

Abstract
The cell’s own antioxidant response element (ARE) can be used to evaluate the complications of diabetes mellitus. The hypothesis that a synthetic ARE could be used as a genetic switch, or biosensor, to turn on and off therapeutic genes is tested herein. Mitochondrial oxidative stress (MOS) has been hypothesized as one of the earliest insults in diabetes. Fluorescent probes used to monitor MOS revealed that the addition of glucose at physiological levels to cultures of endothelial cells was able to induce MOS above normal levels and in a dose dependant manner. Additional data showed that increased glucose levels activated the ARE-GFP in a dose dependant manner. These data support the hypothesis that the induction of MOS is more sensitive to hyperglycemia than the induction of the ARE. Delivery of an ARE-GFP construct with nanoparticles to the eye was successful using sub-retinal injection. This ARE-GFP/nanoparticle construct was functional and reported the activation of the ARE in diabetic rat retinal pigment epithelium (RPE). These data support the use of nanoparticle delivered biosensors for monitoring the oxidative status of tissues in vivo.
INTRODUCTION
In the United States, the leading cause of new blindness in people 20–74 years of age is diabetic retinopathy. Prior work has demonstrated that both endothelial cells and pericytes undergo apoptosis in human diabetic retina and in a rat model of diabetes (Mizutani et al. 1996). This results eventually in the acellular capillaries that occur early diabetic in retinopathy. These collagenous tubes no longer move blood and adjacent retina becomes hypoxic. Hypoxic retina in turn produces vascular endothelial cell growth factor (VEGF), which stimulates increased vascular permeability and eventually retinal neovascularization. Prevention of acellular capillary formation could prevent proliferative diabetic retinopathy.
An alternative to hyperglycemia-induced cell death causing acellular capillaries is that vaso-occlusions in retina and choroid contribute to capillary loss and neural dysfunction in retina. Adhesion of leukocytes to vascular endothelium could initiate capillary occlusion (Lutty and Ogura 2003). Diabetic leukocytes demonstrate increased adhesion (Setiadi et al. 1987) and are more rigid than leukocytes from non-diabetics (Kantar et al. 1991). Therefore, once adherent they could potentially obstruct narrow capillary lumens, especially those with poor vasotonia due to thickened basement membrane and loss of pericytes, which are present in diabetic retinopathy (Yanoff 1969). In diabetics, increased numbers of polymorphonuclear leukocytes (PMN’s) are in the activated state, a state in which they express molecules modulating their adherence to endothelial cells (Wierusz-Wysocki et al. 1987). If activated PMN adhere to endothelial cells, oxygen radical-mediated injury to endothelial cells occurs, which could result in apoptosis of endothelial cells. Activated diabetic PMN produce more superoxide radicals than non-diabetic PMN (Wierusz-Wysocki et al. 1987). In an experimental rat model of diabetes, there was increased adherence and diapedesis of leukocytes (Schröder et al. 1991).
Brownlee proposed that the major molecular mechanisms implicated in glucose-mediated vascular injury reflect a single hyperglycemia-induced process of overproduction of superoxide by the mitochondrial electron transport chain (Brownlee 2001). Mitochondria play a central role in apoptosis triggered by chemical stimuli. Following exposure of the cells to an apoptogenic agent, the mitochondrial outer membrane destabilizes. Reactive oxygen species (ROS) are involved in the destabilization of the mitochondrial membrane. Hyperglycemia can be considered an apoptogenic agent in that it induces oxidative stress that results in mitochondrial membrane depolarization and induction of apoptosis. This has been documented in peripheral nerve by Vincent et al (Vincent et al. 2002) and in endothelial cells by Mizutani (Mizutani et al. 1996). Over expression of uncoupling proteins prevents glucose-induced transient mitochondrial membrane hyperpolarization, reactive oxygen species, and induction of apoptosis (Vincent et al. 2004).
Most diseases have a component of oxidative stress, but cancer researchers have pioneered the use of antioxidants for therapeutics. Induction of the ARE has been a theme of cancer prevention and ARE investigation, since the discovery of strong ARE modulators, e.g. tert-butylhydroquinone and sulforaphane (Talalay et al. 1995; Kraft et al. 2004). Previous work has shown that the ARE can be functional while tethered to a nanoparticle and this system can be used to detect the cellular ARE response to pathogenic stimuli in vitro (Prow et al. 2004; Prow et al. 2006; Prow et al. 2006). This study focuses on the use of nanoparticle-delivered ARE to regulate the expression of reporter genes to specifically detect MOS in diabetes. The data show that endothelial cells in culture do experience MOS with increased glucose and that further increases in glucose activate the ARE. Diabetic rats were used to evaluate nanoparticle-delivered biosensors for oxidative stress. These experiments showed that the nanoparticle-ARE construct was functional in vivo and revealed that diabetes is a powerful inducer of the ARE.
