

Abstract
Adenosine kinase (Ado kinase) from Mycobacterium tuberculosis is structurally and biochemically unique from other known Ado kinases. This purine salvage enzyme catalyzes the first step in the conversion of the adenosine analog, 2-methyl-Ado (methyl-Ado), into a metabolite with antitubercular activity. Methyl-Ado has provided proof of concept that the purine salvage pathway from M. tuberculosis may be utilized for the development of antitubercular compounds with novel mechanisms of action. In order to utilize this enzyme, it is necessary to understand the topography of the active site to rationally design compounds that are more potent and selective substrates for Ado kinase. A previous structure-activity relationship identified modifications to the base moiety of adenosine (Ado) that result in substrate and inhibitor activity. In an extension of that work, sixty-two Ado analogs with modifications to the ribofuranosyl moiety, modifications to the base and ribofuranosyl moiety, or modifications to the glycosidic bond position have been analyzed as substrates and inhibitors of M. tuberculosis Ado kinase. A subset of these compounds was further analyzed in human Ado kinase for the sake of comparison. Although no modifications to the ribose moiety resulted in compounds as active as Ado, the best substrates identified were carbocyclic-Ado, 8-aza-carbocyclic-Ado, and 9-[α-L-lyxofuranosyl]-adenine with 38%, 4.3%, and 3.8% of the activity of Ado respectively. The most potent inhibitor identified, 5′-amino-5′-deoxy-Ado, had a Ki = 0.8 μM and a competitive mode of inhibition. MIC studies demonstrated that poor substrates could still have potent antitubercular activity.
1. Introduction
Adenosine kinase (Ado kinase) is a purine salvage enzyme that catalyzes the reaction Ado + ATP ↔ AMP + ADP in a magnesium-dependent manner. Ado kinase belongs to the PfkB family of carbohydrate and nucleoside kinases, a group of proteins that catalyze the transfer of the γphosphate of ATP to their substrates. This family includes ribokinase, hexokinases, and phosphofructokinase among its members [1]. Ado kinase activity is found in most eukaryotes, fungi, plants, and parasites, but it is seldom found in bacteria. Inhibition of Ado kinase has been the focus of pharmacological research in humans as well as the parasites Toxoplasma gondii and Cryptosporidium parvum. In humans, inhibition of Ado kinase results in an increase in adenosine (Ado) concentrations, and has potent anticonvulsive, analgesic, cardioprotective, and neuroprotective effects [2–4]. The pharmacologic potential for Ado kinase inhibitors against T. gondii and C. parvum parasites is due to that fact that these are both purine auxotrophs that lack a de novo synthesis pathway for purines [5, 6]. Since Ado kinase provides the main source of purine nucleotides in these two parasites, inhibition of this enzyme leads to death by purine starvation.
Unlike T. gondii and C. parvum, Mycobacterium spp including M. tuberculosis have de novo synthesis pathways for nucleotides. Ado kinase activity in Mycobacterium leprae was reported as early as 1983 [7–9], however no further work was done to characterize the protein in Mycobacterium spp until 2003 [10]. Since Mycobacterium spp can synthesize purines de novo, it is unclear why they also have this salvage activity when other intracellular bacteria do not. One plausible explanation is that nucleotide synthesis is rate-limiting for the growth of Mycobacterium spp [11], therefore, purine and pyrimidine salvage augments the sluggish de novo synthesis pathway. Ado concentrations in normal human bronchoepithelial lining are much higher than circulating levels of Ado (60 μM and <0.2 μM, respectively) [12] and Ado is produced at high levels in the lung in response to lung hypoxia, injury, infection, and inflammation [12–16]. In asthma patients, Ado concentrations reach almost 200 μM in bronchoepithelial fluids, largely due to mast cell stimulation and degranulation [12, 13]. It is likely that the proinflammatory state incited by active tuberculosis infection results in similarly high levels of Ado, and that Ado kinase is present in M. tuberculosis to take full advantage of this purine reservoir. While we may never definitively answer the question as to why M. tuberculosis has this activity, the necessity of Ado kinase to the Mycobacterium genus is evidenced by the presence of this gene in Mycobacterium leprae, which is widely considered to have a minimal genome, retaining only the genes that are necessary for survival [17].
Characterization of M. tuberculosis Ado kinase revealed that this enzyme was unique among known Ado kinases [10]. Perhaps most relevantly, it differed significantly from human Ado kinase. The sequence homology that M. tuberculosis Ado kinase shared with other Ado kinases was low enough to prevent identification of the gene on this basis alone. Indeed, M. tuberculosis Ado kinase shared more sequence homology with ribokinases than other Ado kinases [10]. Most Ado kinases are monomeric proteins with a molecular mass between 35 and 40 kDa, and their activity is usually stimulated in the presence of inorganic phosphate (Pi) [15, 18, 19]. In contrast, Ado kinase from M. tuberculosis is a homodimer comprised of identical 35 kDa subunits [10, 20, 21]. The M. tuberculosis enzyme is activated in the presence of potassium but not Pi, characteristics that are more similar to ribokinases than other Ado kinases [22]. Kinetic studies with Ado, ATP, and methyl-Ado revealed that the Michaelis-Menton parameters for these reactants were significantly different between human and M. tuberculosis homologs [10].
Unique specificity for reactants indicated that there may be differences in the active sites for M. tuberculosis and human Ado kinases, which could be exploited for the development of antitubercular drugs. Nucleoside analogs occupy a unique mechanistic niche, usually requiring conversion from an inactive prodrug to an active metabolite that inhibits an essential enzyme involved in nucleic acid utilization. Interruption of nucleotide anabolism then leads to the death of the cell. Ado kinase activity may be utilized for selective phosphorylation of nucleoside analogs to metabolites active against M. tuberculosis. Nucleoside analogs are currently in clinical use as antiviral drugs, anticancer drugs, and gout treatments where they have a history of safety and efficacy, but they are not currently used as antibacterial drugs.
The need for antitubercular drugs with novel mechanisms of action has been heightened by the emergence of multi-drug resistant and extensively drug-resistant strains of M. tuberculosis, particularly in Africa and Eastern European countries [23–26]. Nucleoside analogs would complement the antitubercular drug arsenal because the mechanism of action employed by these compounds differs enough from existing antitubercular drugs that cross-resistance should be avoided. The adenosine analog, 2-methyl-Ado (methyl-Ado), provided proof of concept that nucleoside analogs have therapeutic potential against M. tuberculosis, with a 95% minimum inhibitory concentration (MIC) of 3 μg/ml and a 50% inhibitory concentration of 80 μg/ml in CEM cells [27–29]. Ado kinase played a central role in the mechanism of action of methyl-Ado [10, 29] , and knowledge about the substrate specificity for Ado kinase is required in order to successfully design other nucleoside analogs, which are more potent and selective than methyl-Ado.
In order to complement ongoing crystallographic studies of M. tuberculosis Ado kinase [20, 21], structure-activity studies have been performed in order to identify the requirements for the binding of Ado analogs to the Ado-binding domain [30, 31]. These studies probed the adenine (Ade) moiety of the Ado-binding domain of M. tuberculosis Ado kinases with a total of eighty-one Ado analogs with modifications to the adenine base [30, 31]. Compounds were analyzed as both substrates and inhibitors of the enzyme, and selected compounds were further analyzed in human Ado kinase. The previous structure-activity relationships (SAR) revealed the presence of hydrophobic pockets at the N1, N7 and C8 positions of Ado, where interactions with these pockets were predictive for inhibition of the enzyme [30, 31]. Furthermore, they provided a measure of the limits for the size of exocyclic modifications at the 2, 3, 7, and 8-positions that were able to bind Ado-analogs [30, 31]. These results will guide the synthesis of more compounds that may be selectively phosphorylated by the M. tuberculosis homolog. In addition to information about the topography of the active site, the relative levels of activity and inhibition provided an approximation of the Michaelis-Menton parameters for each compound based on the Cheng-Prusoff relationship between Ki and I50 for the enzyme [32]. The current work complements the previous SAR by evaluating sixty-two Ado analogs with modifications to the ribose moiety alone, or in combination with substitutions to the base, in order to provide a complete picture of the Ado-binding domain of M. tuberculosis Ado kinase.
2. Methods and materials
2.1 Chemicals
The nucleoside analogs used in this study were obtained from several different sources. Ado, 2′-deoxy-Ado (9), 2′-O-methyl-Ado (10), and 9-[β-D-arabinofuranosyl]-adenine (15) were purchased from Sigma-Aldrich (St. Louis, Mo). 9-[β-L-ribofuranosyl]-adenine (52) was kindly provided by Dr. Mahmoud el Kouni (University of Alabama at Birmingham, Birmingham, Al.). [2,8]-3H-Ado used in the inhibition studies was purchased from Moravek Biochemicals (Brea, Ca.). All other compounds were provided by the chemical repository at Southern Research Institute (Birmingham, Alabama). Information regarding the synthesis of these compounds can be provided by contacting the corresponding author. Each compound was solubilized in water or DMSO as necessary.
2.2 Substrate assays
Substrate assays for M. tuberculosis Ado kinase consisted of 50 mM Tris-HCl (pH 8.0), 10 mM KCl, 10 mM MgCl2, 5 mM ATP, 0.01% BSA, 10 μM deoxycoformycin, and 100 μM of the appropriate test compound. Protein extracts were prepared and enzyme was purified as previously described [10]. Substrates for human Ado kinase were assayed similarly with the following changes: assay conditions consisted of 50 mM HEPES (pH 6.0), 40 mM KCl, 1 mM MgCl2, 1 mM ATP, 0.1% BSA, 10 μM deoxycoformycin, and 100 μM of the appropriate test compound. Human Ado kinase was prepared as previously described from clone 20-1 (a generous gift from Dr. Jozef Spychala, UNC Chapel Hill, Chapel Hill, NC) [1, 31].
