrieli, and K.M. Murphy. 2006. Th17: an effector CD4 T cell lineage with regulatory T cell ties. Immunity. 24:677–688. [DOI] [PubMed] [Google Scholar]
- 15.Akimzhanov, A.M., X.O. Yang, and C. Dong. 2007. Chromatin remodeling of interleukin-17 (IL-17)-IL-17F cytokine gene locus during inflammatory helper T cell differentiation. J. Biol. Chem. 282:5969–5972. [DOI] [PubMed] [Google Scholar]
- 16.Chang, S.H., and C. Dong. 2007. A novel heterodimeric cytokine consisting of IL-17 and IL-17F regulates inflammatory responses. Cell Res. 17:435–440. [DOI] [PubMed] [Google Scholar]
- 17.Wright, J.F., Y. Guo, A. Quazi, D.P. Luxenberg, F. Bennett, J.F. Ross, Y. Qiu, M.J. Whitters, K.N. Tomkinson, K. Dunussi-Joannopoulos, et al. 2007. Identification of an interleukin 17F/17A heterodimer in activated human CD4+ T cells. J. Biol. Chem. 282:13447–13455. [DOI] [PubMed] [Google Scholar]
- 18.Hymowitz, S.G., E.H. Filvaroff, J.P. Yin, J. Lee, L. Cai, P. Risser, M. Maruoka, W. Mao, J. Foster, R.F. Kelley, et al. 2001. IL-17s adopt a cystine knot fold: structure and activity of a novel cytokine, IL-17F, and implications for receptor binding. EMBO J. 20:5332–5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McAllister, F., A. Henry, J.L. Kreindler, P.J. Dubin, L. Ulrich, C. Steele, J.D. Finder, J.M. Pilewski, B.M. Carreno, S.J. Goldman, et al. 2005. Role of IL-17A, IL-17F, and the IL-17 receptor in regulating growth-related oncogene-alpha and granulocyte colony-stimulating factor in bronchial epithelium: implications for airway inflammation in cystic fibrosis. J. Immunol. 175:404–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuestner, R.E., D.W. Taft, A. Haran, C.S. Brandt, T. Brender, K. Lum, B. Harder, S. Okada, C.D. Ostrander, J.L. Kreindler, et al. 2007. Identification of the IL-17 receptor related molecule IL-17RC as the receptor for IL-17F. J. Immunol. 179:5462–5473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hizawa, N., M. Kawaguchi, S.K. Huang, and M. Nishimura. 2006. Role of interleukin-17F in chronic inflammatory and allergic lung disease. Clin. Exp. Allergy. 36:1109–1114. [DOI] [PubMed] [Google Scholar]
- 22.Kawaguchi, M., L.F. Onuchic, X.D. Li, D.M. Essayan, J. Schroeder, H.Q. Xiao, M.C. Liu, G. Krishnaswamy, G. Germino, and S.K. Huang. 2001. Identification of a novel cytokine, ML-1, and its expression in subjects with asthma. J. Immunol. 167:4430–4435. [DOI] [PubMed] [Google Scholar]
- 23.Hurst, S.D., T. Muchamuel, D.M. Gorman, J.M. Gilbert, T. Clifford, S. Kwan, S. Menon, B. Seymour, C. Jackson, T.T. Kung, et al. 2002. New IL-17 family members promote Th1 or Th2 responses in the lung: in vivo function of the novel cytokine IL-25. J. Immunol. 169:443–453. [DOI] [PubMed] [Google Scholar]
- 24.Oda, N., P.B. Canelos, D.M. Essayan, B.A. Plunkett, A.C. Myers, and S.K. Huang. 2005. Interleukin-17F induces pulmonary neutrophilia and amplifies antigen-induced allergic response. Am. J. Respir. Crit. Care Med. 171:12–18. [DOI] [PubMed] [Google Scholar]
- 25.Stark, M.A., Y. Huo, T.L. Burcin, M.A. Morris, T.S. Olson, and K. Ley. 2005. Phagocytosis of apoptotic neutrophils regulates granulopoiesis via IL-23 and IL-17. Immunity. 22:285–294. [DOI] [PubMed] [Google Scholar]
- 26.Lockhart, E., A.M. Green, and J.L. Flynn. 2006. IL-17 production is dominated by gammadelta T cells rather than CD4 T cells during Mycobacterium tuberculosis infection. J. Immunol. 177:4662–4669. [DOI] [PubMed] [Google Scholar]
- 27.Ivanov, I.I., B.S. McKenzie, L. Zhou, C.E. Tadokoro, A. Lepelley, J.J. Lafaille, D.J. Cua, and D.R. Littman. 2006. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 126:1121–1133. [DOI] [PubMed] [Google Scholar]
- 28.Nurieva, R., X.O. Yang, G. Martinez, Y. Zhang, A.D. Panopoulos, L. Ma, K. Schluns, Q. Tian, S.S. Watowich, A.M. Jetten, and C. Dong. 2007. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature. 448:480–483. [DOI] [PubMed] [Google Scholar]
- 29.Yang, X.O., B.P. Pappu, R. Nurieva, A. Akimzhanov, H.S. Kang, Y. Chung, L. Ma, B. Shah, A.D. Panopoulos, K.S. Schluns, et al. 2008. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity. 28:29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ziegler, S.F., and Y.-J. Liu. 2006. Thymic stromal lymphopoietin in normal and pathogenic T cell development and function. Nat. Immunol. 7:709–714. [DOI] [PubMed] [Google Scholar]