MATERIALS AND METHODS
Cell Culture
Human retinal microvascular endothelial cells (HREC, Cell Systems, Seattle, WA) were maintained in Dulbecco’s modified eagle medium /Ham’s F12 (DMEM/F12) medium containing 10% FBS and 1% penicillin-streptomycin-fungizone at 37°C in a 5% CO2 environment. Endothelial cells were passaged by washing with phosphate buffered saline followed by trypsin digestion (0.25% trypsin). The cells were then washed with complete medium before plating. Endothelial cells were passaged less than 3 times for these experiments. All cell culture reagents were purchased from Invitrogen (Carlsbad, CA).
In vitro Transfection and Polyclonal Selection
The cells were transfected after being cultured for at least 24 hours and only when they were 70–80% confluent. Cells were transfected with Lipofectamine 2000 (Invitrogen). At the time of transfection, the cells were washed three times with DMEM/F12. The DNA (4 µg for a 6 well plate) was diluted in 250 µl of Opti-MEM and mixed gently. The Lipofectamine 2000 was mixed gently before use. Lipofectamine 2000 (5 µl for a 6 well plate) was mixed with 250 µl of Opti-MEM in a separate tube. The DNA and Lipofectamine 2000 tubes were incubated at room temperature for 5 minutes. After 5 minutes, the diluted DNA and Lipofectamine 2000 were combined and mixed gently. This mixture was incubated at room temperature for 20 minutes. The media was removed from the culture and the DNA/Lipofectamine 2000 complex was added. Growth media was replaced after 4–6 hours and the cells incubated for 24 hours prior to experimentation or selection. Selection was accomplished by supplementing growth media with 500 µg/ml G418 (Invitrogen) for one week and maintenance with 250 µg/ml G418 thereafter.
Flow Cytometry
Flow cytometry was used to assess the expression of ARE-driven EGFP in vitro. These adherent cells were trypsinized and then washed prior to flow cytometry. Cells were run live in all cases. Flow cytometry was done on a BD FACScan in the JHU School of Medicine Flow Cytometry and Cell Sorting Core Facility, using channel FL1 for EGFP fluorescence. WinMDI was used to analyze the data. Experiments were run in triplicate in 6 well plates. After trypsinizing, the three wells were combined to increase cell number and then analyzed. A minimum of 5,000 events were captured for each file.
Nanoparticle Construction
Biosensor-tethered nanoparticles were constructed as described previously (Prow et al. 2006; Prow et al. 2006). Biotin-labelled PCR products were generated from a pARE-GFP template generously donated by William Fahl (University of Wisconsin, Madison, WI) and Ming Zhu (Arizona Cancer Center, Tuscon, AZ). The forward oligonucleotide had a single biotin at the 5’ end and the sequences were as follows: forward 5'-TAG TTA TTA ATA GTA ATC-3', reverse 5'-TAC ATT GAT GAG TTT GGA-3' (Integrated DNA Technologies, Inc., Coralville, IA). Ten millilitres of PCR reaction was generated using Red Taq (Sigma, St. Louis, MO) under the manufactures suggested concentration of reagents. The PCR was carried out in 96 well plates. A typical PCR cycle consisted of denaturing at 94°C for 30 s, annealing at 65°C for 30 s and extension for 2 min at 72°C. Biotin-labelled PCR products were tethered to streptavidin-coated magnetic nanoparticles (MNP, Miltenyi Biotech, Inc., Auburn, CA; Figure 1). The magnetic nanoparticles were incubated with the biotin-labelled PCR fragments at about 30 ng of 1.5 kb TAP per 1 µl of nanoparticles. The mixture was allowed to incubate at room temperature for 30 min. During that time, the magnetic column (Miltenyi Biotech, Inc.) was prepared by washing once with 100 µl of the included nucleic acid buffer and three times with 100 µl Opti-MEM (Invitrogen). Once washed, the column was loaded with the DNA nanoparticle mixture. The column was then washed three times with 100 µl Opti-MEM. The nanoparticles were eluted by removing the column from the magnet and adding 100 µl of Opti-MEM. This solution was then ready for injection.
Fig. 1. Overview of the ARE-GFP and MOS in diabetes.