Reactions were started by the addition of enzyme and incubated in a 37°C water bath. Aliquots of 50 μl were taken at 0, 20, 40, and 60-min intervals and reactions were stopped at each timepoint by the addition of 50 μl of 1 M perchloric acid. Samples were neutralized to pH 7 and precipitated salts were removed by centrifugation. Product formation was detected by HPLC using Bio Basic anion exchange column (Thermo Electron Corp., Bellefonte, Pa.) with a 30-min linear salt and pH gradient from 6 mM ammonium phosphate (pH 2.8) to 900 mM ammonium phosphate (pH 6). Peaks were detected as they eluted from the column by absorbance at their λmax, typically between 260 and 320 nm. All enzyme reactions were linear during the incubation period and substrate conversions were maintained at less than ten percent. Controls were run to ensure that DMSO utilized for solubility did not interfere with enzymatic activity.
2.3 Inhibition assays
Assay mixtures were identical to those for substrate assays with the addition of 0.1 μM [3H] Ado (4 μCi/ml) and 100 μM of the test compound. Inhibition assays were performed as described previously [31]. Briefly, reactions were started by the addition of enzyme, incubated for one hour at 37°C, and stopped by the addition of 10 μl of 0.1 M EDTA. At appropriate timepoints, 50 μl aliquots were applied to a DE-81 cellulose disk and allowed to dry. Disks were batch-washed three times with 1 mM ammonium acetate (pH 5.0), rinsed with 95% ethanol, dried, and transferred to scintillation vials with 10 ml of Complete Counting Cocktail (Research Products International, Mount Prospect, Illinois). Radioactivity was detected with a Packard Tri-Carb model 1900 TR liquid scintillation analyzer, and enzymatic activity was calculated from the amount of radioactivity that bound to the DE-81 disks. Compounds were ranked by their ability to inhibit the phosphorylation of 0.1 μM Ado, and compounds that inhibited by 90% or greater were re-assessed at 10 μM. This iterative process was continued with serial 10-fold dilutions of a test compound until the compound no longer inhibited Ado phosphorylation by ≥90%.
Inhibition constants were determined by assaying various concentrations of inhibitor in the presence of increasing concentrations of Ado. Double-reciprocal plots were created and replots of the slopes of the double-reciprocal plots versus concentration of inhibitor were used to determine the Ki. Sigma plot version 8.02 enzyme kinetics module version 1.1.1 was used to analyze the data.
2.4 Determination of MIC
MIC values were evaluated in M. tuberculosis strains H37Ra, SRICK1, and SRICK1-pVV16/Rv2202c using a colorimetric microdilution broth assay as previously described [10, 31].
2.5 Molecular modeling
All molecular modeling studies were performed on a Pentium 4 computer running the Ubuntu operating system. Pymol (DeLano Scientific LLC, San Carlos, CA) and DOCK 6.1 (Kuntz, I.D., DOCK, UCSF, San Francisco, CA) were used for construction and visualization of the models.
Protein database entries 2PKM (Adenosine Kinase complexed with 9-[α-L-lyxofuranosyl]-adenine) and 2PKN (Adenosine kinase complexed with non-hydrolysable ATP) were used as starting coordinates. For adenosine, the stereochemistry of carbon 4 was inverted, and carbon 5 was rotated prior to a single round of minimization of the ligand in the binding site pocket. The modeling of non-hydrolysable ATP analog, β,γ-methyleneadenosine-5′-triphosphate (AMP-PCP), into the closed active site was achieved by fitting the region of the ATP binding pocket of the open structure 2PKN (16 atom pairs, RMSD 0.18Å) to that of 2PKM, and subjecting the transformed coordinates of AMP-PCP to a single round of rigid body minimization.
3. Results
Sixty-two compounds were tested that had single or multiple changes to the sugar moiety of Ado, or combinations of changes to the sugar and base (Table 1). Five compounds with modifications to the adenine base on Ado (1a–1e) were included for the ease of interpreting results involving multiple modifications. The structure and numbering convention for Ado is shown in Figure 1 and several of the sugar structures used in this work are shown in Figure 2.
Table 1.
Effect of modifications of the ribofuranosyl moiety of Ado on Ado kinase activity
| Compound name | M. tuberculosis Specific activity (nmol/mg-min) | Inhibitiona | Kib (μM) | Human Specific activity (nmol/mg-min) | Inhibitiona | |
|---|---|---|---|---|---|---|
| 1 | Adenosine (9-[β-D-ribofuranosyl]-adenine) | 4000 ± 450 | 2400 ± 750 | |||
| 1a | 2-fluoro-adenosinec | 2070 ± 430 | +++ | 0.5 c | 1800 ± 220 | + |
| 1b | 2-chloro-adenosinec | 460 ± 60 | ++ | 16 ± 3 | ||
| 1c | 2-methyl-adenosinec | 74 ± 2 | + | 4 ± 0.6 | ||
| 1d | 6-methyl-purine ribosidec | 110 ± 6 | ++ | 980 ± 100 | ||
| 1e | 8-aza-adenosinec | 160 ± 50 | ++ | 910 ± 400 | ||
| 4′-oxygen position | ||||||
| 2 | 2-chloro-4′-thio-adenosine | <1 | + | |||
| 3 | Carbocyclic adenosine (aristeromycin) | 1500 ± 230 | + | 860 ± 220 | ||
| 4 | 2-amino-carbocyclic-adenosine | 17 ± 3 | - | <2 | ||
| 5 | 7-deaza-carbocyclic-adenosine | <1 | + | |||
| 6 | 8-aza-carbocyclic-Ado | 170 ± 20 | + | 620 ± 194 | + | |
| 7 | 2-amino-8-aza-2′-deoxy-carbocyclic-adenosine | <6 | − | |||
| 8 | 2-amino-6-hydroxymethyl-8-aza-carbocyclic-adenosine | <2 | − | |||
| 2′-position | ||||||
| 9 | 2′-deoxy-adenosine | 13 ± 2 | + | 73 ± 11 | ||
| 10 | 2′-O-methyl-adenosine | <1 | + | |||
| 11 | 2-chloro-2′-deoxy-adenosine | <1 | + | |||
| 12 | 2-chloro-2′-deoxy-2′-fluoro-adenosine | <1 | ++ | |||
| 13 | 2′-deoxy-2,2′-difluoro-adenosine | 29 ± 3 | + | <1 | ||
| 14 | 8-aza-2′-deoxy-adenosine | <2 | + | |||
| 15 | 9-[β-D-arabinofuranosyl]-adenine (araA) | 23 ± 2.3 | − | 6 ± 0 | ||
| 16 | 2-fluoro-araA | 36 ± 3 | + | <1 | ||
| 17 | 2-chloro-2′-deoxy-2′-fluoro-araA | <1 | + | |||
| 18 | 1′,4′-anhydro-2′-deoxy-D-araA | <1 | − | |||
| 19 | 1′,4′-anhydro-2′-deoxy-6-chloro-D-araA | <3 | − | |||
| 3′-position | ||||||
| 20 | 3′-deoxy-adenosine (cordycepin) | <1 | − | |||
| 21 | 3′-deoxy-3′-amino-adenosine | <1 | − | − | ||
| 22 | 3′-deoxy-3′-azido-adenosine | <1 | − | + | ||
| 23 | 9-[β-D-xylofuranosyl]-adenine | 1 ± 0.1 | − | |||
| 24 | 9-[β-D-xylofuranosyl]-6-methylpurine | <1 | − | |||
| 25 | 9-[β-D-xylofuranosyl]-6-oxy-8-azapurine | <7 | − | |||
| 26 | 9-[β-D-3-ethynyl-ribofuranosyl]-2-methyladenine | <1 | ||||
| 2′- and 3′-positions | ||||||
| 27 | 9-[β-D-2-azido-2-deoxy-xylofuranosyl]-adenine | <1 | − | |||
| 28 | 2-chloro-2′,3′-O-isopropylidene-adenosine | <1 | − | |||
| 29 | 8-aza-2′,3′-O-isopropylidene-adenosine | <3 | − | |||
| 5′-position | ||||||
| 30 | 5′-deoxy-adenosine | <1 | ++ | |||
| 31 | 5′-carboxamido-adenosine | <1 | + | |||