- 31.Maezawa, Y., H. Nakajima, K. Suzuki, T. Tamachi, K. Ikeda, J. Inoue, Y. Saito, and I. Iwamoto. 2006. Involvement of TNF receptor-associated factor 6 in IL-25 receptor signaling. J. Immunol. 176:1013–1018. [DOI] [PubMed] [Google Scholar]
- 32.Zhu, Z., R.J. Homer, Z. Wang, Q. Chen, G.P. Geba, J. Wang, Y. Zhang, and J.A. Elias. 1999. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J. Clin. Invest. 103:779–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakae, S., Y. Komiyama, A. Nambu, K. Sudo, M. Iwase, I. Homma, K. Sekikawa, M. Asano, and Y. Iwakura. 2002. Antigen-specific T cell sensitization is impaired in IL-17-deficient mice, causing suppression of allergic cellular and humoral responses. Immunity. 17:375–387. [DOI] [PubMed] [Google Scholar]
- 34.Kiss, A., M. Montes, S. Susarla, E.A. Jaensson, S.M. Drouin, R.A. Wetsel, Z. Yao, R. Martin, N. Hamzeh, R. Adelagun, et al. 2007. A new mechanism regulating the initiation of allergic airway inflammation. J. Allergy Clin. Immunol. 120:334–342. [DOI] [PubMed] [Google Scholar]
- 35.Schnyder-Candrian, S., D. Togbe, I. Couillin, I. Mercier, F. Brombacher, V. Quesniaux, F. Fossiez, B. Ryffel, and B. Schnyder. 2006. Interleukin-17 is a negative regulator of established allergic asthma. J. Exp. Med. 203:2715–2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shinkai, K., M. Mohrs, and R.M. Locksley. 2002. Helper T cells regulate type-2 innate immunity in vivo. Nature. 420:825–829. [DOI] [PubMed] [Google Scholar]
- 37.Ogawa, A., A. Andoh, Y. Araki, T. Bamba, and Y. Fujiyama. 2004. Neutralization of interleukin-17 aggravates dextran sulfate sodium-induced colitis in mice. Clin. Immunol. 110:55–62. [DOI] [PubMed] [Google Scholar]
- 38.Melgar, S., A. Karlsson, and E. Michaelsson. 2005. Acute colitis induced by dextran sulfate sodium progresses to chronicity in C57BL/6 but not in BALB/c mice: correlation between symptoms and inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 288:G1328–G1338. [DOI] [PubMed] [Google Scholar]
- 39.Chung, Y., X. Yang, S.H. Chang, L. Ma, Q. Tian, and C. Dong. 2006. Expression and regulation of IL-22 in the IL-17-producing CD4+ T lymphocytes. Cell Res. 16:902–907. [DOI] [PubMed] [Google Scholar]
- 40.Yang, X.O., A.D. Panopoulos, R. Nurieva, S.H. Chang, D. Wang, S.S. Watowich, and C. Dong. 2007. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J. Biol. Chem. 282:9358–9363. [DOI] [PubMed] [Google Scholar]
- 41.Dong, C., A.E. Juedes, U.A. Temann, S. Shresta, J.P. Allison, N.H. Ruddle, and R.A. Flavell. 2001. ICOS co-stimulatory receptor is essential for T-cell activation and function. Nature. 409:97–101. [DOI] [PubMed] [Google Scholar]
- 42.Nurieva, R.I., J. Duong, H. Kishikawa, U. Dianzani, J.M. Rojo, I. Ho, R.A. Flavell, and C. Dong. 2003. Transcriptional regulation of th2 differentiation by inducible costimulator. Immunity. 18:801–811. [DOI] [PubMed] [Google Scholar]
- 43.Zheng, Y., D.M. Danilenko, P. Valdez, I. Kasman, J. Eastham-Anderson, J. Wu, and W. Ouyang. 2007. Interleukin-22, a TH17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature. 445:648–651. [DOI] [PubMed] [Google Scholar]
- 44.Harrington, L.E., R.D. Hatton, P.R. Mangan, H. Turner, T.L. Murphy, K.M. Murphy, and C.T. Weaver. 2005. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 6:1123–1132. [DOI] [PubMed] [Google Scholar]
- 45.Kawaguchi, M., D. Takahashi, N. Hizawa, S. Suzuki, S. Matsukura, F. Kokubu, Y. Maeda, Y. Fukui, S. Konno, S.K. Huang, et al. 2006. IL-17F sequence variant (His161Arg) is associated with protection against asthma and antagonizes wild-type IL-17F activity. J. Allergy Clin. Immunol. 117:795–801. [DOI] [PubMed] [Google Scholar]
- 46.Sun, M., S. Richards, D.V. Prasad, X.M. Mai, A. Rudensky, and C. Dong. 2002. Characterization of mouse and human B7-H3 genes. J. Immunol. 168:6294–6297. [DOI] [PubMed] [Google Scholar]
- 47.Laky, K., L. Lefrancois, and L. Puddington. 1997. Age-dependent intestinal lymphoproliferative disorder due to stem cell factor receptor deficiency: parameters in small and large intestine. J. Immunol. 158:1417–1427. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}