(A) In the normal state, Nrf2 (green triangle) is retained in the cytoplasm by Keap1 (red octagon). In diabetes, the mitochondria undergoes MOS (red mitochondria) (Panel B). Next, the MOS induces cellular reactive oxygen species (ROS) (yellow), both of which can inactivate Keap1 and allow Nrf2 to bind the ARE in the nucleus (Panel B). MOS and ROS activated ARE and other cell stress pathways induces the expression of antioxidant enzymes and the EGFP reporter protein from the ARE-GFP (arrow, green, Panel C). The antioxidant enzymes attempt to neutralize the ROS and some of the MOS (light red mitochondria) (Panel C) and hopefully return the cell to normal levels of ROS (Panel D). If not controlled, these events are thought to reoccur and cycle from Panels B to D.
MOS Assay
The concentration of the CM-H2TMRos (Invitrogen) probe was empirically determined to be 2 µM. The probe was prepared in complete media. For hand counting studies, the cells were cultured on cover slip chamber slides. The media containing the fluorescent probe was pre-warmed and the cells incubated with the probe for 30 minutes under normal culture conditions. Once the cells were loaded, they were washed briefly in complete media and incubated for an additional 10 minutes. The cells were monitored with an inverted fluorescence microscope equipped with a Cy3 filter set to determine the optimal dye concentration and incubation times. The cell media was changed to media without phenol red prior to evaluation with confocal microscopy.
Animal Studies
Male Lewis rats aged 8 weeks, 180–220g, were maintained at 22°C, 30–70% humidity, and acclimatized to a 12 hour light cycle. A total of 12 rats were randomly assigned to two groups: Streptozotocin (STZ, St. Louis, MO) and vehicle treated. The rats were fed normal chow diet that contained 5% fat, 19% protein, and 5% crude fiber. The Streptozotocin group were treated with an intraperitoneal injection of Streptozotocin (30 mg/kg diluted in 0.1 mol/L sodium citrate buffer, pH 4.5). The vehicle treated animals were injected with the buffer only. Plasma glucose testing was done after overnight fasting with a One Touch Ultra glucometer (Lifescan, Burnaby, BC, Canada) 3 days post STZ injection). Animals with a blood glucose level of 300g/dL were considered diabetic.
Sub-retinal injection
All procedures were performed in accordance with the ARVO statement for use of animals in ophthalmologic and vision research. Prior to injection, the animals were anesthetized with a 1:1 dilution of 20 mg/ml Xylazine and 100 mg/ml Ketamine (Phoenix Scientific, Inc. St. Louis, MO). The pupils were dilated with 1% Tropicamide and 2.5% Phenylephrine HCl (Akorn, Inc. Somerset, NJ) and 0.5% Tetracaine HCl (Phoenix Pharmaceutical, Inc., St. Joseph, MD) was applied for topical anesthesia. Sub-retinal injections were done with a 1 cc syringe using a blunt, plastic, 30 gauge tube. Once the retina was reached, 50 µl was injected slowly resulting in a bleb-shaped elevation in retina. Sub-retinal injections were given 2 disk diameters inferior to the optic nerve. Bacitracin Zinc and Polymyxin B Sulfate ophthalmic ointment (Akorn, Inc. Somerset, NJ) was applied to both eyes immediately after surgery. The animals were sacrificed seven days after sub-retinal injection by a lethal intra-venous injection of 1 ml Beuthanasia (Schering-Plough, Inc., Kenilworth, NJ). The eyes were then removed, injected with 1 ml of 2% Paraformaldehyde in 0.1 M Phosphate buffer, and placed in 30 ml of 2% Paraformaldehyde in 0.1 M Phosphate buffer for one hour.
Confocal Micrsocopy
Confocal analysis was done on a Zeiss 510 META confocal microscope (Carl Zeiss MicroImaging, Inc., Thornwood, NY) in the Wilmer Imaging Core Facility. Excitation wavelengths include 405, 488, 514, 543, and 633 nm depending on the fluorescent probes used, e.g. 405 nm excitation and 420 nm to 480 nm emission bandpass for Hoechst 33342, 488 nm excitation and 505 nm to 530 nm emission bandpass for EGFP, and 543 nm for CM-H2TMRos with 560 nm to 615 nm emission bandpass. Appropriate emission wavelengths were used for each fluorochrome used. Cells were analyzed with either 5, 10, 20, 40, 63, or 100X objectives. Multispectral confocal microscopy was used as a validation tool for both biosensor targeting to separate overlapping spectra and autofluorescence from bona fide labelling. The method of spectral deconvolution was developed at JPL/Cal Tech (Pasadena, CA) and was implemented in a new generation of multispectral confocal microscope (Model 510 META, Zeiss, Inc.). The “emission fingerprinting” algorithm works by fitting the spectral components over low or non-overlapping portions of the combined spectrum of a multicolor image. The components are appropriately weighted so that the combination of color components matches the overall spectrum from the image pixel-by-pixel in each image plane.