| 32 | 5′-amino-5′-deoxy-adenosine | ? | +++ | 0.8 c | <1 | +++ |
| 33 | 9-[β-D-glucofuranosyl]-2-methyladenine | <1 | ||||
| 34 | 9-[α-L-lyxofuranosyl]-adenine | 150 ± 24 | + | 360 ± 150 | ||
| 35 | 9-[α-L-lyxofuranosyl]-2-fluoro-adenine | 130 ± 16 | + | 0.8 ± 0.2 | ||
| 36 | 9-[β-D-5-methyl-(allo)-ribofuranosyl]-2-fluoro-adenine | 4 ± 0.3 | ++ | <0.5 | ||
| 37 | 9-[β-D-5-methyl-(talo)-ribofuranosyl]-2-fluoro-adenine | 1.5 ± 0.06 | + | <0.7 | ||
| 38 | 9-[β-D-5-methyl-(allo)-ribofuranosyl]-6-methyl-purine | <2 | ++ | |||
| 39 | 9-[β-D-5-methyl-(talo)-ribofuranosyl]-6-methyl-purine | <1 | + | |||
| 40 | 8,5′-(R)-cycloadenosine | <1 | − | |||
| 41 | 8-aza-5′-deoxy-5′-methylsulfonyl-adenosine | <2 | − | |||
| Multiple positions | ||||||
| 42 | 9-[β-L-ribofuranosyl]-adenine | <1 | − | |||
| 43 | 9-[α-D-ribofuranosyl]-adenine | <1 | + | |||
| 44 | 9-[α-D-lyxofuranosyl]-adenine | <1 | − | |||
| 45 | 7-[α-D-ribofuranosyl]-adenine | <2 | + | |||
| 46 | 7-[α-D-arabinofuranosyl]-adenine | <2 | + | |||
| 47 | 3-[β-D-ribofuranosyl]-adenine | 28 ± 2 | ++ | 268 ± 41 | ||
| 48 | 8-aza-8-[β-D-ribofuranosyl]-adenine | <1 | + | |||
| 49 | 2′,3′,5′-tri-O-acetyl-adenosine | <1 | + | |||
| 50 | 9-(2-hydroxyethoxymethyl)-adenine (Acyclo-adenosine) | <1 | − | − | ||
| 51 | 9-[2-[(2-hydroxyethyl)-methylamino]-ethoxymethyl]-adenine | <1 | − | − | ||
| Pyranoses | ||||||
| 52 | 9-[β-D-ribopyranosyl]-adenine | <1 | − | |||
| 53 | 9-[β-D-allopyranosyl]-adenine | <1 | − | |||
| 54 | 9-[β-D-fructopyranosyl]-adenine | <1 | − | |||
| 55 | 1-(adenyl-9-yl)-1-doxy-β-D-glucopyranuramide | <1 | − | |||
| 56 | 9-(2-deoxy-β-D-erythro-pentopyranosyl)-adenine | <1 | + | |||
| 57 | 9-(2-deoxy-α-D-erythro-pentopyranosyl)-adenine | <1 | − | |||
| 58 | 9-(3-azido-3,4-dideoxy-β-D-erythro-pentopyranosyl)-adenine | <1 | − | |||
| 59 | 9-[β-L-galactopyranosyl]-adenine | <1 | − | |||
| 60 | 9-[6-O-α-D-galactopyranosyl-β-D-glucopyranosyl]-adenine | <1 | − | |||
| 61 | 9-[α-D-mannopyranosyl]-adenine | <1 | − | |||
| 62 | 9-[α-D-talopyranosyl]-adenine | <1 | − | |||
| 63 | 6-(adenyl-9-yl)-tetrahydropyran-2-methanol | <1 | − | |||
Inhibition of 0.1 μM adenosine phosphorylation is indicated as follows: ‘−’, <10% inhibition at 100 μM of compound; ‘+’, 10–90% inhibition at 100 μM; ‘++’, 10–90% inhibition at 10 μM; ‘+++’, 10–90% inhibition at 1 μM.
The manner of inhibition is denoted by a ‘c’ for competitive inhibition.
These previous results were included for ease of comparison with the current study [32].
Figure 1.
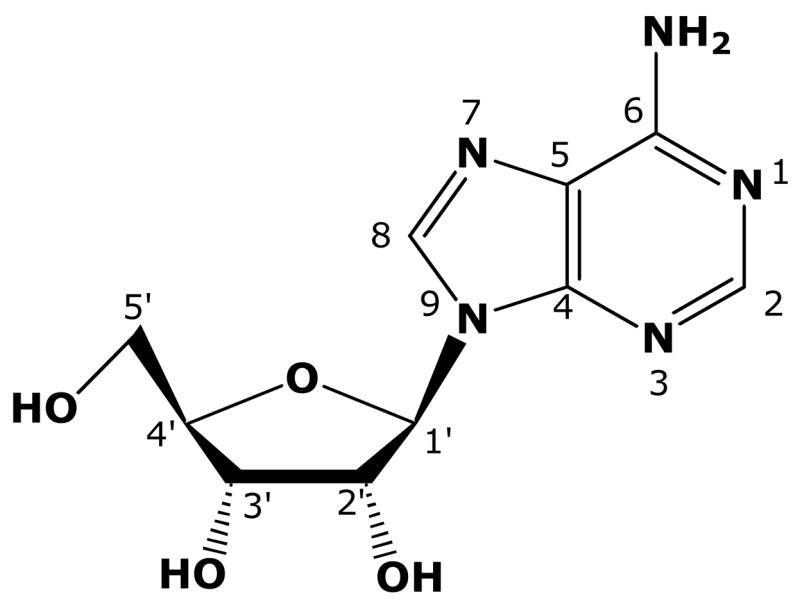
Structure and numbering convention for Ado.
Figure 2.

Structures of sugar moieties utilized in this structure-activity relationship.
3.1 Ribofuranosyl conformation
M. tuberculosis Ado kinase preferred the β-D-ribofuranosyl conformation or modifications that closely resembled this conformation such as carbocyclic-Ado (3, Figure 2e). The L-enantiomer, 9-[β-L-ribofuranosyl]-adenine (Fig 2b, 42), was neither a substrate, nor an inhibitor, indicating that it was not recognized by the active site. Furthermore, the α-isomer, 9-[α-D-ribofuranosyl]-adenine (Fig 2c, 43), was not a substrate, but was a weak inhibitor, indicating that it at least bound poorly to the active site. Ado kinase only phosphorylated sugars in the furan conformation and excluded all twelve pyranoses tested (52–63). Furthermore, the acyclo-adenosine analogs, 9-(2-hydroxyethoxymethyl)-adenine (acyclo-adenosine, 50) and 9-[2-[(2-hydroxyethyl)-methylamino]-ethoxymethyl]-adenine (51), were not substrates or inhibitors of M. tuberculosis Ado kinase. Following is a summary of the results of changes to the ribofuranosyl moiety.
3.2 2′-position
Consistent with previous observations with human Ado kinase [33–35], 2′-deoxy-Ado (9) was a poor substrate for M. tuberculosis Ado kinase. The enzyme had a preference for the 2′-hydroxyl moiety to be present and trans to the adenine moiety as illustrated by the <99% decrease in activity seen with both 2′-deoxy-adenosine (9) and 9-[β-D-arabinofuranosyl]-adenine (araA, 15, Fig 2f). Several compounds with 2′-modifications were substrates, albeit at low levels. The most active substrates were 2′-deoxy- 2,2′-difluoro-Ado (13), araA (15) and 2-fluoro-araA (16). When substitutions to the base are combined with substitutions at the 2-position as in the case of 2-fluoro-araA (16) and 2′-deoxy-2,2′-difluoro-Ado (13), the resulting activity is closer to the activity seen with a single change at the 2′-position than an intermediate value. Also, addition of a methyl group to the 2′-hydroxyl group (10) resulted in activity of <1 nmol/mg-min, indicating that substitutions to the 2′-hydroxyl are poorly tolerated.
2-Chloro-2′-deoxy-2′-fluoro-Ado (12) was the only compound with a 2′-substitution that demonstrated noteworthy inhibition with 13% of inhibition at 10 μM. This inhibition was similar to that previously determined for 2-chloro-adenosine [31]. All other compounds with modifications at the 2′-position were either poor inhibitors or did not inhibit M. tuberculosis Ado kinase at all.
3.3 3′-position
For M. tuberculosis Ado kinase, the presence of a 3′-hydroxyl group trans to the adenine moiety was a requirement for substrate activity. Modification of the 3′-hydroxyl group had a greater impact on substrate activity than modification of the 2′-hydroxyl group for this enzyme. Replacing the 3′-hydroxyl group with an amino or azido group (21 and 22) abolished activity and 3′-deoxy-Ado (20) had no measurable activity; each of these had substrate activity of <1 nmol/mg-min. Whereas, changing the 3′-hydroxyl group to the cis-conformation, as in the case of 9-[β-D-xylofuranosyl]-Ade (Fig 2g, 23), resulted in a specific activity of 1 nmol/mg-min. Furthermore, 9-[β-D-3-ethynyl-ribofuranosyl]-2-methyladenine, which has a 3′-ethynyl group (26, 3′-C≡CH ↑) was not a substrate for M. tuberculosis Ado kinase.
These results stood in contrast to human and mammalian Ado kinases that were more permissive to modifications to the 3′-position, such as 3′-deoxyadenosine or 3′-aminoadenosine, than of modifications to the 2′-position, such as araA and 2′-deoxyadenosine [33, 35–37].
No compounds with combinations of 2′ and 3′ substitutions (27–29) were either substrates or inhibitors of M. tuberculosis Ado kinase and none of the multiple changes involving the base and the 3′-position resulted in active compounds (24–26). No compounds with substitutions or modifications at the 3′-position were inhibitors. The presence of a 3′-hydroxyl group trans to the adenine moiety is important both for substrate recognition and the binding of inhibitors, suggesting that steric hindrance may play a role at this site.
3. 4 4′-oxygen
The 4′-oxygen position was the most flexible among all of the substitutions made to the ribose moiety for both human and M. tuberculosis Ado kinases. Carbocyclic-Ado (aristeromycin, Fig 2e, 3) maintained about the same activity in both human and M. tuberculosis homologs (37% and 38% of the activity of Ado respectively). Although 2- amino-Ado was similarly active in both Ado kinases [31], 2-amino-carbocyclic-Ado (4) was at least a 20-fold better substrate for M. tuberculosis Ado kinase than human. The double-substituted 8-aza-carbocyclic-adenosine (6) was less active than carbocyclic-Ado (3), but was the second-best substrate in this series with a specific activity of 170 nmol/mg-min. Although 8-aza-carbocyclic-adenosine (6) exhibited good substrate activity in M. tuberculosis, this compound was more active in human Ado kinase. Activities measured with human Ado kinase were consistent with previous reports [35, 36, 38, 39].
Figure 3. BioBasic anion exchange chromatography of an Ado kinase assay with 5′-amino-5′-deoxyadenosine as a substrate.
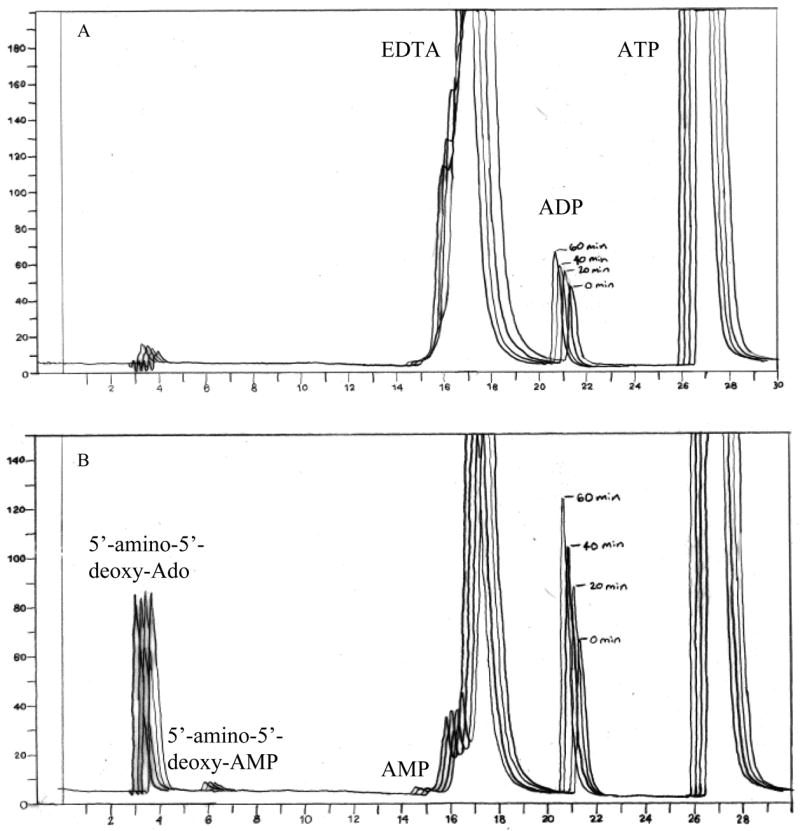
Activity assays were performed as described in the materials and methods section. Two μg of protein were used per 50 μl sample. Reactions were stopped by the addition of 10 mM EDTA prior to perchloric acid extraction. Aliquots of 50 μl were taken at 0, 20, 40, and 60-min. A) A negative control assay contains everything except 5′-amino-5′-deoxyadenosine. B) A typical assay with 5′-amino-5′-deoxyadenosine.
Substitution of the 4′-oxygen with a 4′-sulfur atom, as is the case with 2-chloro-4′-thio-adenosine (2, <1nmol/mg-min), resulted in at least a 460-fold decrease in substrate activity over its parent, 2-chloro-adenosine (1b, 460 nmol/mg-min) [31]. Since sulfur atoms have similar electronegativity and have valence electrons more similar to oxygen than carbon atoms, and carbocyclic-adenosine (3) is a better substrate than the 4′-thio-adenosine analog (2), it is unlikely that the loss of activity is due to an inability to bind this site. This loss of activity is more likely due to the larger size of the sulfur atom, which may negatively effect the puckering of the ribose moiety. Therefore, the size of the substitution at the 4′-position may be a determinate of substrate activity for this site.
All of the carbocyclic compounds (3–8) were poor inhibitors, inhibiting Ado phosphorylation by no more than 81% at 100 μM.
3.5 5′-position
The 5′-position was also permissive to substitution, possibly because it is the site of catalysis and therefore needs room for the phosphorylation reaction to take place. The cis-conformation of the 5′-C was not an absolute requirement for either human or M. tuberculosis Ado kinase as demonstrated by 9-[α-L-lyxofuranosyl]-adenine (Fig 2d, 34) with 4% and 15 % of the activity of Ado in M. tuberculosis and human enzymes respectively. This compound differs from Ado only in that the 5′-C is trans to the adenine moiety. The addition of a 2-fluorine atom to this compound (9-[α-L-lyxofuranosyl]-2-fluoro-adenine, 35) resulted in a compound with similar activity in M. tuberculosis Ado kinase, but greatly diminished activity in the human homolog (0.8 nmol/mg-min). One report exists where 9-[α-L-lyxofuranosyl]-adenine (34) was determined to be an inhibitor, but not a substrate for human Ado kinase [4]. However, the substrate activity that we measured for 9-[α-L-lyxofuranosyl]-adenine (34) in human Ado kinase was similar to previous reports in human and mammalian Ado kinase [35, 36]. Other compounds with modifications to the 5′-position, including 5′-amino-5′-deoxy-adenosine (32), 9-[β-D-5-methyl-(allo)-ribofuranosyl]-2-fluoro-adenine (36), and 9-[β-D-5-methyl-(talo) ribofuranosyl]-2-fluoro-adenine (37) resulted in very low substrate activity for human Ado kinase, maintaining at most 0.3% of the activity of Ado. Although they were both poor substrates for M. tuberculosis Ado kinase, 9-[β-D-5-methyl-(allo)-ribofuranosyl]-2-fluoro-adenine (36) was about twice as active as 9-[β-D-5-methyl-(talo)-ribofuranosyl]-2-fluoro-adenine (37). The difference between these two stereoisomers is that the 5′-methyl group in the allo-conformation points towards the back of the plane of the ribose moiety in the direction of the 4′-oxygen, while in the talo-conformation it points forward toward the 2′-hydroxyl group. Neither of these compounds had measurable substrate activity in human Ado kinase.
While it is known not to be a substrate for mammalian Ado kinases [40–42], there was strong evidence that 5′-amino-5′-deoxy-Ado (32) was a substrate for M. tuberculosis Ado kinase. With the M. tuberculosis enzyme, a peak appeared in the region consistent with the formation of a monophosphate, but the peak area did not increase linearly with time. The ADP peak increased with time. Since ADP is also a product of the reaction, the increase of this product provided indirect evidence that 5′-amino-5′-deoxy-Ado (32) was a substrate for M. tuberculosis Ado kinase. The appearance of the monophosphate peak and the increase of the ADP peak were magnesium-dependent and did not appear in the presence of iodotubercidin, a potent inhibitor of Ado kinase, indicating that product formation was due to Ado kinase activity and not due to a possible contaminating enzyme. Although a peak appeared in the region where one would expect a nucleoside monophosphate, we have not yet verified that the peak was 5′-amino-5′-deoxy-AMP (Fig 3). The nucleoside apparently fluctuated between the parent compound and the monophosphate. It was not further metabolized because the sum of these two peaks was constant with time. The fluctuation of the monophosphate peak led us to believe that the product of the reaction was unstable. Since the ADP peak increased with time, we ruled out a reversible reaction which would have consumed this product as well. It was uncertain whether the disappearance of the product was due to innate instability or assay conditions, since we terminated the reaction by the addition of 1M perchloric acid. Terminating the reaction by the addition of EDTA or iodotubercidin did not change the results, and the product never disappeared altogether. Therefore, we believe that the product is unstable under our assay conditions.
The activity of 5′-amino-5′-deoxy-Ado (32) is noteworthy because this compound is known not to be a substrate for human Ado kinase; indeed, it is a potent inhibitor of Ado kinases [40–43]. The fact that the 5′-hydroxyl group is the site of catalysis for this enzyme makes this compound more intriguing than changes to other sites. If the peak that we have observed is the monophosphate product, it indicates that there are major differences at the catalytic site between human and M. tuberculosis Ado kinases. Further studies of 5′-amino-5′-deoxy-adenosine (32) and its phosphorylated product are warranted in light of these unusual results.
The 5′-position provided the most potent inhibitors of all of the sugar substitutions. Of these, 5′-amino-5′-deoxy-Ado (32) was the best competitive inhibitor with a Ki of 0.8 ± 0.4 μM. This compound was more potent at inhibiting Ado phosphorylation than 5′-deoxy-Ado (30). Furthermore, compounds that had the addition of a 5′-methyl group in the talo-conformation (37 and 39) were about 10-times less potent as inhibitors than their allo-conformers (36 and 38).
3.6 Glycosidic bond position
Four compounds were evaluated which had multiple changes, at least one of which involved the position of the glycosidic bond. Three of these, 7-[α-D-ribofuranosyl]-adenine (45, Fig 2c), 7-[α-D-arabinofuranosyl]-adenine (46 similar to Fig 2c, but with the 2′-OH ↑), and 8-aza-8-[β-D-ribofuranosyl]-adenine (48) had no measurable substrate activity and all were poor inhibitors of Ado kinase. The fourth compound, 3-[β-D-ribofuranosyl]-adenine (47), demonstrated both substrate and inhibitor activity with a specific activity of 28 nmol/mg-min (0.7% of the activity of Ado). This compound, however, was also a much better substrate for human Ado kinase, with a specific activity of 268 nmol/mg-min (11% of the activity of Ado).