RESULTS
MOS and ARE Activity in Human Retinal Endothelial Cells Exposed to Hyperglycemia
Theoretically, mitochondrial oxidative stress should be detected first through CM-H2TMRos and then ARE-mediated GFP expression visually detected. According to this model, if the cell does not recover from hyperglycemia or MOS/ROS, there tends to be accumulation of GFP, followed by apoptosis, or programmed cell death (PCD). At this stage, the mitochondria is depolarized and, therefore, does not accumulate CM-H2TMRos resulting in low or no mitochondrial fluorescence. These events are shown through confocal microscopy in Fig. 2. Cultured endothelial cells were exposed to three different concentrations of glucose, 5, 10, and 30 mM. Normal glucose levels for these cells are 5 mM and there are no visible signs of MOS nor ARE activity. However MOS positive cells were present when endothelial cells were exposed to 10 and 30 mM glucose for 3 days. The ARE was activated in cells treated with 30 mM glucose for 3 days.
Fig. 2. MOS and ARE activity in hyperglycemic HREC.
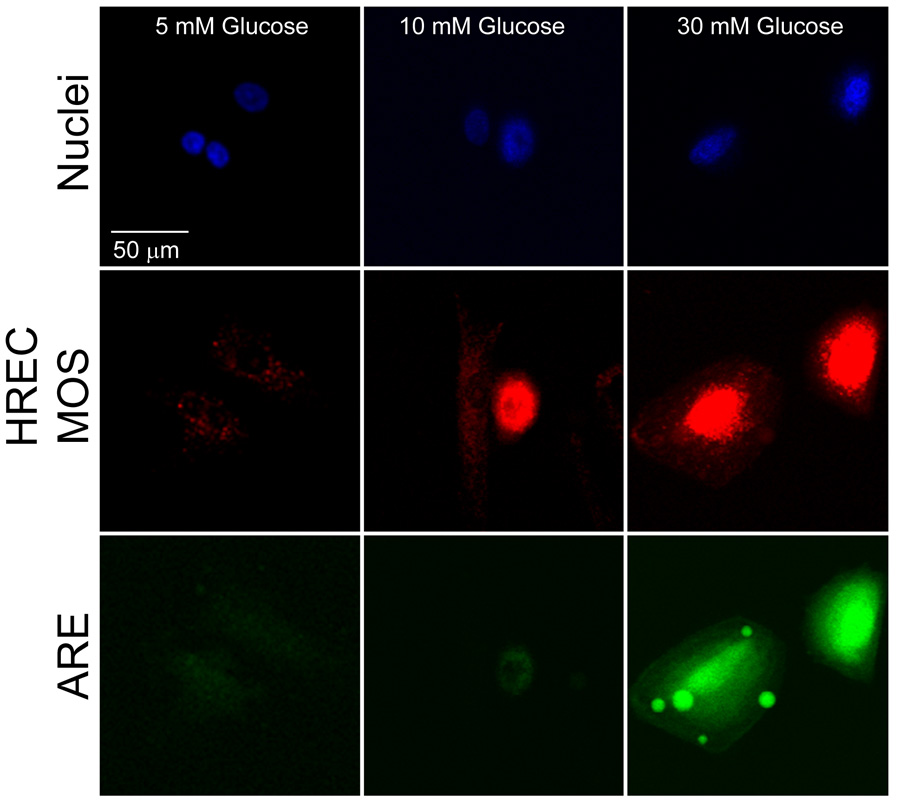
These confocal images are of a ARE-GFP polyclonal HREC line exposed to different doses of glucose (5, 10, and 30 mM) for three days. The cells were stained with Hoechst 33342 (top panels, Nuclei, blue) and CM-H2TMRos (middle panels, MOS, red). ARE-GFP activity is also shown (lower panels, ARE, green).