Previous studies of Ado analogs with substitutions at the 3-position of Ado indicated that steric hindrance may inhibit the binding of substrates to the active site [30]. The result that we found with 3-[β-D-ribofuranosyl]-adenine (47) seems to contradict this theory. One possible explanation for this difference may be that the series of compounds evaluated in the previous study were 3-deaza-adenosine analogs. In the current study, the nitrogen atom at the 3-position remains intact, indicating that this nitrogen likely plays an important role in substrate recognition. That 3-[β-D-ribofuranosyl]-adenine (47) fits well into the active site is supported not only by its substrate activity, but also by its ability to inhibit Ado kinase at the 10 μM level.
3.7 MIC assays
MIC values were determined in M. tuberculosis H37Ra, in an Ado kinase-deficient strain derived from H37Ra (SRICK1), and in SRICK1 complemented with the adoK gene (SRICK1::adoK) using three nucleoside analogs (Table 2). Each of the compounds selected had modifications at the 5′-position with substrate activity ranging from 4 to 130 nmol/mg-min. Of the compounds tested, 9-[β-D-5-methyl-(allo)-ribofuranosyl]-2-fluoro-adenine (36) was the most promising in terms of antimycobacterial activity, with activity similar to the ethambutol control. The specific activity of the compound as measured by our assays was not predictive for efficacy against bacteria. Indeed, 9-[α-L-lyxofuranosyl]-2-fluoro-adenine (35) was not as effective against the intact organism as 9-[β-D-5-methyl-(allo)-ribofuranosyl]-2-fluoro-adenine (36), even though 9-[α-L-lyxofuranosyl]-2-fluoro-adenine (35) was a much better substrate for Ado kinase. Although they were active at different levels, both compounds exerted their antimycobacterial activity in an Ado kinase-dependent manner as evidenced by the lack of activity in SRICK1, and restoration of activity in SRICK1::adoK. The lack of antitubercular activity seen with 5′-amino-5′-deoxy-adenosine (32) is consistent with its unusual substrate activity; this compound was included specifically because of its unusual activity and modifications at the site of catalysis.
Table 2.
MIC results with selected nucleosides
| Compound Name | specific activity (nmol/mg-min) | H37Ra | SRICK1 | SRICK1::adoK |
|---|---|---|---|---|
| MICa(μg/ml) | ||||
| 5′-amino-5′-deoxy-Ado | NDb | >100 | >100 | >100 |
| 9-[β-D-5-methyl-(allo)-ribofuranosyl]-2-fluoro-adenine | 4 | 1–10 | >50 | 0.1–1 |
| 9-[α-L-lyxofuranosyl]-2-fluoro-adenine | 130 | 10–100 | >100 | 10–100 |
MIC assays were performed at least twice for each compound.
ND specific activity was not determined for this compound.
4. Discussion
M. tuberculosis Ado kinase is of interest because it is the first prokaryotic Ado kinase to be characterized and preliminary characterization demonstrated that this enzyme is unique among Ado kinases [10]. In addition to its role in purine salvage, Ado kinase can also be used as a selective filter for phosphorylation of subversive substrates with antimycobacterial activity. This SAR was performed as part of an effort to rationally design compounds that will be subversive substrates for Ado kinase from M. tuberculosis. When modeled against a high-resolution crystal structure, these studies will identify the required elements of a pharmacophore for Ado kinase and to lead future compound design. A previous SAR explored the Ade moiety of Ado and provided insight into the requirements for the binding of substrates and inhibitors to the active site [31]. The current work complemented the previous study by similarly studying structural modifications to the ribose moiety of Ado that would result in phosphorylated products or inhibition of enzymatic activity.
Very few modifications to the ribofuranosyl moiety resulted in active substrates or inhibitors; the furan conformation had to be maintained as none of the sugars in the pyran conformation (52–63) or acyclo-adenosine analogs (50 and 51) were substrates. In contrast to the SAR involving modifications to the adenine base in which 32 of the 80 compounds tested (40%) had measurable substrate activity and 69 of the 80 were inhibitors (86%), in the current study involving modifications to the ribofuranosyl moiety only 14 out of 63 (22%) of the compounds tested were substrates and 28 of 63 (44%) were inhibitors. The strict conservation of the ribofuranosyl moiety may be due to the structural similarity that this enzyme shares with ribokinase, and further bolsters the argument that this activity may have arisen in Mycobacterium spp as a result of convergent evolution [10, 21, 22]. Ribokinase is a member of the PfkB family of carbohydrate kinases along with Ado kinase, inosine-guanosine kinase, fructokinase, 1-phosphofructokinase, 6-phosphofructokinase, and other enzymes [44, 45]. Like Ado kinase from M. tuberculosis, ribokinase exists as a homodimer, and optimum kinase activity occurs in the presence of Mg+2 and K+, with an optimum pH range similar to that of M. tuberculosis Ado kinase, pH 8–9 [10]. The best substrates for ribokinase are D-ribose and 2-deoxy-D-ribose, with kcat values of 86 and 27 s-1, respectively [46]. Other substrates for ribokinase include D-arabinose, D-xylose, and D-fructose, with respective kcat values of 0.64, 0.92, and 0.24 s−1. The overwhelming preference of ribokinase for D-ribose mirrors the selectivity of M. tuberculosis Ado kinase for β-D-ribofuranoses.
The high-resolution crystal structure of M. tuberculosis Ado kinase that was recently published helped to explain some of the observations made in the current work (Figure 4) [20]. For example, the crystal structure revealed a pocket with a diameter of ~6.5Å adjacent to the 2-C of Ado that permits substitutions at the 2-position like 2-fluoro-Ado (1a) and 2-methyl-Ado (1c) (Figure 4B). Furthermore, the conservation of the ribofuranosyl moiety has its origins in the protein structure. Asp12, and Asn52 hydrogen bond to the O3′, while Phe102 may provide steric hindrance that prohibits the addition of a moiety at the 3-position on the cis side of the base like 9-[β-D-xylofuranosyl]-adenine (23) (Figure 4A). Hydrogen bonding may occur at the 2′-OH due to interactions with Gly48 and Asp12 when the 2′-OH is trans to the base, however this SAR demonstrated the ability to make small additions at the 2′-C cis to the adenine base like 9-[β-D-arabinofuranosyl]-adenine (araA, 15), 2-fluoro-araA (16), and 2′-deoxy-2,2′-difluoro-adenosine (12). In these and other cases, the crystal structure supports the results that we observed with our SAR studies.
Figure 4. Ligands docked in the active site of Ado kinase.
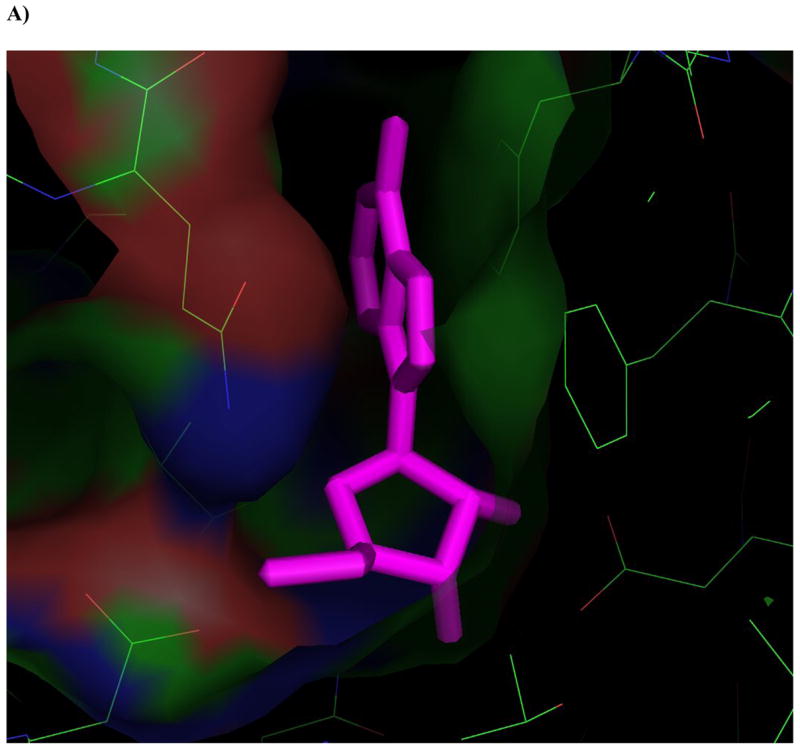