Dose dependant activation of the ARE and MOS in hyperglycemia
If the the ARE can be used for detection oxidative stress in vivo activation of the ARE during diabetes should be glucose dose-dependant. Figure 3 indicates that ARE activation is dose dependant with respect to glucose level and closely follows MOS as measured by CM-H2TMRos fluorescence in HREC. There is also a dose dependant decrease in the number of cells per field of view (FOV). This indicates that the cells are sensitive to hyperglycemia-mediated MOS and can not mitigate its pathogenic effects. The cell count data has been normalized to the 5 mM glucose group (normal growth media). The large error bars in the hyperglycemia groups (10–55 mM glucose) are indicative of the reduced number of cells that could be counted. Additionally, these cells were derived from a single flask of ARE-GFP transfected HREC that were briefly selected with G418. Therefore, these cells are a polyclonal line and have varying capacities to use the ARE.
Fig. 3. Dose dependant activation of the ARE and MOS in hyperglycemia.

A polyclonal HREC cell line transfected with ARE-GFP were incubated in growth media with 5, 10, 30, and 55 mM glucose) for 3 days. After 3 days the cells were stained with Hoechst 33342 and CM-H2TMRos to label nuclei and MOS positive cells, respectively. Cell counts (black bars) indicated hyperglycemic-toxicity, all cell counts are shown as a % of the negative control (5 mM glucose). The percentage of apoptotic cells, as determined by nuclear morphology, are shown in the white bars. ARE-GFP activity is shown as the percentage of cells with high levels of GFP fluorescence (hatched bars). The percentage of MOS positive cells are shown with cross hatched bars.
Relationship between MOS and ARE activation in cultured endothelial cells
Data from endothelial cells transiently transfected with pARE-GFP were stained with CM-H2TMRos, treated with different levels of glucose for 24 hours and analyzed by flow cytometry (Fig 4). Each bar represents the data gathered from 3 wells of endothelial cells that were combined just before analysis. Cells that had levels of fluorescence that were greater that the 95 percentile of the 10 mM glucose group were considered positive. The number of MOS positive cells in the 30 mM glucose group was over 1.3 fold more than the 10 mM group and the 55 mM group showed the greatest fold increase of all of the groups at over 2 fold. The vast majority of the GFP positive cells were also positive for MOS. This supports the hypothesis that MOS is associated with ARE activation.
Fig. 4. Relationship between MOS and ARE activity in vitro.
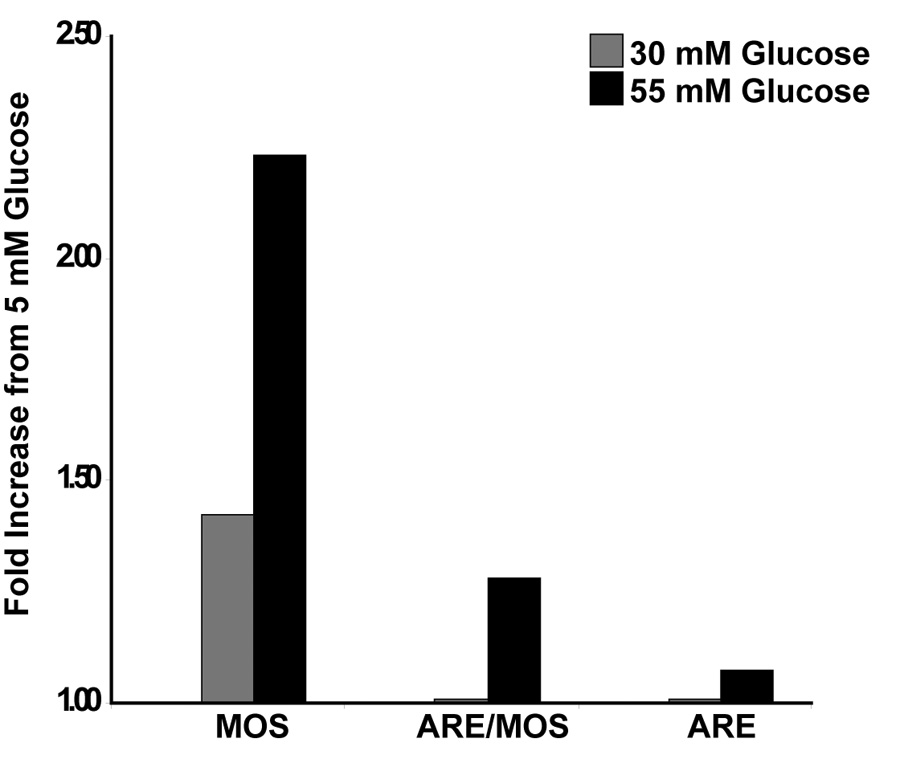
HREC were transiently transfected with ARE-GFP and incubated in 10, 30, and 55 mM glucose for 24 hours. Flow cytometry was used to determine the fold increase in the number of MOS, ARE/MOS, and ARE positive cells compared to the 10 mM glucose group. The gray and black bars indicate 30 and 55 mM glucose levels, respectively.