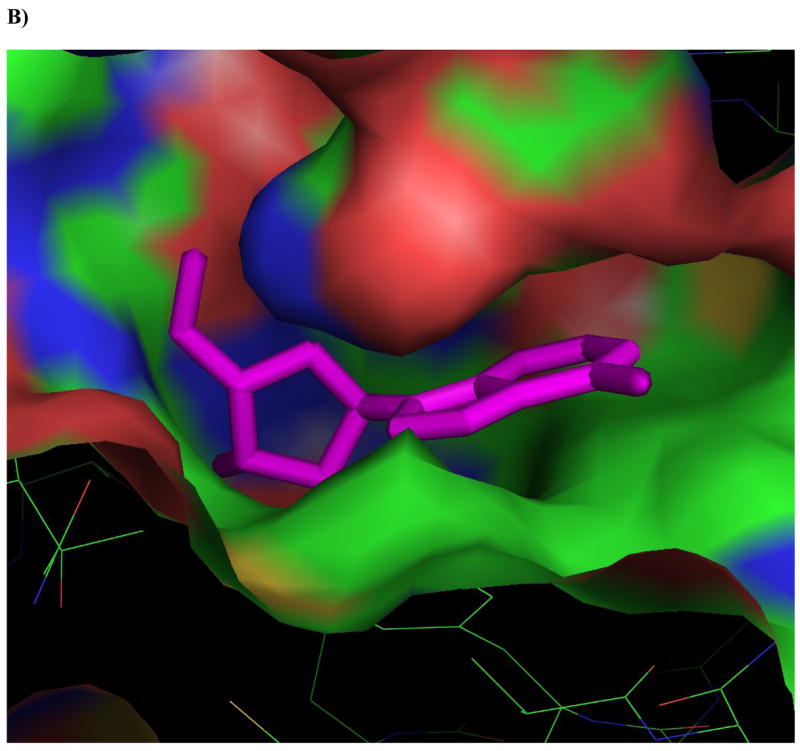
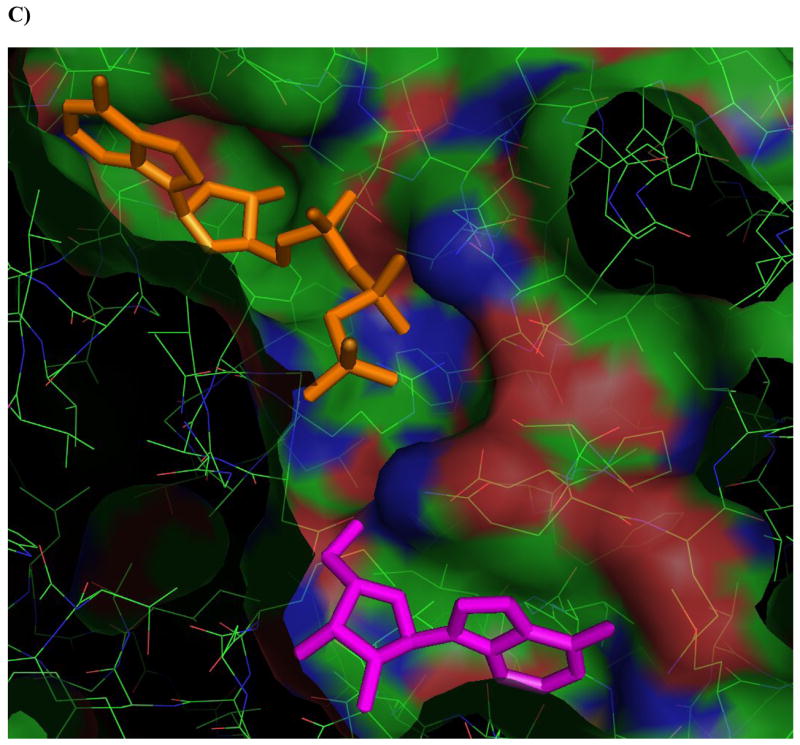
A) Adenosine (magenta) modeled into the active site from the perspective of the ribofuranosyl moiety. Phe 102 (right of center) forms the upper surface of the pocket where the ribofuranosyl moiety nests, while Asp 12 forms hydrogen bonds with the 2′ and 3′ hydroxyl groups (lower right). B) One of the better substrates, 9-[α-L-lyxofuranosyl]-adenine (34, Fig 2d) from a top-down perspective of looking downward from C1 and N7 of the adenine base. The area above this region is open and available for modifications. The yellow lining the pocket near the 2-position of the Ade base is Met 121, this is the pocket in which a 2-methyl or 2-fluoro group would rest. The 2′ and 3′-hydroxyl groups are clearly nested within the ribofuranosyl pocket, and there is plenty of room to rotate the 5′-hydroxyl so that it is trans to the adenine base, as in the case of 9-[α-L-lyxofuranosyl]-adenine (shown). C) This image depicts 9-[α-L-lyxofuranosyl]-adenine (magenta) modeled with AMP-PCP (orange), a non-hydrolysable form of ATP, in the active site of Ado kinase, as they would appear when Ado kinase is in a closed conformation. The γ-phosphate of AMP-PCP is nestled within the anion hole in the active site.
The actual mechanism by which the γ-phosphate of ATP is transferred to Ado has yet to be elucidated. In other Ado kinases, including human, this transfer involves the deprotonation of the 5′-hydroxyl group by an aspartic acid residue, followed by a water-mediated nucleophilic attack of the γ-phosphate by the negatively charged O-5′ atom, and the products, AMP and ADP are formed via an SN2 reaction [20, 47]. Although the same mechanism has not yet been empirically proven for Ado kinase from M. tuberculosis, all of the players are in place, including the aspartic acid residue (Asp 257, in M. tuberculosis Ado kinase), two water molecules, and an anion hole near the γ-phosphate of ATP (Figure 4C) [20]. One major difference in the mechanism of phosphorylation between the human and M. tuberculosis enzymes involves the binding of ATP to the active site. In the human enzyme, the binding of Ado to the active site induces a conformational change that optimizes the configuration of the anion hole to accommodate ATP. There is no evidence that M. tuberculosis Ado kinase undergoes a similar conformational change upon binding ATP [20].
Integrating the results of this SAR with previous studies of modifications to the Ade moiety, a recurring result was that a combination of a substitution such as a fluorine atom or amino group at the 2-position with a modification at another site on either the base or sugar moieties resulted in compounds that were more selective for M. tuberculosis than human Ado kinase (Table 3). This held true even when the substitution was a fluorine atom, although as a single substitution 2-fluoro-Ado was more active in human than in M. tuberculosis Ado kinase [31]. The compound sets carbocyclic-Ado (3) vs 2-amino-carbocyclic-Ado (4), araA (15) vs 2-fluoro-araA (16), and 9-[α-L-lyxofuranosyl]-adenine (34) vs 9-[α-L-lyxofuranosyl]-2-fluoro-adenine (35) illustrate this point best. For compounds that exhibit substrate activity in both enzymes, addition of a small, exocyclic substitution at the 2-position improves selectivity for the M. tuberculosis enzyme and reduces the possibility that the compound will be a substrate for Ado deaminase. Two of the major challenges of developing Ado analogs as antitubercular drugs are attempting to circumvent catabolism by host enzymes like Ado deaminase, and to avoid host toxicity. One example of this is the liberation of 2-fluoro-adenine by methylthioadenosine phosphorylase (MTAP). Some of our most promising compounds, including 9-[α-L-lyxofuranosyl]-2-fluoro-adenine (35) and 9- [β-D-5-methyl-(talo)-ribofuranosyl]-2-fluoro-adenine (37), are known substrates for this host-associated protein [48]. In addition to MTAP, there are a multitude of host and bacterial enzymes that utilize nucleosides and nucleotides that could result in inactivation of the compound or host toxicity.
Table 3.
Effect of exocyclic 2-substituted Ado analogs on enzyme selectivity
| Ado kinase Specific Activity (% of Ado control) | |||
|---|---|---|---|
| Compound | M. tuberculosis | Human | Ratioa |
| Single substitutions: | |||
| Adenosine | 100 | 100 | 1 |
| 2-Fluoro-Adob | 52 | 75 | 0.7 |
| 2-Chloro-Adob | 12 | 0.7 | 17 |
| 2-Amino-Adob | 6 | 5 | 1.2 |
| 2-Methyl-Adob | 1.9 | 0.2 | 9.5 |
| Effect of multiple substitutions: | |||
| 3-deaza-Adoc | 0.03 | <0.04 | >0.8 |
| 2-Fluoro-3-deaza-Adoc | 1.7 | <0.04 | 42 |
| 8-aza-Adob | 4 | 38 | 0.1 |
| 2-Fluoro-8-aza-Adob | 3.8 | 9 | 0.4 |
| Carbocyclic-Ado | 38 | 36 | 1.1 |
| 2-Amino-carbocyclic-Ado | 0.4 | <0.08 | >5 |
| araA | 0.6 | 0.2 | 3 |
| 2-Fluoro-araA | 0.9 | <0.04 | >22 |
| 9-[α-L-Lyxofuranosyl]-adenine | 3.8 | 15 | 0.2 |
| 2-Fluoro-9-[α-L-lyxofuranosyl]-adenine | 3.2 | 0.03 | 107 |
| Formycin Ab | 95 | 25 | 3.8 |
| 2-Fluoro-formycin Ab | 55 | <0.04 | >1400 |
| 2-Amino-formycin Ab | 45 | 0.6 | 75 |
The ratio is calculated as (% of Ado control) for M. tuberculosis/human. A ratio of >1 is more selective for M. tuberculosis Ado kinase while a ratio of <1 is more selective for human Ado kinase.
Values are adapted from a previous study of modifications to the adenine base of Ado [32].
Values are adapted from a previous study of 3-deaza-Ado analogs [31].
Although the ribofuranosyl moiety offered fewer opportunities for modification than the adenine moiety, the results are still promising for drug development (results are summarized in Table 4). Even compounds that are good substrates may not have antitubercular activity if they do not inhibit a critical downstream target, or if they are metabolized by other enzymes in the purine pathway. Conversely, MIC studies illustrated that compounds need not be excellent substrates in order to exert an antimycobacterial effect in an Ado kinase-dependent manner. Indeed, 9-[α-L-lyxofuranosyl]-2-fluoro-Ade (35) and 9-[β-D-5-methyl-(allo)-ribofuranosyl]-2-fluoro-Ade (38) both had Ado kinase-dependent antimycobacterial activity even though their substrate activity was low. As long as the phosphorylated product is potent enough to inhibit a critical downstream target, a poor Ado kinase substrate may still have antitubercular activity. For active substrates, MIC studies like the one presented here will give insight into whether or not Ado kinase is involved in the mechanism of action of the compound. This SAR was critical for completing studies of the Ado binding domain of M. tuberculosis Ado kinase, and permitting the design and synthesis of more active substrates.
Table 4.
Summary of results of modifications to the ribofuranosyl moiety
| Furan conformation There was an absolute requirement for the sugar moiety to be in a furanosyl-like conformation. Neither of the acyclo-adenosine analogs tested (50–51), nor any of the twelve pyranoses assayed (52–63) were substrates for Ado kinase. It is unlikely that many of these compounds were recognized by the active site despite the presence of the adenine base as evidenced by the fact that only 9- (2-deoxy-β-D-erythro-pentopyranosyl)-adenine (56) poorly inhibited the phosphorylation of 0.1 μM Ado at 100 μM. None of the other pyranoses or acyclo-Ado analogs were either substrates for, or inhibitors of, M. tuberculosis Ado kinase. Only modifications that maintained a β-D-ribofuranosyl-like conformation were substrates for M. tuberculosis Ado kinase. |
| 2′ A trans-2′-hydroxyl group was preferred, although a cis-2′-hydroxyl group (araA, 15) and 2′-deoxy-Ado (9) were also poor substrates. Compounds with 2′-modifications were poor inhibitors unless they also had a second substitution at the 2-position. |
| 3′ A trans-3′-hydroxyl group was nearly a requirement for substrate and inhibitor recognition, although 9-[β-D-xylofuranosyl]-adenine (23), which has a cis-3′-hydroxyl group, was a poor substrate with 1 nmol/mg-min of activity. Modifications at this position may be limited by steric hindrance. |
| 4′-O The 4′-oxygen was the most flexible of the substitutions to the ribose moiety. Carbocyclic-Ado (3) was a good substrate for both human and M. tuberculosis Ado kinases. The selectivity for carbocyclic-Ado analogs can be improved with the addition of an exocyclic substitution at the 2-position of the adenine base. |
| 5′ Substitutions to the 5-position were promising in terms of substrate activity and antitubercular activity. The best substrates in this category were 9-[α-L- lyxofuranosyl]-adenine (34) and 9-[α-L-lyxofuranosyl]-2-fluoroadenine (35), both maintained about 3% of the activity of Ado. Compounds with an additional 5′-methyl group in the allo and talo conformations (36–39) revealed that the allo- conformations resulted in better substrates and inhibitors of Ado kinase. Although it has yet to be confirmed, 5′-amino-5′-deoxy-Ado (32) appeared to be a substrate for this enzyme. This compound was also the best inhibitor of all of the substitutions to the ribose moiety. |
| Glycosidic bond position The preferred position for the N-glycosidic bond was at the 9-position of Ado. However, activity was measured with 3-[β-D-ribofuranosyl]-adenine (47) as well. Ado kinase did not recognize compounds as substrates when the bond was moved to the 7 or 8-positions on Ado (45, 46, and 48). |
Acknowledgments
The authors would like to thank Paula W. Allan (Southern Research Institute, Birmingham, Alabama) for her technical assistance, Dr. Mahmoud el Kouni from the department of pharmacology and toxicology at the University of Alabama at Birmingham (Birmingham, Alabama) for his valuable advice and contribution of compounds, Dr. Joseph Spychala from the department of pharmacology at University of North Carolina (Chapel Hill, North Carolina) for generously providing the human adenosine kinase clone, Jim Riordan and David Poon from Southern Research chemical repository for their assistance with identifying and validating the chemical structures of nucleoside analogs from Southern Research Institute, Dr. Fardos Naguib from the department of pharmacology and toxicology at the University of Alabama at Birmingham and Lucile White from Southern Research Institute for their assistance in interpreting our Ki results. This work was supported by NIH research grant AI43241.
Abbreviations
- Ade
adenine
- Ado
adenosine
- AD
adenosine deaminase
- araA
9-[β-D-arabinofuranosyl]-adenine
- AMP-PCP
β,γ-methyleneadenosine-5′-triphosphate
- BSA
bovine serum albumin
- DTT
dithiothreitol
- formycin A
8-aza-9-deaza-Ado
- IPTG
isopropyl-β-D-thiogalactopyranoside
- methyl-Ado
2-methyl-adenosine
- MTAP
methylthioadenosine phosphorylase
- MIC
95% minimum inhibitory concentration
- PBS
phosphate buffered saline
- Pi
inorganic phosphate
- PMSF
phenylmethylsulfonyl fluoride
- SAR
structure-activity relationship
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Spychala J, Datta NS, Takakbayashi K, Datta M, Fox IH, Gribbin T, et al. Cloning of human adenosine kinase cDNA: sequencec similarity to microbial ribokianses and fructokinases. Proceedings of the National Academy of Sciences USA. 1996;93:1232–7. doi: 10.1073/pnas.93.3.1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boison D. Adenosine kinase, epilepsy and stroke: mechanisms and therapies. Trends Pharmacol Sci. 2006;27:652–8. doi: 10.1016/j.tips.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 3.Pignataro G, Simon RP, Boison D. Transgenic overexpression of adenosine kinase aggravates cell death in ischemia. J Cereb Blood Flow Metab. 2007;27:1–5. doi: 10.1038/sj.jcbfm.9600334. [DOI] [PubMed] [Google Scholar]
- 4.Ugarkar BG, Castellino AJ, DaRe JS, Ramirez-Weinhouse M, Kopcho JJ, Rosengren S, et al. Adenosine kinase inhibitors. 3 Synthesis, SAR, and antiinflammatory activity of a series of l-lyxofuranosyl nucleosides. J Med Chem. 2003;46:4750–60. doi: 10.1021/jm030230z. [DOI] [PubMed] [Google Scholar]
- 5.Galazka J, Striepen B, Ullman B. Adenosine kinase from Cryptosporidium parvum. Mol Biochem Parasitol. 2006;149:223–30. doi: 10.1016/j.molbiopara.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 6.Kim YA, Sharon A, Chu CK, Rais RH, Al Safarjalani ON, Naguib FN, et al. Synthesis, biological evaluation and molecular modeling studies of N6-benzyladenosine analogues as potential anti-toxoplasma agents. Biochem Pharmacol. 2007;73:1558–72. doi: 10.1016/j.bcp.2007.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khanolkar SR, Wheeler PR. Purine metabolism in Mycobacterium leprae grown in armadillo liver. FEMS Microbiology Letters. 1983;20:273–8. [Google Scholar]
- 8.Wheeler PR. Enzymes for purine synthesis and scavenging in pathogenic mycobacteria and their distribution in Mycobacterium leprae. Journal of General Microbiology. 1987;133:3013–8. doi: 10.1099/00221287-133-11-3013. [DOI] [PubMed] [Google Scholar]
- 9.Wheeler PR. Biosynthesis and scavenging of purines by pathogenic mycobacteria Including Mycobacterium leprae. Journal of General Microbiology. 1987;133:2999–3011. doi: 10.1099/00221287-133-11-2999. [DOI] [PubMed] [Google Scholar]
- 10.Long MC, Escuyer V, Parker WB. Identification and characterization of a unique adenosine kinase from Mycobacterium tuberculosis. Journal of Bacteriology. 2003;185:6548–55. doi: 10.1128/JB.185.22.6548-6555.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wheeler PR, Ratledge C. Metabolism of Mycobacterium tuberculosis. In: Bloom BR, editor. Tuberculosis pathogenesis, protection, and control. Washington, DC: ASM Press; 1994. pp. 353–85. [Google Scholar]
- 12.Meade CJ, Dumont I, Worrall L. Why do asthmatic subjects respond so strongly to inhaled adenosine? Life Sci. 2001;69:1225–40. doi: 10.1016/s0024-3205(01)01231-0. [DOI] [PubMed] [Google Scholar]
- 13.Blackburn MR. Too much of a good thing: adenosine overload in adenosine-deaminase-deficient mice. Trends Pharmacol Sci. 2003;24:66–70. doi: 10.1016/S0165-6147(02)00045-7. [DOI] [PubMed] [Google Scholar]
- 14.Blackburn MR, Lee CG, Young HW, Zhu Z, Chunn JL, Kang MJ, et al. Adenosine mediates IL-13-induced inflammation and remodeling in the lung and interacts in an IL-13-adenosine amplification pathway. J Clin Invest. 2003;112:332–44. doi: 10.1172/JCI16815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Markowitz M, Morales-Ramirez JO, Nguyen BY, Kovacs CM, Steigbigel RT, Cooper DA, et al. Antiretroviral activity, pharmacokinetics, and tolerability of MK-0518, a novel inhibitor of HIV-1 integrase, dosed as monotherapy for 10 days in treatment-naive HIV-1-infected individuals. J Acquir Immune Defic Syndr. 2006;43:509–15. doi: 10.1097/QAI.0b013e31802b4956. [DOI] [PubMed] [Google Scholar]