In vivo evaluation of nanoparticle-delivered ARE-GFP
Lewis rats were made diabetic with a single dose of Streptozotocin. Diabetes was monitored by blood glucose levels. Diabetic rats were maintained for one week prior to nanoparticle injection. The rats were maintained for three days after sub-retinal injection with MNP-ARE-GFP. At 72 hours the rats were sacrificed and the eyecups fixed overnight in 2% Paraformaldehyde. The eyecups were washed and then flattened and photographed with confocal microscopy (Fig. 5). The bleb site was immediately visible under transmitted light. The bleb site of non-diabetic animals did not have many EGFP positive cells. This suggested that non-diabetic retinas did not have highly activated ARE systems. Diabetic animals had many more EGFP positive cells. These cells appeared to be in a sheet and the morphology implied that some of the EGFP positive cells were RPE.
Fig. 5. ARE-tethered nanoparticle activity in normal and diabetic rats.

Non-diabetic (top panels, Non-Diabetic) and diabetic (bottom panels, Diabetic) Lewis rats were injected, via sub-retinal route, with ARE-GFP-tethered nanoparticles. After three days the eyecups were photographed with confocal microscopy. The flattened z-stacks are shown in the left panels (2D) and 3D projections are shown in the right panels (3D).
DISCUSSION
Although oxidative stress is accepted as a major effect of hyperglycemia, oral and systemic antioxidants are ineffective in treating this. The reason may be that most destructive ROS are generated in mitochondria and traditional antioxidants do not reach this site. Our data takes advantage of the cells own antioxidant response to oxidative stress, the ARE, to detect the presence of ROS in vivo.
It is well documented that pancreatic beta cells exposed to hyperglycemia produce ROS and that this causes dysfunction of the beta cells in that glucose-induced insulin secretion is suppressed (Sakai et al. 2003). Raza and associates demonstrated that the pancreas and brain were more severely effected by mitochondrial ROS than liver in STZ rats (Raza et al. 2004). Kraus et al found that endogenous superoxide generated by hyperglycemia activates uncoupling protein 2 diverting energy away from ATP synthesis and impairing glucose stimulated insulin secretion (Krauss et al. 2003). Brownlee suggests that the inhibition of mitochondrial superoxide overproduction in b-cells exposed to hyperglycemia could prevent a positive feed-forward loop of glucotoxicity that drives impaired glucose tolerance toward type 2 diabetes (Brownlee 2003).
Oxidative stress has been implicated in a great variety of disease states, including diabetes (Giardino et al. 1996; Brownlee 2003; Wiernsperger 2003). The cell protects itself from oxidative insult through a variety of signaling pathways that converge onto the multitude of promoter sequences found in the genomic DNA. From bacteria to human beings, oxidative stress can be sensed and responded to by a series of proteins including powerful antioxidants and support enzymes. Oxidative stress is an induced state whose levels are communicated through signaling pathways resulting the induction of antioxidant genes as a protective measure in cells (Prochaska et al. 1985; Cory and Szentivanyi 1987; Prochaska and Talalay 1988). These responses are coordinated through a complex signaling network involving the antioxidant response element (Friling et al. 1992; Wasserman and Fahl 1997). Recent work in the area of oxidative stress has yielded anti-oxidant response element based sensors that are capable of initiating reporter gene expression in response to the presence of oxidative stress inducing chemicals (Zhu and Fahl 2000; Zhu et al. 2001; Zhu and Fahl 2001).
The ARE is composed of a cis-acting enhancer region known as the ARE that is directly controlled by several factors that include the transcription factor Nrf2, a repressor Keap1, and small Maf proteins (Fig. 1).This system is endogenously activated through the actions of oxidative stress on the Keap1 protein (Dinkova-Kostova et al. 2002). This protein normally represses the Nrf2 activator by localizing Nrf2 in the cytoplasm. The activated ARE enhances the expression of any gene downstream of its sequence. Although the ARE can induce a variety of antioxidant and supporting genes, the literature has repeatedly focused on glutathione s-transferase and NAD(P)H:quinone oxidoreductases (Jaiswal 2000; Day et al. 2003).