- 16.Zhong H, Shlykov SG, Molina JG, Sanborn BM, Jacobson MA, Tilley SL, et al. Activation of murine lung mast cells by the adenosine A3 receptor. J Immunol. 2003;171:338–45. doi: 10.4049/jimmunol.171.1.338. [DOI] [PubMed] [Google Scholar]
- 17.Sassetti CM, Rubin EJ. Genetic requirements for mycobacterial survival during infection. Proc Natl Acad Sci U S A. 2003;100:12989–94. doi: 10.1073/pnas.2134250100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maj M, Singh B, Gupta RS. The influence of inorganic phosphate on the activity of adenosine kinase. Biochim Biophys Acta. 2000;1476:33–42. doi: 10.1016/s0167-4838(99)00220-4. [DOI] [PubMed] [Google Scholar]
- 19.Maj MC, Singh B, Gupta RS. Pentavalent ions dependency is a conserved property of adenosine kinase from diverse sources: identification of a novel motif implicated in phosphate and magnesium ion binding and substrate inhibition. Biochemistry. 2002;41:4059–69. doi: 10.1021/bi0119161. [DOI] [PubMed] [Google Scholar]
- 20.Reddy MC, Palaninathan SK, Shetty ND, Owen JL, Watson MD, Sacchettini JC. High Resolution Crystal Structures of Mycobacterium tuberculosis Adenosine Kinase: Insights into the mechanism and specificity of this novel prokaryotic enzyme. J Biol Chem. 2007;282:27334–42. doi: 10.1074/jbc.M703290200. [DOI] [PubMed] [Google Scholar]
- 21.Wang Y, Long MC, Ranganathan S, Escuyer V, Parker WB, Li R. Overexpression, purification and crystrallographic analysis of a unique adenosine kinase from Mycobacterium tuberculosis. Acta Crystallographica Section F. 2005;F61:553–7. doi: 10.1107/S1744309105013473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Andersson CE, Mowbray SL. Activation of ribokinase by monovalent cations. J Mol Biol. 2002;315:409–19. doi: 10.1006/jmbi.2001.5248. [DOI] [PubMed] [Google Scholar]
- 23.Drobniewski F, Balabanova Y, Nikolayevsky V, Ruddy M, Kuznetzov S, Zakharova S, et al. Drug-resistant tuberculosis, clinical virulence, and the dominance of the Beijing strain family in Russia. Jama. 2005;293:2726–31. doi: 10.1001/jama.293.22.2726. [DOI] [PubMed] [Google Scholar]
- 24.Espinal MA, Laszlo A, Simonsen L, Boulahbal F, Kim SJ, Reniero A, et al. Global trends in resistance to antituberculosis drugs. World Health Organization-International Union against Tuberculosis and Lung Disease Working Group on Anti-Tuberculosis Drug Resistance Surveillance. N Engl J Med. 2001;344:1294–303. doi: 10.1056/NEJM200104263441706. [DOI] [PubMed] [Google Scholar]
- 25.Nettleman MD. Multidrug-resistant tuberculosis news from the front. Journal of the American Medical Association. 2005;293:2788–90. doi: 10.1001/jama.293.22.2788. [DOI] [PubMed] [Google Scholar]
- 26.The Centers for Disease Control and Prevention (CDC) Emergence of Mycobacterium tuberculosis with extensive resistance to second-line drugs--worldwide, 2000–2004. MMWR Morb Mortal Wkly Rep. 2006;55:301–5. [PubMed] [Google Scholar]
- 27.Grinsztejn B, Nguyen BY, Katlama C, Gatell JM, Lazzarin A, Vittecoq D, et al. Safety and efficacy of the HIV-1 integrase inhibitor raltegravir (MK-0518) in treatment-experienced patients with multidrug-resistant virus: a phase II randomised controlled trial. Lancet. 2007;369:1261–9. doi: 10.1016/S0140-6736(07)60597-2. [DOI] [PubMed] [Google Scholar]
- 28.Barrow EW, Westbrook L, Bansal N, Suling WJ, Maddry JA, Parker WB, et al. Antimycobacterial activity of 2-methyl-adenosine. Journal of Antimicrobial Chemotherapy. 2003;52:801–8. doi: 10.1093/jac/dkg444. [DOI] [PubMed] [Google Scholar]
- 29.Parker WB, Barrow EW, Allan PW, Shaddix SC, Long MC, Barrow WW, et al. Metabolism of 2-methyladenosine in Mycobacterium tuberculosis. Tuberculosis. 2004;84:327–36. doi: 10.1016/j.tube.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 30.Long MC, Allan PW, Luo MZ, Liu MC, Sartorelli AC, Parker WB. Evaluation of 3-deaza-adenosine analogues as ligands for adenosine kinase and inhibitors of Mycobacterium tuberculosis growth. J Antimicrob Chemother. 2007;59:118–21. doi: 10.1093/jac/dkl448. [DOI] [PubMed] [Google Scholar]
- 31.Long MC, Parker WB. Structure-activity relationship for nucleoside analogs as inhibitors or substrates of adenosine kinase from Mycobacterium tuberculosis. I Modifications to the adenine moiety. Biochem Pharmacol. 2006;71:1671–82. doi: 10.1016/j.bcp.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 32.Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 33.Hurley MC, Lin B, Fox IH. Regulation of deoxyadenosine and nucleoside analog phosphorylation by human placental adenosine kinase. J Biol Chem. 1985;260:15675–81. [PubMed] [Google Scholar]
- 34.Schnebli HP, Hill DL, Bennett LL., Jr Purification and properties of adenosine kinase from human tumor cells of type H. Ep. No.2. J Biol Chem. 1967;242:1997–2004. [PubMed] [Google Scholar]
- 35.Miller RL, Adamczyk DL, Miller WH, Koszalka GW, Rideout JL, Beacham LM, 3rd, et al. Adenosine kinase from rabbit liver. II Substrate and inhibitor specificity. J Biol Chem. 1979;254:2346–52. [PubMed] [Google Scholar]
- 36.Bennett LL, Jr, Hill DL. Structural requirements for activity of nucleosides as substrates for adenosine kinase: orientation of substituents on the pentofuranosyl ring. Mol Pharmacol. 1975;11:803–8. [PubMed] [Google Scholar]
- 37.Carrasco L, Vazquez D. Molecular bases for the action and selectivity of nucleoside antibiotics. Med Res Rev. 1984;4:471–512. doi: 10.1002/med.2610040403. [DOI] [PubMed] [Google Scholar]
- 38.Bennett LL, Jr, Allan PW, Hill DL. Metabolic studies with carbocyclic analogs of purine nucleosides. Mol Pharmacol. 1968;4:208–17. [PubMed] [Google Scholar]
- 39.Bennett LL, Jr, Bowdon BJ, Allan PW, Rose LM. Evidence that the carbocyclic analog of adenosine has different mechanisms of cytotoxicity to cells with adenosine kinase activity and to cells lacking this enzyme. Biochem Pharmacol. 1986;35:4106–9. doi: 10.1016/0006-2952(86)90036-5. [DOI] [PubMed] [Google Scholar]
- 40.Cottam HB, Wasson DB, Shih HC, Raychaudhuri A, Di Pasquale G, Carson DA. New adenosine kinase inhibitors with oral antiinflammatory activity: synthesis and biological evaluation. J Med Chem. 1993;36:3424–30. doi: 10.1021/jm00074a024. [DOI] [PubMed] [Google Scholar]
- 41.Miller RL, Adamczyk DL, Miller WH. Adenosine kinase from rabbit liver. I Purification by affinity chromatography and properties. J Biol Chem. 1979;254:2339–45. [PubMed] [Google Scholar]
- 42.Newby AC. The interaction of inhibitors with adenosine metabolising enzymes in intact isolated cells. Biochem Pharmacol. 1981;30:2611–5. doi: 10.1016/0006-2952(81)90589-x. [DOI] [PubMed] [Google Scholar]
- 43.Iltzsch MH, Uber SS, Tankersley KO, el Kouni MH. Structure-activity relationship for the binding of nucleoside ligands to adenosine kinase from Toxoplasma gondii. Biochem Pharmacol. 1995;49:1501–12. doi: 10.1016/0006-2952(95)00029-y. [DOI] [PubMed] [Google Scholar]
- 44.Bork P, Sander C, Valencia A. Convergent evolution of similar enzymatic function on different protein folds: the hexokinase, ribokinase, and galactokinase families of sugar kinases. Protein Sci. 1993;2:31–40. doi: 10.1002/pro.5560020104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maj MC, Gupta RS. The effect of inorganic phosphate on the activity of bacterial ribokinase. J Protein Chem. 2001;20:139–44. doi: 10.1023/a:1011081508171. [DOI] [PubMed] [Google Scholar]
- 46.Chuvikovsky DV, Esipov RS, Skoblov YS, Chupova LA, Muravyova TI, Miroshnikov AI, et al. Ribokinase from E. coli: expression, purification, and substrate specificity. Bioorg Med Chem. 2006;14:6327–32. doi: 10.1016/j.bmc.2006.05.057. [DOI] [PubMed] [Google Scholar]
- 47.Schumacher MA, Scott DM, Mathews II, Ealick SE, Roos DS, Ullman B, et al. Crystal structures of Toxoplasma gondii adenosine kinase reveal a novel catalytic mechanism and prodrug binding. J Mol Biol. 2000;298:875–93. doi: 10.1006/jmbi.2000.3753. [DOI] [PubMed] [Google Scholar]
- 48.Parker WB, Allan PW, Ealick SE, Sorscher EJ, Hassan AE, Silamkoti AV, et al. Design and evaluation of 5′-modified nucleoside analogs as prodrugs for an E. coli purine nucleoside phosphorylase mutant Nucleosides Nucleotides. Nucleic Acids. 2005;24:387–92. doi: 10.1081/ncn-200059807. [DOI] [PubMed] [Google Scholar]