The reactive oxygen species-induced biosensor is a promoter based system that relies on cellular machinery for the detection of reactive oxygen species. The cell detects reactive oxygen species and activates transcription factors which bind to specific promoter regions. Transcription downstream of these response elements, in this case a fluorescent reporter gene, is then activated. This construct is composed of a number of antioxidant response element repeats followed by a minimal Thymidine Kinase promoter, ahead of a reporter gene, enhanced green fluorescent protein or EGFP. The sensitivity of the biosensor is dramatically affected by the number of ARE repeats, (Zhu and Fahl 2000; Zhu et al. 2001).
This study utilizes fluorescent dye and the ARE to detect the cellular response to hyperglycemia in vitro. These data show that the cell is quickly affected by the presence of hyperglycemia. Fluorescent dye sensitive to oxidation showed that one of the early events in hyperglycemia induced pathology appears to be MOS. This supports the hypothesis that MOS is a key player in hyperglycemia-induced pathology. In our previous work, we have demonstrated the use of a biosensor-tethered nanoparticle to detect the cellular response to ROS insult in vitro. Fig. 5 illustrates the use of that same technology in vivo. The ARE-tethered nanoparticle was capable of detecting the cellular response to the diabetic condition in the eye. These data show that the cells in the retina are responding in a way that strongly suggests ROS and provide the potential to detect ARE activity in the diabetic retina.
Footnotes
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414(6865):813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- Brownlee M. A radical explanation for glucose-induced beta cell dysfunction. J Clin Invest. 2003;112(12):1788–1790. doi: 10.1172/JCI20501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cory JG, Szentivanyi A. Cancer biology and therapeutics. New York: Plenum Press; 1987. [Google Scholar]
- Day RM, Suzuki YJ, et al. Regulation of glutathione by oxidative stress in bovine pulmonary artery endothelial cells. Antioxid Redox Signal. 2003;5(6):699–704. doi: 10.1089/152308603770379991. [DOI] [PubMed] [Google Scholar]
- Dinkova-Kostova AT, Holtzclaw WD, et al. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci U S A. 2002;99(18):11908–11913. doi: 10.1073/pnas.172398899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friling RS, Bergelson S, et al. Two adjacent AP-1-like binding sites form the electrophile-responsive element of the murine glutathione S-transferase Ya subunit gene. Proc Natl Acad Sci U S A. 1992;89(2):668–672. doi: 10.1073/pnas.89.2.668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giardino I, Edelstein D, et al. BCL-2 expression or antioxidants prevent hyperglycemia-induced formation of intracellular advanced glycation endproducts in bovine endothelial cells. J Clin Invest. 1996;97(6):1422–1428. doi: 10.1172/JCI118563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal AK. Regulation of genes encoding NAD(P)H:quinone oxidoreductases. Free Radic Biol Med. 2000;29(3–4):254–262. doi: 10.1016/s0891-5849(00)00306-3. [DOI] [PubMed] [Google Scholar]
- Kantar A, Giorgi P, et al. Alterations in membrane fluidity of diabetic polymorphonuclear leukocytes. Biochem. Med. Metab. Biol. 1991;46:422–426. doi: 10.1016/0885-4505(91)90090-8. [DOI] [PubMed] [Google Scholar]
- Kraft AD, Johnson DA, et al. Nuclear factor E2-related factor 2-dependent antioxidant response element activation by tert-butylhydroquinone and sulforaphane occurring preferentially in astrocytes conditions neurons against oxidative insult. J Neurosci. 2004;24(5):1101–1112. doi: 10.1523/JNEUROSCI.3817-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krauss S, Zhang CY, et al. Superoxide-mediated activation of uncoupling protein 2 causes pancreatic beta cell dysfunction. J Clin Invest. 2003;112(12):1831–1842. doi: 10.1172/JCI19774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutty GA, Ogura Y. Involvement of leukocytes in diabetic retinopathy and choroidopathy. In: Schmid-SchÖnbein GW, Granger ND, editors. Molecular Mechansims of Microvascular Disorders. New York: Springer Verlag; 2003. [Google Scholar]
- Mizutani M, Kern TS, et al. Accelerated death of retinal microvascular cells in human and experimental diabetic retinopathy. J Clin Invest. 1996;97(12):2883–2890. doi: 10.1172/JCI118746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prochaska HJ, De Long MJ, et al. On the mechanisms of induction of cancer-protective enzymes: a unifying proposal. Proc Natl Acad Sci U S A. 1985;82(23):8232–8236. doi: 10.1073/pnas.82.23.8232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prochaska HJ, Talalay P. Regulatory mechanisms of monofunctional and bifunctional anticarcinogenic enzyme inducers in murine liver. Cancer Res. 1988;48(17):4776–4782. [PubMed] [Google Scholar]
- Prow T, Grebe R, et al. Nanoparticle tethered antioxidant response element as a biosensor for oxygen induced toxicity in retinal endothelial cells. Mol Vis. 2006;12:616–625. [PubMed] [Google Scholar]
- Prow T, Smith JN, et al. Construction, gene delivery, and expression of DNA tethered nanoparticles. Mol Vis. 2006;12:606–615. [PubMed] [Google Scholar]
- Prow TW, Kotov NA, et al. Nanoparticles, molecular biosensors, and multispectral confocal microscopy. J Mol Histol. 2004;35(6):555–564. doi: 10.1007/s10735-004-2196-4. [DOI] [PubMed] [Google Scholar]
- Raza H, Ahmed I, et al. Tissue specific expression and immunohistochemical localization of glutathione S-transferase in streptozotocin induced diabetic rats: modulation by Momordica charantia (karela) extract. Life Sci. 2004;74(12):1503–1511. doi: 10.1016/j.lfs.2003.08.023. [DOI] [PubMed] [Google Scholar]
- Sakai K, Matsumoto K, et al. Mitochondrial reactive oxygen species reduce insulin secretion by pancreatic beta-cells. Biochem Biophys Res Commun. 2003;300(1):216–222. doi: 10.1016/s0006-291x(02)02832-2. [DOI] [PubMed] [Google Scholar]
- Schröder S, Palinski W, et al. Activated monocytes and granulocytes, capillary nonperfusion, and neovascularization in diabetic retinopathy. Am. J. Pathol. 1991;139:81–100. [PMC free article] [PubMed] [Google Scholar]
- Setiadi H, Wautier J, et al. Increased adhesion to fibronectin and MO-1 expression by diabetic monocytes. J. Immunol. 1987;138:3230–3234. [PubMed] [Google Scholar]
- Talalay P, Fahey JW, et al. Chemoprotection against cancer by phase 2 enzyme induction. Toxicol Lett. 1995;82–83:173–179. doi: 10.1016/0378-4274(95)03553-2. [DOI] [PubMed] [Google Scholar]
- Vincent AM, Brownlee M, et al. Oxidative stress and programmed cell death in diabetic neuropathy. Ann N Y Acad Sci. 2002;959:368–383. doi: 10.1111/j.1749-6632.2002.tb02108.x. [DOI] [PubMed] [Google Scholar]
- Vincent AM, Olzmann JA, et al. Uncoupling proteins prevent glucose-induced neuronal oxidative stress and programmed cell death. Diabetes. 2004;53(3):726–734. doi: 10.2337/diabetes.53.3.726. [DOI] [PubMed] [Google Scholar]
- Wasserman WW, Fahl WE. Functional antioxidant responsive elements. Proc Natl Acad Sci U S A. 1997;94(10):5361–5366. doi: 10.1073/pnas.94.10.5361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiernsperger NF. Oxidative stress as a therapeutic target in diabetes: revisiting the controversy. Diabetes Metab. 2003;29(6):579–585. doi: 10.1016/s1262-3636(07)70072-1. [DOI] [PubMed] [Google Scholar]
- Wierusz-Wysocki B, Wysocki H, et al. Evidence of polymorphonuclear neutrophils (PMN) activation in patients with insulin-dependent diabetes mellitus. J. Leukocyte Biol. 1987;42:519–523. doi: 10.1002/jlb.42.5.519. [DOI] [PubMed] [Google Scholar]
- Yanoff M. Ocular pathology of diabetes mellitus. Am. J. Ophthalmol. 1969;67:21–38. doi: 10.1016/0002-9394(69)90004-x. [DOI] [PubMed] [Google Scholar]
- Zhu M, Chapman WG, et al. Polymorphic electrophile response elements in the mouse glutathione S-transferase GSTa1 gene that confer increased induction. Cancer Lett. 2001;164(2):113–118. doi: 10.1016/s0304-3835(00)00664-9. [DOI] [PubMed] [Google Scholar]
- Zhu M, Fahl WE. Development of a green fluorescent protein microplate assay for the screening of chemopreventive agents. Anal Biochem. 2000;287(2):210–217. doi: 10.1006/abio.2000.4875. [DOI] [PubMed] [Google Scholar]
- Zhu M, Fahl WE. Functional characterization of transcription regulators that interact with the electrophile response element. Biochem Biophys Res Commun. 2001;289(1):212–219. doi: 10.1006/bbrc.2001.5944. [DOI] [PubMed] [Google Scholar]